

Abstract
Inflammatory signaling and oxidative stress are two major components in the pathogenesis of alcoholic hepatitis. Alcohol consumption results in translocation of gut bacteria into the portal system along with lipopolysaccharides that interact with toll-like receptors and results in the production of inflammatory and immunogenic mediators such as tumor necrosis factor-alpha (TNF-α) and interferons. Chronic consumption of alcohol causes priming of this process in which there is enhanced production of cytokines, interferon, interleukins, and TNF-α. Oxidative stress, genetic predisposition, and the unfolded protein response are other contributory mechanisms. Novel therapies aimed at these pathways may prevent, decrease, or delay the complications of alcoholic hepatitis.
Keywords: Alcoholic hepatitis, Lipopolysaccharide, Toll like receptors, Oxidative stress, Endotoxin
INTRODUCTION
It is important to understand the pathogenesis of acute alcoholic hepatitis (AH) in order to develop new treatment strategies for management of this potentially fatal condition.
MECHANISMS OF ALCOHOL INDUCED STEATOSIS
Macrovesicular steatosis occurs in all drinkers within a few weeks of drinking and is completely reversible on abstinence. A combination of increased lipogenesis and impaired fatty acid breakdown results in steatosis which worsens as the ALD progresses. Mechanisms of ethanol-induced steatosis are: (1) increase in fatty acid synthesis through transcription factor sterol regulatory-element-binding protein (SREBP-1) which codes for lipogenic enzymes; (2) ethanol promotes lipid metabolism through inhibition of peroxisome-proliferator–activated receptor α (PPAR-α) and AMP kinase and stimulation of sterol regulatory element-binding protein 1. This in turn, increases fatty acid oxidation and leads to lipid storage remodeling; (3) lowering of circulating adiponectin levels which further decreases AMPK; (4) increase in insulin resistance in adipocytes that disrupts insulin signaling to phosphoinositide 3-kinase (PI3K)13; and (5) interference with fatty acid beta oxidation in mitochondria and peroxisomes through increase in NADH/NAD ratio[1].
LIVER INJURY DUE TO ALCOHOL METABOLITES
Hepatocytes convert ethanol to acetaldehyde through three mechanisms: Alcohol dehydrogenase, cytochrome P-450 isoenzyme-1 (CYP2E1), and catalase[2]. Ethanol oxidation by CYP2E1 itself creates hydroxyethyl free radicals that interact with hepatocyte nucleic acids and proteins to make antigenic adducts such as malondialdehyde and acetaldehyde that elicit an immune response and cause direct oxidative injury of DNA and proteins[3].
Against the background of steatosis and existing liver damage, heavy and continued drinking in some patients causes AH. Two main mechanisms are involved: inflammation and oxidative stress.
PATHOGENETIC PATHWAYS LEADING TO HEPATIC INFLAMMATION
AH is a condition with features similar to systemic inflammatory response syndrome: fever, tender enlarged liver, leukocytosis, and increased hepatic blood flow. Initiating events include expression of gut-derived lipopolysaccharides (LPS), interaction of LPS with TLR4 receptors, activation of inflammatory signaling pathways, cytokine release, and Kupffer cells (Figure 1).
Figure 1.

Overview of the pathway of alcoholic injury resulting in the direct production of oxidative stress and the LPS-TLR pathway that culminates in production of cytokines and other inflammatory processes that result in hepatitis of the liver.
LPS, a component of the outer membrane of gram-negative bacteria interacts with immune cells and triggers inflammatory reactions with release of cytokines. Chronic alcohol exposure increases gut permeability and facilitates translocation of endotoxins from the intestinal lumen to the portal circulation (Figure 2). Increased levels of endotoxins and increase in gut permeability have been shown in patients with alcoholic liver disease[4]. Further, pretreatment with antibiotics or lactobacillus in animal models decreases the LPS-endotoxin and the severity of liver injury.[5,6]
Figure 2.
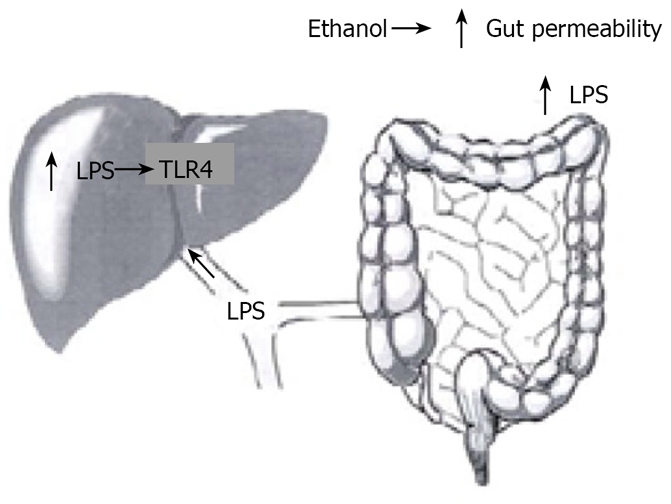
Link between gut derived bacterial lipopolysacaride and Toll like receptor-4 stimulation within the liver.
Toll-like receptors (TLRs), important components of the innate immune system, function as pattern recognition receptors which recognize and bind proteins and toxins released by pathogens. There are many TLRs, but in alcoholic liver disease TLR4 is most relevant[7,8]. TLR4 signals activate early growth response 1 (EGR1, a transcription factor), nuclear factor-kappaB (NF-κB), and TLR4 adaptor (toll-interleukin-1-receptor-domain-containg adapter-inducing interferon-beta or TRIF)[9]. CD14 and TLR4 deficient mice are protected from alcoholic liver injury[10]. Interestingly, TLR4 is expressed by a number of other cells including hepatocytes, hepatic stellate cells, and sinusoidal epithelial cells which may further contribute to ALD[7,8,11]. Thus, TLR4 up regulation, in response to endotoxins, prompts Kupffer cells to release large amounts of TNF-α and NF-κB.
TLR4 acts through a pathway common to other TLRs, myeloid differentiation factor 88 (MyD88) pathway, as well as a specific and unique pathway to TLR4, TRIF signaling (the MyD88 independent pathway)[12]. In the MyD88 pathway, interleukin 1 receptor associated kinase (IRAK) and possibly TNF receptor associated kinase 6 (TRAF6) are recruited to the TLR4 complex by MyD88 and eventually express TNF-α and apoptosis transcription factor AP-1. The TRIF pathway (My88-independent) activates IRF-3, NF-κB and eventually IFN-β and TNF-α production[13]. IRF-3 may also bind to the promoter region and up regulate transcription of TNF-α[13].
NF-κB is a transcription factor that is translocated to the nucleus in response to stress signals and binds to the promoter region of pro-inflammatory genes[14]. Chronic alcohol intake primes the liver through continued NF-κB activation and increased TNF-α production in response to LPS, and murine models show increased binding of NF-κB to the DNA[15]. Monocytes from chronic alcoholic patients show increased NF-κB activation in comparison to controls[16]. Additionally, monocytes cultured from chronic alcoholics show increased amounts of TNF in response to LPS stimulation[17]. Studies have shown that in rats chronically fed ethanol, injected LPS results in a much higher plasma level of TNF-α compared to controls[18,19].
Other pathways which are stimulated in this process are: (1) the signal transducer and activator transcription factor (STAT) pathway activated by interleukin-6 (IL-6) and interferons-LPS causes STAT3 to induce IL-10 production during acute alcohol exposure in monocytes[20]; (2) LPS stimulation of MAPK family members which include JNK (c-jun-N-terminal kinase), ERK (extracellular receptor activated kinases), and p38 culminates in NF-κB production and mRNA stabilization[21]; and (3) Changes in immune function accompany LPS-induced hepatic inflammation- CD4+ and CD8+ T lymphocyte infiltrates are found in 40% of patients with ALD[22]. It is thought that abnormal proteins may leak into the portal system and collect in germinal centers where the antigens are presented to CD4+ T-cells resulting in antibody creation against the proteins. Hepatic stellate cells can also present such antigens to CD4+ cells, and dendrites that scavenge dead hepatocytes may present antigens to CD8+ cells, thus providing numerous possible pathways by which oxidative stress can stimulate the immune system through both humoral and cellular mechanisms.
All these pathways result in recruitment of inflammatory cells (polymorphs and mononuclear cells) and cause necroinflammation. This is followed by Kupffer cell activation, hepatocyte ballooning, and apoptosis.
OXIDATIVE STRESS
Oxidative stress occurs when there is an imbalance between antioxidants and reactive oxidizing species[3]. Oxidative stress is indirectly measured through markers such as protein oxidation, lipid oxidation, DNA oxidation, and depletion of antioxidants. In AH, there is decreased production and increased depletion of antioxidants alongside an increase in production of reactive oxygen species(ROS), reactive nitrogen species, and peroxidized lipids.
Markers of oxidative stress increase with acute alcohol ingestion and in persons with AH[23]. Activated Kupffer cells and hepatocytes act as sources of free radicals in response to alcohol[24]. As alluded to earlier, CYP2E1 may increase 5-20 fold in patients with AH, leading to increased electron leakage and release of ROS causing oxidative stress, adduct formation and immunogenic responses[25]. Oxidative stress leads to damage of the mitochondria, endoplasmic reticulum (ER) stress and subsequent apoptosis, and increased lipid synthesis[3,26]. The ROS cause lipid peroxidation and the formation of Mallory bodies through breakdown of proteins. Other sources of oxidative stress include granulocytes from the inflammatory process catalyzed by the enzymes NADPH oxidase and myeloperoxidase which cause production of ROS, hepatic iron accumulation, and a decrease in antioxidants[3,27]. ROS can also activate ERK1/2, p38 MAPK kinases, and NF-κB which stimulates TNF-α, complementing the LPS-induced pathways of TNF-α production and creating a vicious cycle of inflammation and oxidative stress[28].
Mitochondria rely on transport of the powerful antioxidant glutathione from the cytosol. Alcohol consumption inhibits methionine conversion to S-adenosylmethionine (SAMe), the precursor for glutathione synthesis. Additionally, alcohol inhibits methionine synthase and folate which act as cofactors for methyl group transfers. Disruption of these pathways impairs the conversion of homocysteine to methionine, resulting in the accumulation of homocysteine, further disabling the redox balance of cells in favor of oxidative stress[2].
MISCELLANEOUS MECHANISMS AND PATHWAYS
Proteasomes degrade defective proteins and help in regulation of cell processes such as gene regulation and cell division. When the proteasomes do not work properly, proteins accumulate and enhance progression of liver disease[29]. In animal models of chronic alcohol feeding, proteasome activity decreases and serum levels of ubiquitin, involved in the degradation of defective proteins, increases[30]. Ubiquitin also accumulates in hepatocytes and appears as Mallory bodies on liver histology[31].
The unfolded protein response (UPR) is a series of signals triggered by the accumulation of unfolded proteins and is due to stress placed on ER[32]. UPR reduces protein synthesis and favors protein degradation as well as activation of Nfr2-mediated apoptosis signals[32]. UPR is aggravated by the accumulation of protein adducts and ROS as well as by the depletion of antioxidants such as glutathione[32,33].
SAMe is the primary methyl donor and precursor to glutathione. In alcoholic hepatitis, there is a decrease in hepatic methionine levels and a 50-60% decrease in the activity of methionine adenosyl transferase, the enzyme which converts methionine to SAMe[34]. This causes a decrease in glutathione synthesis which results in impaired clearance of oxidative species such as 4-hydroxynonenal (marker of lipid peroxidation) and mitochondrial injury. Human and animal studies have suggested that SAMe replacement results in increased GSH and decreased markers of oxidation[35].
Footnotes
Peer reviewers: Valentina Medici, MD, PhD, Department of Internal Medicine, University of California Davis, 4150 V Street, Suite 3500, Sacramento, CA 95817, United States; Neeraj Saxena, PhD, Assistant Professor of Medicine, Department of Medicine, Division of Digestive Diseases, Room 255, Whitehead Biomedical Research Bldg.615 Michael Street, Atlanta, GA 30322, United States
S- Editor Zhang HN L- Editor Hughes D E- Editor Zhang L
References
- 1.Lieber CS. New concepts of the pathogenesis of alcoholic liver disease lead to novel treatments. Curr Gastroenterol Rep. 2004;6:60–65. doi: 10.1007/s11894-004-0027-0. [DOI] [PubMed] [Google Scholar]
- 2.Lieber CS. Ethanol metabolism, cirrhosis and alcoholism. Clin Chim Acta. 1997;257:59–84. doi: 10.1016/s0009-8981(96)06434-0. [DOI] [PubMed] [Google Scholar]
- 3.Albano E. New concepts in the pathogenesis of alcoholic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2:749–759. doi: 10.1586/17474124.2.6.749. [DOI] [PubMed] [Google Scholar]
- 4.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 5.Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc Soc Exp Biol Med. 1994;205:243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- 6.Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 7.Petrasek J, Mandrekar P, Szabo G. Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract. 2010;2010 doi: 10.1155/2010/710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szabo G, Mandrekar P, Dolganiuc A. Innate immune response and hepatic inflammation. Semin Liver Dis. 2007;27:339–350. doi: 10.1055/s-2007-991511. [DOI] [PubMed] [Google Scholar]
- 9.Szabo G. New insights into the the molecular mechanisms of alcoholic hepatitis: a potential role for NF-kappaB activation? J Lab Clin Med. 2000;135:367–369. doi: 10.1067/mlc.2000.106452. [DOI] [PubMed] [Google Scholar]
- 10.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 11.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 13.Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, Kolls JK. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol. 2008;181:3049–3056. doi: 10.4049/jimmunol.181.5.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh S. Regulation of inducible gene expression by the transcription factor NF-kappaB. Immunol Res. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- 15.Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, Tahan SR, Su GL. Activation of nuclear factor kappa B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology. 1999;30:934–943. doi: 10.1002/hep.510300402. [DOI] [PubMed] [Google Scholar]
- 16.Hill DB, Barve S, Joshi-Barve S, McClain C. Increased monocyte nuclear factor-kappaB activation and tumor necrosis factor production in alcoholic hepatitis. J Lab Clin Med. 2000;135:387–395. doi: 10.1067/mlc.2000.106451. [DOI] [PubMed] [Google Scholar]
- 17.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 18.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–G502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 19.Pennington HL, Hall PM, Wilce PA, Worrall S. Ethanol feeding enhances inflammatory cytokine expression in lipopolysaccharide-induced hepatitis. J Gastroenterol Hepatol. 1997;12:305–313. doi: 10.1111/j.1440-1746.1997.tb00426.x. [DOI] [PubMed] [Google Scholar]
- 20.Norkina O, Dolganiuc A, Shapiro T, Kodys K, Mandrekar P, Szabo G. Acute alcohol activates STAT3, AP-1, and Sp-1 transcription factors via the family of Src kinases to promote IL-10 production in human monocytes. J Leukoc Biol. 2007;82:752–762. doi: 10.1189/jlb.0207099. [DOI] [PubMed] [Google Scholar]
- 21.Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-kappaB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 22.Colombat M, Charlotte F, Ratziu V, Poynard T. Portal lymphocytic infiltrate in alcoholic liver disease. Hum Pathol. 2002;33:1170–1174. doi: 10.1053/hupa.2002.129414. [DOI] [PubMed] [Google Scholar]
- 23.Meagher EA, Barry OP, Burke A, Lucey MR, Lawson JA, Rokach J, FitzGerald GA. Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest. 1999;104:805–813. doi: 10.1172/JCI5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nanji AA. Role of Kupffer cells in alcoholic hepatitis. Alcohol. 2002;27:13–15. doi: 10.1016/s0741-8329(02)00207-0. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–1494. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 26.Yin M, Gäbele E, Wheeler MD, Connor H, Bradford BU, Dikalova A, Rusyn I, Mason R, Thurman RG. Alcohol-induced free radicals in mice: direct toxicants or signaling molecules? Hepatology. 2001;34:935–942. doi: 10.1053/jhep.2001.28888. [DOI] [PubMed] [Google Scholar]
- 27.Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778–790. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- 28.Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79:1348–1356. doi: 10.1189/jlb.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donohue TM. The ubiquitin-proteasome system and its role in ethanol-induced disorders. Addict Biol. 2002;7:15–28. doi: 10.1080/135562101200100562. [DOI] [PubMed] [Google Scholar]
- 30.Bardag-Gorce F, Vu J, Nan L, Riley N, Li J, French SW. Proteasome inhibition induces cytokeratin accumulation in vivo. Exp Mol Pathol. 2004;76:83–89. doi: 10.1016/j.yexmp.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Bardag-Gorce F, van Leeuwen FW, Nguyen V, French BA, Li J, Riley N, McPhaul LW, Lue YH, French SW. The role of the ubiquitin-proteasome pathway in the formation of mallory bodies. Exp Mol Pathol. 2002;73:75–83. doi: 10.1006/exmp.2002.2451. [DOI] [PubMed] [Google Scholar]
- 32.Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 33.Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2004;28:173–181. doi: 10.1097/01.ALC.0000108654.77178.03. [DOI] [PubMed] [Google Scholar]
- 35.Lieber CS. S-Adenosyl-L-methionine and alcoholic liver disease in animal models: implications for early intervention in human beings. Alcohol. 2002;27:173–177. doi: 10.1016/s0741-8329(02)00230-6. [DOI] [PubMed] [Google Scholar]