

Conspectus
The peroxidation of lipids in biological membranes has been implicated in both the onset and development of most degenerative diseases. The primary products of this autoxidation process are usually lipid hydroperoxides. They form as a consequence of a free radical chain reaction: lipid peroxyl radicals propagate the chain by rate-limiting H-atom abstraction from another lipid. Studies of the mechanism of lipid peroxidation are a specific part of a wider effort to understand the more general phenomenon of hydrocarbon autoxidation, which dates back some 70 years. However, the autoxidation of lipids is generally much more complicated than that of other hydrocarbons because of additional reaction pathways afforded by a variety of uniquely positioned unsaturated bonds. Indeed, polyunsaturation is an important aspect of many of the most relevant of physiological lipids, such as linoleate and arachidonate. In this Account, we present our current understanding of the mechanism of unsaturated lipid peroxidation, effectively updating our Account on the same topic published 25 years ago. Our more recent work has, in large part, been stimulated by the discovery of the non-conjugated linoleate hydroperoxide as a product under certain autoxidation conditions. The identification of this long-elusive bis-allylic hydroperoxide prompted our kinetic characterization of the reaction leading to its formation. The product distributions obtained from autoxidations of newly synthesized model compounds, which vary in either the substitution of the bis-allylic moiety or the configuration of the double bonds, have provided key insights into the overall mechanism. These insights have in turn been reinforced by the results of theoretical calculations. The picture that emerges is one wherein the delocalized carbon-centered radicals, which arise as intermediates in these reactions, first associate with dioxygen to form pre-reaction complexes. These complexes then collapse through transition state structures that maximize the orbital interactions between the incipient diene moieties and the peroxyl radical singly occupied molecular orbital (SOMO). The energies of these transition states are influenced by steric effects; thus, there are distinct changes in product distribution in the autoxidation of dienes having different substitution patterns. The radical:dioxygen complexes are also intermediates in the isomerization of allylperoxyl and pentadienylperoxyls, helping explain the high regio- and stereochemical fidelity of these processes. We have taken advantage of the rapid fragmentation of non-conjugated peroxyl radicals to develop a powerful peroxyl radical clock methodology, which can be used to determine rate constants for reactions of peroxyl radicals with molecules having rate constants ranging from 1 to 107 M−1s−1. We can make use of this methodology to address various questions, both fundamental and applied, relating to lipid peroxidation and its inhibition by radical-trapping antioxidants.
Introduction
Unsaturated organic molecules with weak C-H bonds are particularly prone to undergo autoxidation, a process that proceeds by a free radical chain mechanism. Autoxidation of polyunsaturated fatty acid esters and sterols, known as lipid peroxidation, has attracted increased research attention over the last few decades. One reason for this interest is due to the unique role that lipid-derived peroxides play in biology, both as modulators of enzymes and as intermediates in biosynthetic processes. The study of free radical autoxidation has a history dating from the 1940s2 and its relevance to issues in chemistry and biology continues to grow. “Oxidative stress” has importance in pathologies as diverse as aging, cancer, as well as in cardiovascular and neurodegenerative diseases.
The primary products of autoxidation are peroxides or hydroperoxides, but these compounds are frequently unstable and decompose to aldehydes, ketones and other reactive substructures. The mechanism for the process, shown in Scheme 1, involves a very fast3 reaction of a chain-carrying carbon radical with oxygen and a slow rate-determining propagation step in which a peroxyl radical abstracts hydrogen from an organic substrate, (Eq. 2)4,5 Peroxyl radicals can also undergo propagation by addition reactions to alkenes,6 (Eq. 3). Styrene, for example, readily forms an alternating addition co-polymer with oxygen. If the intermediate peroxyl radicals are able to undergo a 5-exo cyclization onto an alkene, then cyclic peroxide products can form.7 Phenolic inhibitors of autoxidation (antioxidants) compete for peroxyl radicals by H-atom transfer, see Scheme 1B. The generated aryloxy radical in the transformation described by Eq. 4 traps a second peroxyl and gives non-radical products, ending the chain (Eq. 5).
Scheme 1.

Peroxidation of homoconjugated dienes
The autoxidation of linoleic acid, 1, or linoleate esters in solution or in liposomes has been the focus of studies by our group over the course of three decades. Linoleate is the simplest lipid containing the prototypical homoconjugated diene substructure of polyunsaturated fatty acids and esters and the four hydroperoxide products, 5a, 5b, 6a and 6b are the established primary products. The early proposals for the mechanism of linoleate autoxidation centered on the pentadienyl radical 2 that is formed by abstraction of the weak C-H bond in linoleate, see Scheme 2. This radical is presumed to add oxygen at either end to give conjugated diene peroxyl radicals that lead, ultimately, to the conjugated diene hydroperoxides. The distribution of the Z,E and E,E products can be understood based upon a mechanism that involves competitive H-atom abstraction by peroxyls and reversible addition of oxygen to intermediate pentadienyls.1,8
Scheme 2.

A longstanding problem with the mechanism shown in Scheme 2 is the fact that no linoleate products are observed that result from oxygen addition at the center position of the pentadienyl radical. ESR studies as well as calculations suggest that the spin density at the center carbon of simple pentadienyl radicals is higher than at the terminal carbons and one therefore expects that the bis-allylic hydroperoxide 7 should form.9,10 However, extensive efforts in our laboratories to find 7 in linoleate autoxidation product mixtures failed. Most of these failed studies involved autoxidation of methyl linoleate at low temperatures in the presence of good hydrogen atom donors such as 1,4-cyclohexadiene or phenols. A particularly revealing experiment, however, came from Alan Brash, a colleague in the Division of Clinical Pharmacology at Vanderbilt. Alan studies lipoxygenase enzymes and transformations of their hydroperoxide products among other things, and on this occasion Alan required samples of an allylic 8-hydroperoxide of linoleate, reported as a minor product of linoleate autoxidation. He carefully reexamined a sample containing a mixture of methyl linoleate and 5 to 10% of α–tocopherol that had been stored at −20 °C for approximately four years following oxidation studies. A search for non-conjugated diene monohydroperoxides by HPLC found none of the desired 8-hydroperoxide. However, under the prominent peak of one of the conjugated diene products, Alan uncovered significant amounts of the bis-allylic 11-hydroperoxy-linoleate 7, which he proceeded to characterize by UV, GC-MS and NMR.11
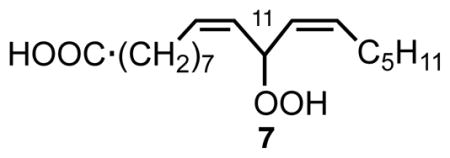
The Brash experiment stimulated us to expand our studies on the co-oxidation of linoleate esters with α–TOH to assess the effect of the antioxidant on the formation of 7. We found that 7 is indeed formed in the co-autoxidation of methyl linoleate and α–TOH and the amount formed depends on the concentration of α–TOH present during the oxidation, as shown in Figure 1.12 The Figure shows that substantial amounts of 7 form, but only in co-oxidations having α–TOH concentrations of 0.1 to 1M. In fact, 7 is the major product at the kinetic limit, consistent with the greater spin density at the bis-allylic carbon of the pentadienyl radical.
Figure 1.
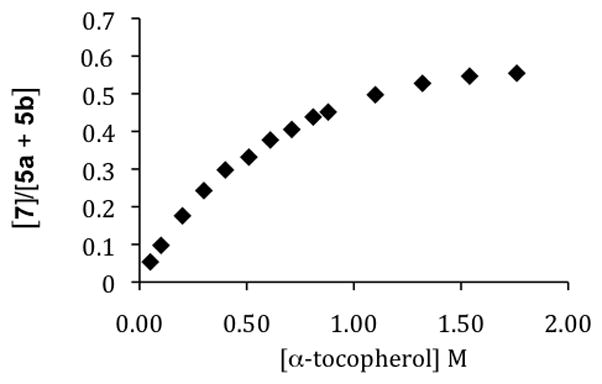
Methyl linoleate Product Composition vs. α-Tocopherol.
Antioxidants, most commonly substituted phenols (ArOH), effectively intercept peroxyls by transferring the phenolic H-atom to a propagating peroxyl radical with a rate constant, kinh, that is faster than the rate of chain propagation, kp13 (Scheme 1, eq 4 and 2). The propagation rate constant for peroxidation of linoleate (62 M−1s−1)6 is some 5 · 104 less than the inhibition rate constant for α-TOH, 3.5 · 106 M−1s−1 at 37°C.13–15 The kinetic analysis of this system is based upon a reasonably straightforward competition experiment similar to studies we have carried out for other chain oxidations. The expression that links product ratio vs. [α-TOH] is shown in Scheme 3. A fit of data shown in Figure 1 gives values for the oxygen partition factor α and kβ, the rate for loss of oxygen from the deconjugated peroxyl radical (α = .45; kβ = 2.6 · 106 s−1 at 37°C). The study outlined here establishes the rate constant for conversion of radical 8 to the conjugated diene peroxyls by timing it with a known bimolecular rate process, the transfer of an H-atom from α-TOH to 8. The unimolecular process indicated by kβ will be used in work described elsewhere in this Account to clock H-atom transfer rates of other antioxidants. This process is shown in Scheme 3 as a reversible addition of oxygen to the intermediate pentadienyl radical.
Scheme 3.

At high concentrations of α-TOH, peroxyl radical 8 is trapped to give 7 while at lower concentrations of antioxidant, rearrangement of the peroxyl occurs and this leads to the conjugated diene peroxyls. The conjugated diene peroxyls abstract hydrogen and ultimately give the conjugated diene hydroperoxides 5a and 5b. The Brash experiment provided the lead to unraveling this complex mechanistic picture and in retrospect its design could not have been more favorable. High concentrations of α-TOH in neat linoleate are conditions that favor formation of 7 relative to the conjugated diene products. In fact, one wonders whether substantially more of product 7 was formed over the course of 4 years at −20°C than was generated during the initial experiment at room temperature.
The mechanism that we proposed (Scheme 3) to account for the effect of α-TOH on linoleate products raised many questions. We immediately sought more information on the relative stabilities of the different peroxyl radicals that are formed as intermediates in these reactions.16,17 Using theoretical approaches, (ROB3P86/6-311G(d,p)//B3P86/-6311G(d,p)),18 we found that the non-conjugated peroxyl radical 8 has a C-OO bond that is almost 8 kcal/mol weaker than that in 3a and 3b, accounting for its ready fragmentation and why it could not be observed in the absence of such a large concentration of a strong peroxyl radical-trapping agent as α-TOH. Likewise, 3a and 3b had weaker (1.5 kcal/mol) C-OO bonds relative to 4a and 4b, accounting for their isomerization under conditions where they could equilibrate to thermodynamic products. While this provided a thermodynamic rationale for the product distribution, clearly a kinetic rationale was necessary to explain what first controls the site of oxygen addition to delocalized pentadienyl radicals.
To address this question, a series of dienes was studied in which a pentyl group was fixed on one end of the diene but the group at the other end of the structure was systematically changed. The results of this series of free radical oxidations (carried out in the presence and absence of α-TOH) led to the conclusion that the distribution of hydroperoxides formed from these dienes depends on the size of substituents at C-1 and C-5 of the substructure.19
The distribution of products can be understood based on simple steric effects of the substituents on the transition state for reaction of intermediate pentadienyl radicals with oxygen as illustrated in Scheme 4 with Newman projections of the transition states for oxygen addition at C-1 and C-5. We suggest that these transition states, along with the two shown for addition of oxygen at C-3, are preferred structures based on experiments discussed subsequently in this Account. When R is methyl, oxygen partitions to the intermediate pentadienyl radical in a C-1=0.41 to C-3=0.34 to C-5=0.25 ratio when the reaction is carried out in the presence of ~1M α-TOH. However, when R=tBu, that ratio is a C-1=0.06 to C-3=0.49 to C-5=0.45. In short, Taft Es parameters of substituents at C-1 and C-5 correlate with the distribution of products formed at C-1, C-3 and C-5 for reactions carried out in the presence of α-TOH. A bulky substituent at C-1 drives more oxygen addition to C-3 and C-5, whereas bulky substituents at both C-1 and C-5 lead to more products formed from addition at C-3. Good linear free energy plots for the product distribution vs. size of the R group support the notion that steric effects control the site of oxygen addition.
Scheme 4.

The effect of α-TOH on the ratio of non-conjugated to conjugated diene products formed from the dienes shown in Scheme 4 is understood based upon the mechanism outlined in Scheme 3. Non-conjugated diene product increases with α-TOH in a manner similar to the data shown in Figure 1. But the ratio of the two conjugated products formed from oxygen addition at C-1 and C-5 is not affected by α-TOH; the only effect of the antioxidant is to increase the amount of C-3 addition product formed. A good fraction of the conjugated diene products formed must have proceeded through first formation of the non-conjugated peroxyl radical 8, followed by a secondary conversion from this radical to the conjugated products. The fact that the C-1 to C-5 product ratio is unchanged by the concentration of α-TOH present during oxidation supports the notion that the pentadienyl radical reaction with oxygen is the product determining step in both the initial encounter and reaction of this species with oxygen as well as in the rearrangement of 8 to conjugated products, in support of the fragmentation mechanism proposed.
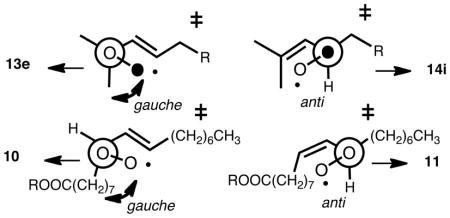
Additional experiments suggest that there is a preferred orientation for oxygen addition to the C-1 and C-5 centers of the delocalized pentadienyl radical such that secondary interactions between oxygen and the radical are maximized. Support for a preferred orientation in the transition state for oxygen addition is found in the oxidation of Z,E diene precursors such as 9 in the presence and absence of α-TOH (Scheme 5). Oxygen addition occurs preferentially at the end of the pentadienyl radical that minimizes gauche interactions in the preferred transition state. Under all conditions of oxidation studied, the Z,E conjugated diene product is favored by over 2:1 compared to the E,E product.
Scheme 5.

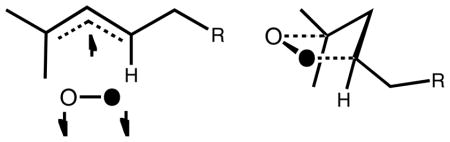
At first glance, it is perhaps unclear why the transition state structures in Schemes 4 and 5 are drawn as they are, and subsequently used to rationalize the reaction outcomes. Recent theoretical calculations have provided support for these transition state structures; shown above as being operative in these reactions. The preference arises due to the significant secondary orbital interaction between the pentadienyl and ground-state triplet dioxygen π MOs as in the doubly occupied HOMO shown in Figure 2.20 In fact, the enthalpy of the transition state structure of pentadienyl + O2 (calculated by ROCCSD(T)/6-311+G(2df,p)//B3LYP/6-311+G(2df,p)) wherein the dioxygen moiety is oriented toward the diene moiety is 4.1 kcal/mol lower than that oriented away from the diene (as in the other TS structures in Scheme 5). Therefore, both steric and electronic arguments are important in understanding product distributions from substituted diene oxidations.
Figure 2.

Secondary orbital interactions lead to a preferred geometry in the transition state structure for the reaction between pentadienyl radicals and molecular oxygen.
Peroxidation of mono-alkenes
While lipids containing the homoconjugated diene structure such as linoleate readily undergo free radical chain oxidation, mono-alkene fatty acid esters such as oleate or sterols such as cholesterol are much less prone to undergo peroxidation. The allylic C-H BDE of oleate is substantially higher than the bis-allylic center on linoleate, accounting for the lower reactivity. Cholesterol is also a mono-alkene but its alkene is trisubstituted and endocyclic, making the sterol somewhat more prone to oxidative attack by peroxyl radicals than is oleate. Six primary products of oleate oxidation are shown in Scheme 6, three resulting from H-atom abstraction at C-8, three from H-atom removal at C-11.21 Thus, H-atom removal at C-11 gives the three hydroperoxides, 10a–12a, while C-8 abstraction gives products 10b–12b. Abstraction of the C-11 hydrogen, for example, leads to the formation of three isomeric allylic peroxyl radicals, two of which are formed from oxygen addition to the first-formed allyl radical, the third resulting from a subsequent allylic rearrangement. These three allyl peroxyl radicals are converted to three allylic hydroperoxides, 10a–12a. An analogous mechanism accounts for the formation of the three 10b–12b products. The peroxyl radical allylic interconversions that account for the formation of the observed products are identified as [2,3] rearrangements in the Scheme. Rearrangements of allyl peroxyl radicals derived from acyclic lipids has been one of our interests and we report below our contributions to this field. We acknowledge here the fundamental work of the late Athel Beckwith on the rearrangement of cholesterol hydroperoxides22–24 as well as his studies in many other areas of free radical mechanistic chemistry over the course of decades.
Scheme 6.

The rearrangement may be considered as either a dissociative fragmentation-addition as shown in Scheme 3 for the rearrangement of the non-conjugated linoleate peroxyl radical 8 or as an associative [2,3] rearrangement in which oxygen and the three-carbon backbone do not separate, as presented in Scheme 7. Free radical promoted rearrangement of the oleate allylic hydroperoxides proceeds through the corresponding peroxyl radicals and the E oleate hydroperoxide (S)-10 rearranges to the corresponding (R)-E product 12 at room temperature with (S)→(R) selectivity substantially greater than 97% and with little incorporation of label when the reactions are run under an (18O)2 atmosphere, Scheme 7A.25,26 The Z hydroperoxide (R)-11 gives product 10 with somewhat reduced selectivity (~90%) and with the configurational course being (R)→(R) in this case, Scheme 7B. Rearrangement of both (S)-10 and (R)-11 takes place with less stereochemical fidelity and with more incorporation of atmospheric oxygen when the temperature is raised to 40°C. The experiments therefore support the associative mechanism, but the incorporation of some atmospheric oxygen into the hydroperoxides at 40°C does indicate that dissociation of the peroxyl radicals and reincorporation of atmospheric oxygen can occur.
Scheme 7.

Subsequent experiments showed that solvent viscosity plays a role in the selectivity of the rearrangements.27,28 Thus, rearrangement of unlabeled 16O (R)-11 in hexane under an atmosphere of (18O)2 gives 16O labeled 10 with high stereofidelity (R:S=90:10). But 13% of the product formed under these conditions is a racemic mixture of 18O labeled 10, formed by dissociation and exchange of hydroperoxide oxygen with the atmosphere. In octadecane under otherwise identical conditions, the stereoselectivity of unlabeled 16O 10 formed increases to 99:1 but with only 3% of racemic product formed with incorporation of atmospheric 18O. Increasing temperature led to the formation of more racemic product that had incorporated atmospheric oxygen.
A particularly illuminating experiment was carried out by Jennifer (Lowe) Allen, who studied the synthesis and rearrangement of the unsymmetrically labeled29 hydroperoxide 13 shown in Scheme 8.30 At 20°C under an atmosphere of (16O)2 13e (e refers to an external oxygen label) rearranges to a mix of products containing 90% 14i (i refers to an internal oxygen label), 0.3% 14e (external label) with the remainder being 14u (u indicates unlabeled hydroperoxide). At 60°C, the rearrangement proceeds with significantly lower (e→i) selectivity and with substantially more incorporation of atmospheric oxygen, 14i:e:u = 64:6:30. Rearrangement at 20°C of the internally labeled 14i back to 13 as shown in Scheme 8B occurs with more incorporation of atmospheric oxygen and with lower regioselectivity of the label (i→e) than is observed in the rearrangement of 13→14.
Scheme 8.

It is of interest to consider the results of the [2,3] rearrangement studies on the oleate hydroperoxides as well as those of 13 and 14 with the studies on steric effects on transition states for oxygen addition and Taft Es parameters in mind. We suggest that the high regioselectivity observed in the rearrangement of 13→14 results from the fact that formation of the transition state leading to 14 is favorable since this structure has no gauche interactions with the alkene substituent. In contrast, the rearrangement of 14→13 proceeds through a transition state that has such a destabilizing gauche interaction. The rearrangement in this direction proceeds with more scrambling of the label and more incorporation of atmospheric oxygen because of this steric effect. The results of rearrangement in the oleate series are also consistent with this picture. Rearrangements that proceed through transition states having the anti arrangement proceed with more stereochemical fidelity and less incorporation of atmospheric oxygen than those rearrangements that proceed by formation of a transition state having a gauche steric interaction.
Again this picture is supported by theoretical calculations that indicate the low energy transition state structures that connect allylperoxyl radicals have a much preferred geometry which places the dioxygen toward the allylic moiety resulting from a secondary orbital interaction between the dioxygen and allyl π MOs. In the case of allyl + O2, the “toward” TS is preferred over the “away” TS by 5.1 kcal/mol (Figure 3).20
Figure 3.

Secondary orbital interactions lead to a preferred geometry in the transition state structure for the reaction between allyl radicals and molecular oxygen.
A picture for rearrangement that emerges from these studies is fragmentation of the allyl peroxyl radical to an allyl radical-oxygen complex of defined geometry that can dissociate to molecular oxygen and the allyl radical. The complex is also one in which its collapse to the rearranged allylperoxyl radical is competitive with oxygen escape, a process that accounts for the observation of regioselective transfer of oxygen label in the rearrangements linking 13 and 14. The structure and energetics of allyl radical-oxygen complex relative to classical allyl peroxyl radical structures are not revealed in these studies, but it seems clear that either a bounded complex or a transition state exists in which the 2 oxygens are fixed relative to the 3-atom allyl constituents.
Theoretical calculations have helped delineate these possibilities. Transition state structures have been determined for both the step-wise and concerted rearrangements of allylperoxyl radicals using various methodologies. At all levels studied, the verdict is consistent: the step-wise reaction proceeds with a much lower barrier (7.5 kcal/mol lower by ROCCSD(T)/6-311+G(3df,2p)//MP2/6-311+G(3df,2p)).20 Furthermore, theory predicts the intervention of a loosely-bound allyl:dioxygen complex in the step-wise rearrangement. The exact energetics of this interaction are difficult to quantify, but the best calculations to date would suggest a stabilization of at least 1.3 kcal/mol at 0 K.
Theoretical calculations also support a role for a complex of the pentadienyl radical and oxygen that precedes the transition states shown in Schemes 4 and 5. These calculations predict that this complex collapses preferentially to give the non-conjugated peroxyl over the conjugated peroxyls, thereby explaining the predominance of 8 at the kinetic limit. However, it should be pointed out that given that these processes have essentially negligible barriers, the predominance of 8 at the kinetic limit may be more plausibly explained on a purely statistical level. Since there are two possible pre-reaction complexes for the reaction of oxygen with pentadienyls (15 and 16), which are essentially isoenergetic when R1 ~ R2, and both can lead to 8, a 1:2:1 product ratio might be expected on purely statistical grounds (Scheme 9) and indeed this is the ratio that is observed in these reactions (cf. Figure 1). The introduction of substituents of different size at R1 and R2 (or double bond configuration) may change the contribution of the pentadienyl:dioxygen complexes at equilibrium and/or increase the barrier for their collapse to yield products (i.e. the “gauche effect” referred to above) leading to different product ratios.
Scheme 9.

Rates of Reaction; Peroxyl Radical Clocks
The commercial importance of autoxidation of organic compounds and the recent interest in peroxidation reactions in biology has focused a substantial effort on the determination of rates of the important reactions in the chain sequence.4,5 The measurement of absolute rate constants for free radical reactions has indeed been fundamental to advances in free radical chemistry during the last half century and many methods for determining absolute rate constants of radical reactions have been established. Approaches based on the rotating sector, flash photolysis, and pulse radiolysis have been used to determine important rate constants for processes that include radical polymerization, and a variety of atom transfer reactions. More recently, the use of free radical clock reactions has been particularly useful to “time” reactions of carbon radicals in circumstances where classical methods prove to be difficult or impossible to apply. The cyclization of the 5-hexenyl radicals or the ring opening reactions of cyclopropylcarbinyl species, for example, have been at the heart of many mechanistic studies and methods based upon these unimolecular reactions have been used to “time” other bimolecular radical-molecule reactions.31
Knowledge of rate constants has been critical to understanding the chemistry of oxygen-centered radicals as well. These species carry the chain for radical reactions carried out under atmospheric oxygen since carbon radicals undergo diffusion-controlled reaction with molecular oxygen to yield peroxyl radicals (eq 1). For this reason, the rate constants for peroxyl radical reactions have been the focus of extensive investigation. Classical methods, in particular the rotating sector approach, have provided an extensive set of important rate constants for peroxyl radical reactions that underpin our understanding of important commercial and biological processes involving the reaction of organic compounds with molecular oxygen.
It occurred to us that radical clocks might provide a useful alternative for determining rate constants for peroxyl radical-molecule reactions. Radical clocks are established by setting up a competition between a unimolecular reaction having a known rate constant and a bimolecular reaction with an unknown rate constant.31 The peroxyl radical clocks that we have developed set up a competition between peroxyl radical unimolecular rearrangements and bimolecular H-atom transfer.32 One example of a peroxyl radical clock is based upon the competing processes for peroxyl radical 8 that give rise to isomeric hydroperoxide products, 5a/5b and 7, Scheme 10. The rate constant kp can be determined from the [7]/[5a+5b] product ratio, since kR is known (kβ = 2.6 · 106 s−1 at 37°C) as is the partition constant for oxygen addition to C-1, C-3 and C-5 of the pentadienyl radical 2.33 The clock based upon peroxyl radical 8 proves useful for measuring bimolecular rate constants kp, for reactions having rate constants on the order of 105–107 M−1s−1 since the unimolecular clock reaction, kR, is so fast.
Scheme 10.

To broaden the range of peroxyl radical rate constants that can be measured by the clock method, two other clocks were established based upon unimolecular peroxyl radical reactions. These systems rely on the chemistry shown in Scheme 11 for the peroxyl radicals 3 and 17. Radical 3 is the first-formed conjugated diene peroxyl radical in linoleate oxidation, giving the Z,E kinetic products 5a and 5b, Scheme 11A. Competing with H-atom transfer and formation of 5a and 5b is rearrangement of 3 leading ultimately to the thermodynamic E,E products 6a and 6b. This rearrangement appears to be an authentic fragmentation-addition reaction since it occurs with scrambling of oxygen with the atmosphere. The rate constant for fragmentation (6.9 · 102 s−1 at 37°C) was established by oxidation of linoleic acid alone since kp for the only H-atom donor present, linoleic acid, is known (62 M−1s−1 at 37°C). Measuring the ratio of the 5 and 6 products for an H atom donor, R-H, gives the rate constant for H-atom transfer from R-H to the linoleate peroxyl radical. Similarly, the clock from allylbenzene shown in Scheme 11B sets up a competition for formation of kinetic and thermodynamic products, 18 and 19, with increasing concentrations of H-atom donors giving more of the kinetic product 19. Radical clocks based on peroxyl radicals 3, 8 and 17 have been used to determine rate constants ranging from as low as 5 M−1s−1 for H-atom transfer from allyl benzene up to 1 · 107 M−1s−1 for a pyrimidinol antioxidant. Radical 3 is used for rate constants in the low rate range, up to 104 M−1s−1, 8 is useful for constants greater than 105 M−1s−1 and 17 for those in the range of 103–106 M−1s−1.
Scheme 11.

While the clock approach described above offered a more convenient means to measure inhibition rate constants than conventionally employed, we understood there were some limitations. Under the conditions of these experiments, the radical derived from the H-atom donor is the chain-carrying radical, and the rate constant for its reaction with the clock precursor (methyl linoleate for 3 or 6 and allylbenzene for 17) is slow (e.g. 0.01 M−1s−1 for α-TO• and methyl linoleate). This presents a challenge for the determination of inhibition rate constants of H-atom donors that yield persistent (i.e. hindered) radicals, such as the most common industrial antioxidants, butylated hydroxytoluene (BHT) and diarylamines. Furthermore, a high concentration of the precursor (e.g. 2.6 M allylbenzene as precursoro 17) must be used to generate sufficient amounts of product in order to obtain reliable data. This precludes the determination of the inhibition rate constants in a variety of media, an unfortunate limitation since solvent effects can be quite large on these reactions.
We have since employed β, γ-unsaturated (or homoconjugated) peroxyesters such as 20 as an alternative source of the carbon-centered radicals leading to 17, setting up the radical clock without having to involve the slow chain-carrying reaction of the inhibitor-derived radical with allylbenzene. In doing so, the reactions can now be done with millimolar concentrations of the clock precursor, allowing competition kinetic experiments to be carried out in virtually any solvent. Decomposition of 20 in benzene containing α-TOH reveals product profiles that are essentially indistinguishable from those obtained by autoxidation of allylbenzene as described above.34 Varying the solvent in which 20 is decomposed has revealed rate constants for the β-fragmentation of 17 that differ significantly; from 3.5 × 105 s−1 in hexane to 1.8 × 104 s−1 in propionitrile, owing presumably to the large permanent dipole moment of peroxyls (~2.5 D).
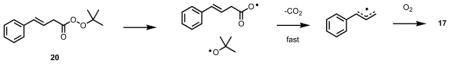
The ability to easily determine inhibition rate constants in different solvents has allowed us to confirm the longstanding prediction of Ingold (which was based on the kinetics of other radical-molecule reactions) that the greater the equilibrium constant for formation of a H-bonded complex between the antioxidant and the solvent, the lower the observed rate constant for peroxyl radical trapping.35 The equilibrium constant is related to the H-bond acidity of the phenol and the H-bond basicity of the solvent, which can be quantified by Abraham’s α2H and β2H parameters, respectively. Using the peroxyester peroxyl radical clock approach, the inhibition rate constant of p-methylphenol (α2H = 0.57) is shown to be much more sensitive to changes in solvent H-bond accepting ability (plotted versus the solvent β2H gives a slope of −4.8), than is trimethylphenol (α2H = 0.37, slope = −3.1), than is BHT (α2H = 0.22, slope = −1.8). We continue to use this methodology to address various kinetic and mechanistic questions in antioxidant chemistry.
In another application of the peroxyl radical clock methodology, we recently used the linoleate conjugated diene peroxyl radical 3 to determine the propagation rate constants for oxidation of several important lipids, including arachidonic (20:4), eicosapentaenoic (20:5), and docosahexaenoic (22:6) acids as well as cholesterol and 7-dehydrocholesterol (7-DHC).36 The rates for the fatty acids depend, as expected, on the number of oxidizable –CH2– centers in the molecule that are flanked by two double bonds. Linoleate has one such center, arachidonate has 3, eicosapentaenoate 4 and docosahexaenoate 5, and the relative propagation rate constants reflect this statistical bias (krel = 1 : 3.2 : 4.0 : 5.4). The rate constants for cholesterol and 7-DHC were also determined because of the importance of these constituents in mammalian biology. Cholesterol is an important component of biological membranes and 7-DHC is an immediate biosynthetic precursor not only to cholesterol but also to vitamin D3. The rate constant determined for oxidative free radical propagation of cholesterol, 11 M−1s−1, falls in line with those determined by other methods for cycloalkenes like cyclohexene and cyclopentene; both have rate constants of about 6 M−1s1. On the other hand, the constant determined for autoxidation of 7-DHC, 2260 M−1s−1, is surprisingly large. Arachidonic acid, which is normally considered to be a highly oxidizable lipid, is an order of magnitude less reactive than 7-DHC. Indeed, the propagation rate constant for 7-DHC makes this substrate, to our knowledge, the most reactive lipid known that can maintain a peroxidative free radical chain reaction.
Acknowledgments
We are grateful to Professor Alan Brash of Vanderbilt for many helpful discussions. NAP acknowledges funding from The NIH (ES013125, HD064727),), the NSF (CHE 0717067), and support from the Center in Molecular Toxicology, T32 Vanderbilt University. Support from the the NSERC of Canada, the Ontario Ministry of Research and Innovation and the Canada Research Chairs program is gratefully acknowledged by DAP.
Biographies
Derek Pratt received his B.Sc, from Carleton University in 1999 and his Ph.D. from Vanderbilt University in 2003. After completing post-doctoral work at the University of Illinois at Urbana-Champaign in 2005, he returned to Canada to take up a faculty position at Queen’s University. In 2010 he moved to the University of Ottawa where he is an Associate Professor and Canada Research Chair in Free Radical Chemistry.
Keri Tallman graduated from The College of Wooster in 1994 with a B.A. in chemistry. After receiving her Ph.D. from Colorado State University in 2000, she carried out post-doctoral work with Ned Porter at Vanderbilt University. She is currently a Research Assistant Professor at Vanderbilt.
Ned Porter graduated from Princeton in 1965 with a B.S. in Chemical Engineering and accepted a position as Assistant Professor at Duke University in 1969, after receiving his Ph.D. with Paul D. Bartlett at Harvard. In 1998 he moved to Vanderbilt University where he is Stevenson Professor of Chemistry. His research interests have centered on the mechanisms of free radical reactions. A continuous research theme has been on the interplay between free radical chemistry, lipids and oxidative stress in biology.
References
- 1.Porter NA. Mechanisms for the autoxidation of polyunsaturated lipids. Acc Chem Res. 1986;19:262–270. [Google Scholar]
- 2.Criegee R. Houben-Weyl Methoden der organischen Chemie. 4. Vol. 8 George Thieme Verlag; Stuttgart, Germany: 1953. [Google Scholar]
- 3.Maillard B, Ingold KU, Scaiano JC. Rate Constants for the Reactions of Free Radicals with Oxygen in Solution. J Am Chem Soc. 1983;105:5095–5099. [Google Scholar]
- 4.Howard JA, Ingold KU. Absolute rate constants for hydrocarbon autoxidation. VI. Alkyl aromatic and olefinic hydrocarbons. Can J Chem. 1967;45:793–802. [Google Scholar]
- 5.Korcek S, Chenier JHB, Howard JA, Ingold KU. Absolute Rate Constants for Hydrocarbon Autoxidation. 21. Activation-Energies for Propagation and Correlation of Propagation Rate Constants with Carbon-Hydrogen Bond Strengths. Can J Chem. 1972;50:2285–2897. [Google Scholar]
- 6.Howard JA. Absolute Rate Constants for Reactions of Oxyl Radicals. In: Williams GH, editor. Adv Free Rad Chem. IV. Logos Press; 1972. pp. 49–174. [Google Scholar]
- 7.Jahn U, Galano JM, Durand T. Beyond prostaglandins-chemistry and biology of cyclic oxygenated metabolites formed by free-radical pathways from polyunsaturated fatty acids. Angew Chem Int Edit. 2008;47:5894–5955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- 8.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 9.Bascetta E, Gunstone FD, Walton JC. An electron spin resonance study of fatty acids and esters. Part 1. Hydrogen abstraction from olefinic and acetylenic long-chain esters. J Chem Soc, Perkin Tran. 1983;2:603–13. [Google Scholar]
- 10.Bascetta E, Gunstone FD, Walton JC. An electron spin resonance study of fatty acids and esters. Part 2. Hydrogen abstraction from saturated acids and their derivatives. J Chem Soc, Perkin Tran. 1984;2:401–409. [Google Scholar]
- 11.Brash AR. Autoxidation of methyl linoleate: identification of the bis-allylic 11-hydroperoxide. Lipids. 2000;35:947–952. doi: 10.1007/s11745-000-0604-0. [DOI] [PubMed] [Google Scholar]
- 12.Tallman KA, Pratt DA, Porter N. Kinetic products of linoleate peroxidation: Rapid beta-fragmentation of nonconjugated peroxyls. J Am Chem Soc. 2001;123:11827–11828. doi: 10.1021/ja0169724. [DOI] [PubMed] [Google Scholar]
- 13.Burton GW, Ingold KU. Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc Chem Res. 1986;19:194–201. [Google Scholar]
- 14.Barclay LRC, Vinqvist MR, Antunes F, Pinto RE. Antioxidant activity of vitamin E determined in a phospholipid membrane by product studies: Avoiding chain transfer reactions by vitamin E radicals. J Am Chem Soc. 1997;119:5764–5765. [Google Scholar]
- 15.Barclay LRC. 1992 Syntex Award Lecture - Model Biomembranes - Quantitative Studies of Peroxidation, Antioxidant Action, Partitioning, and Oxidative Stress. Can J Chem. 1993;71:1–16. [Google Scholar]
- 16.Pratt DA, Mills JH, Porter N. Theoretical calculations of carbon-oxygen bond dissociation enthalpies of peroxyl radicals formed in the autoxidation of lipids. J Am Chem Soc. 2003;125:5801–10. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]
- 17.Pratt DA, Porter N. Role of hyperconjugation in determining carbon-oxygen bond dissociation enthalpies in alkylperoxyl radicals. Org Lett. 2003;5:387–390. doi: 10.1021/ol027094x. [DOI] [PubMed] [Google Scholar]
- 18.DiLabio GA, Pratt DA. Density Functional Theory Based Model Calculations for Accurate Bond Dissociation Enthalpies. 2. Studies of X-X and X-Y (X, Y = C, N, O, S, Halogen) Bonds. J Phys Chem A. 2000;104:1938–1943. [Google Scholar]
- 19.Tallman KA, Rector CL, Porter NA. Substituent Effects on Regioselectivity in the Autoxidation of Nonconjugated Dienes. J Am Chem Soc. 2009;131:5635–5641. doi: 10.1021/ja900040d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu D, Pratt D. Secondary orbital interactions in the propagation steps of lipid peroxidation. Chem Commun. 2010;46:3711–3713. doi: 10.1039/c0cc00019a. [DOI] [PubMed] [Google Scholar]
- 21.Porter NA, Mills KA, Carter RL. A Mechanistic Study of Oleate Autoxidation: Competing Peroxyl H-Atom Abstraction and Rearrangement. J Am Chem Soc. 1994;116:6690–6696. [Google Scholar]
- 22.Beckwith ALJ, Davies AG, Davison IGE, Maccoll A, Mruzek MH. The mechanisms of the rearrangements of allylic hydroperoxides. J Chem Soc, Chem Commun. 1988:475–476. [Google Scholar]
- 23.Beckwith ALJ, Davies AG, Davison IGE, Maccoll A, Mruzek MH. The mechanisms of the rearrangements of allylic hydroperoxides: 5α-hydroperoxy-3β-hydroxycholest-6-ene and 7α-hydroperoxy-3β-hydroxy-cholest-5-ene. J Chem Soc, Perkin Tran. 1989;2:815–824. [Google Scholar]
- 24.Beckwith ALJ, O’Shea DM, Roberts DH. Novel formation of bis-allylic products by autoxidation of substituted 1,4-cyclohexadienes. J Am Chem Soc. 1986;108:6408–09. [Google Scholar]
- 25.Porter N, Kaplan JK, Dussault PH. Stereoselective acyclic 3,2 peroxyl radical rearrangements. J Am Chem Soc. 1990;112:1266–1267. [Google Scholar]
- 26.Porter N, Wujek JS. Allylic hydroperoxide rearrangement. Beta-scission or concerted pathway? J Org Chem. 1987;52:5085–5089. [Google Scholar]
- 27.Mills KA, Caldwell SE, Dubay GR, Porter N. An allyl radical-dioxygen caged pair mechanism for cis-allylperoxyl rearrangements. J Am Chem Soc. 1992;114:9689–9691. [Google Scholar]
- 28.Caldwell S, Porter N. Preparation and Reactions of Unsymmetrically Labeled Hydroperoxides: Solvent Viscosity Dependent Oxygen Scrambling of Cumylperoxyl Free Radicals. J Am Chem Soc. 1995;117:8676–8677. [Google Scholar]
- 29.Allen JL, Paquette KP, Porter N. A Baeyer-Villiger approach to O-18-Labeled peroxides: A protected form of unsymmetrically labeled hydrogen peroxide. J Am Chem Soc. 1998;120:9362–9363. [Google Scholar]
- 30.Lowe J, Porter N. Preparation of an unsymmetrically labeled allylic hydroperoxide and study of its allylic peroxyl radical rearrangement. J Am Chem Soc. 1997;119:11534–11535. [Google Scholar]
- 31.Newcomb M. Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 32.Roschek B, Tallman KA, Rector CL, Gillmore JG, Pratt DA, Punta C, Porter NA. Peroxyl radical clocks. J Org Chem. 2006;71:3527–3532. doi: 10.1021/jo0601462. [DOI] [PubMed] [Google Scholar]
- 33.Tallman KA, Roschek B, Porter NA. Factors influencing the autoxidation of fatty acids: Effect of olefin geometry of the nonconjugated diene. J Am Chem Soc. 2004;126:9240–9247. doi: 10.1021/ja049104q. [DOI] [PubMed] [Google Scholar]
- 34.Jha M, Pratt DA. Kinetic solvent effects on peroxyl radical reactions. Chem Commun. 2008:1252–1254. doi: 10.1039/b800369f. [DOI] [PubMed] [Google Scholar]
- 35.Litwinienko G, Ingold KU. Solvent effects on the rates and mechanisms of reaction ofphenols with free radicals. Acc Chem Res. 2007;40:222–30. doi: 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]
- 36.Xu L, Davis TA, Porter NA. Rate Constants for Peroxidation of Polyunsaturated Fatty Acids and Sterols in Solution and in Liposomes. J Am Chem Soc. 2009;131:13037–13044. doi: 10.1021/ja9029076. [DOI] [PMC free article] [PubMed] [Google Scholar]