

Abstract
Primary hyperoxalurias (PH) are inborn errors in the metabolism of glyoxylate and oxalate. PH type 1, the most common form, is an autosomal recessive disorder caused by a deficiency of the liver-specific enzyme alanine, glyoxylate aminotransferase (AGT) resulting in overproduction and excessive urinary excretion of oxalate. Recurrent urolithiasis and nephrocalcinosis are the hallmarks of the disease. As glomerular filtration rate decreases due to progressive renal damage, oxalate accumulates leading to systemic oxalosis. Diagnosis is often delayed and is based on clinical and sonographic findings, urinary oxalate assessment, DNA analysis, and, if necessary, direct AGT activity measurement in liver biopsy tissue. Early initiation of conservative treatment, including high fluid intake, inhibitors of calcium oxalate crystallization, and pyridoxine in responsive cases, can help to maintain renal function in compliant subjects. In end-stage renal disease patients, the best outcomes have been achieved with combined liver-kidney transplantation which corrects the enzyme defect.
1. Introduction
Hyperoxaluria may be either inherited or acquired. The primary hyperoxalurias (PH) are inborn error of metabolism resulting in increased endogenous production of oxalate leading to excessive urinary oxalate excretion. To date, three distinct hereditary enzymatic deficiencies have been linked to PH, namely, PH type 1 (PH1), type 2 (PH2), and type 3 (PH3), and there is evidence to speculate that further causes are yet to be identified. Due to marked hyperoxaluria, recurrent urolithiasis and progressive nephrocalcinosis are the principal manifestations of PH. As a result of kidney injury, glomerular filtration rate (GFR) declines leading to chronic kidney disease, and ultimately to end-stage renal disease (ESRD) and systemic involvement in PH1, the most severe type of PH. Despite recent improvement in disease spectrum knowledge, diagnostic procedure, and treatment strategies, PH1 still represents a challenging issue for both adult and pediatric nephrologists worldwide.
2. Physiopathology
Primary hyperoxalurias are inborn errors in the metabolism of glyoxylate and oxalate (Figure 1). They are characterized by an excessive production of oxalate, a metabolic endproduct. The most common type of PH, PH1 (MIM #259900) is caused by a deficiency of the liver specific, peroxisomal, pyridoxal phosphate-dependent enzyme alanine : glyoxylate amino transferase (AGT; EC 2.6.1.44) [1, 2]. The second type, PH2 (MIM #260000), is caused by a deficiency in glyoxylate reductase/hydroxypyruvate reductase (GRHPR; EC 1.1.1.26/79) [3–5], a cytosolic enzyme. The recently identified PH type 3 (MIM #613616) [6] is linked to the gene DHDPSL, encoding a mitochondrial enzyme, although the metabolic reactions involved remain to be confirmed. All three are autosomal recessive diseases. AGT catalyses the transamination of glyoxylate to glycine while pyruvate is converted to alanine, whereas GRHPR catalyses the reduction of glyoxylate to glycolate [7]. The failure to detoxify glyoxylate in PH1 and 2 results in its conversion into oxalate by cytosolic lactate dehydrogenase. Since the main source of oxalate removal from the body is urine excretion, the deposition of the calcium oxalate salt, which is very poorly soluble, occurs primarily there either as urolithiasis along the urinary tract or in the kidney, or as interstitial deposition in the kidney leading to nephrocalcinosis [8]. The accompanying overproduction of glycolate in PH1 or L-glycerate in PH2 has no identified consequence. The crystal structures for both AGT and GRHPR have been solved, and some rationalization of the effect of mutations in the AGXT and GRHPR genes has been possible [9, 10].
Figure 1.
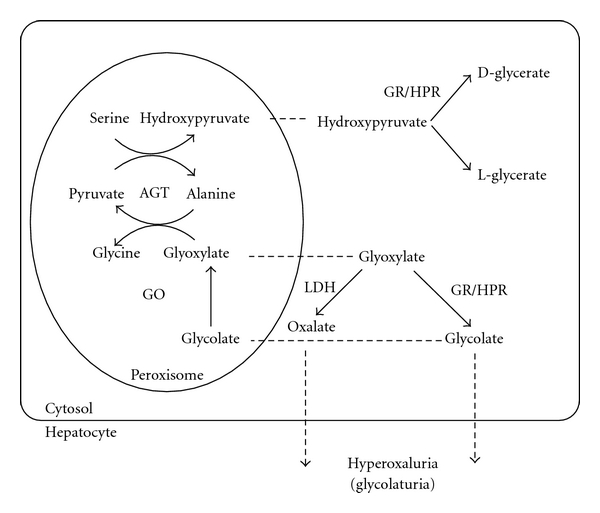
Reactions involved in oxalate, glyoxylate, and glycolate metabolism in human hepatocytes. Abbreviations: AGT alanine: glyoxylate aminotransferase; GR/HPR glyoxylate reductase/hydroxypyruvate reductase; GO glycolate oxydase; LDH lactate dehydrogenase.
3. Epidemiology
PH are rare autosomal-recessive inherited disorders. PH1 is the most common form of PH. The disease has an estimated prevalence ranging from 1 to 3 per million population and an estimated incidence rate of ~1 : 100,000 live births per year in Europe [11–13]. Higher rates are reported from inbred populations [14]. PH accounts for <1% of pediatric ESRD population in registries from USA, UK, and Japan [15–17]. In contrast, PH is more prevalent in countries where consanguineous marriages are common. Due to lack of registries, epidemiological information from developing countries primarily originates from major referral centers. Approximately 10% of Kuwaiti children and 13% of Tunisian children with ESRD have been reported to have PH [18, 19]. Incidence and prevalence of other types of PH are unknown but appear to be much lower than PH1.
4. Clinical Features and Oxalate Burden
Owing to the high urinary oxalate excretion, the urine become supersaturated for calcium oxalate (CaOx) resulting in the formation of crystals within the tubular lumen. PH1, therefore, manifests as severe urolithiasis and/or nephrocalcinosis (Figure 2). Progressive renal parenchyma inflammation and interstitial fibrosis due to nephrocalcinosis and recurrent urolithiasis cause renal impairment, which usually progresses to ESRD over time [8]. Once the GFR falls below 30–50 mL/min per 1.73 m2, reduced renal excretion of oxalate by the kidneys together with continued overproduction by the liver lead to an increase plasma oxalate (Pox) that exceeds the supersaturation point for CaOx (Pox >30 μmol/l). Systemic deposition of CaOx, namely, oxalosis, can then occur in many organs such as bone, retina, skin, soft tissues, heart, vessels, and central nervous system (Figure 3). Severe systemic complications result in high morbidity and poor quality of life and, if treated late or untreated, early death. The bone compartment seems to be the main site for oxalate storage (15 to 910 in PH1 patients versus 2 to 9 μmol of oxalate per gram of bone tissue in non-PH1 ESRD patients) [20]. The skeleton can therefore store huge amounts of oxalate, even though the threshold of GFR at which this occurs is debated. Clinically, PH1 patients can experience severe bone pains and pathological fractures with low-trauma mechanism, as well as EPO-resistant anemia. Joints can also be damaged, with synovitis, chondrocalcinosis and oxalate deposits. Characteristics of oxalate osteopathy on X-ray include dense metaphyseal bands, submarginal metaphyseal lucency, sclerosis of adjacent diaphysis, cystic bone changes, deformities, subperiosteal resorption, blurred trabecular pattern, radio-opaque rims in flat bones and epiphyseal nuclei, as well as increased bone density in vertebra and iliac crest [21, 22]. Symmetrical transverse lines of increased bone density at areas of rapid growth have been related to crystalline precipitation in cartilage calcification sites as well as in hematopoietic and other highly vascularized areas [23, 24]. Bone biopsies from the iliac crest can show specific features: oxalate crystals often surrounded by a granulomatous reaction corresponding to an invasion of bone surface by macrophages [20]. Instead of invasive bone biopsy, bone mineral density (BMD) measurement could be a valuable and noninvasive tool in determining and monitoring oxalate burden in PH1 [25]. Using a new bone imaging technique (e.g., high-resolution peripheral quantitative computed tomography, HR-pQCT), it has been shown that children with PH1 had decreased compartmental volumetric BMD and bone modifications with disorganized trabecular microarchitecture [26].
Figure 2.
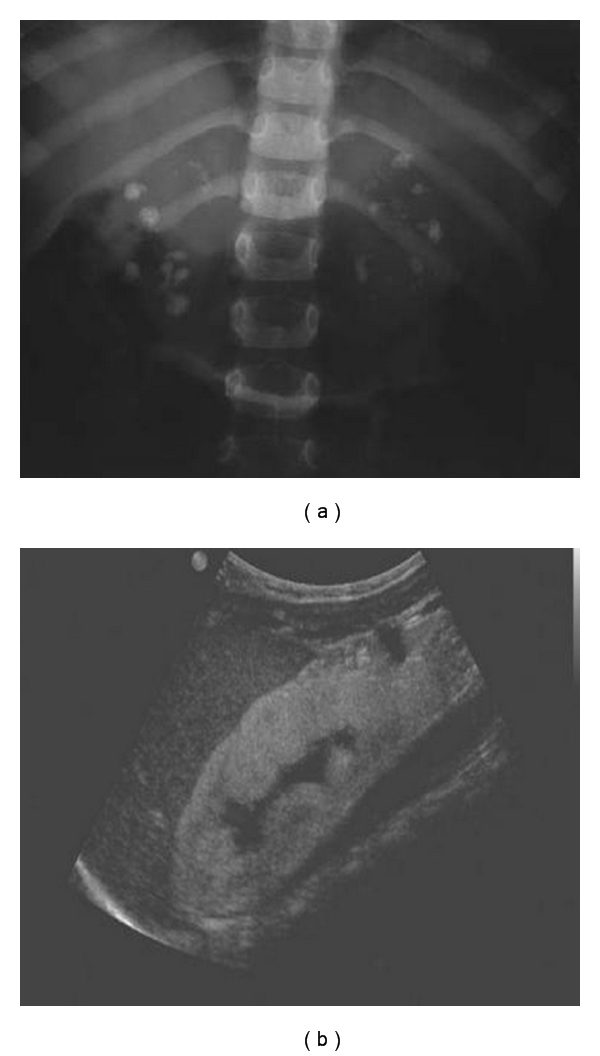
Abdomen X-ray (a) and renal ultrasonography (b) showing urolithiasis and nephrocalcinosis in PH1.
Figure 3.
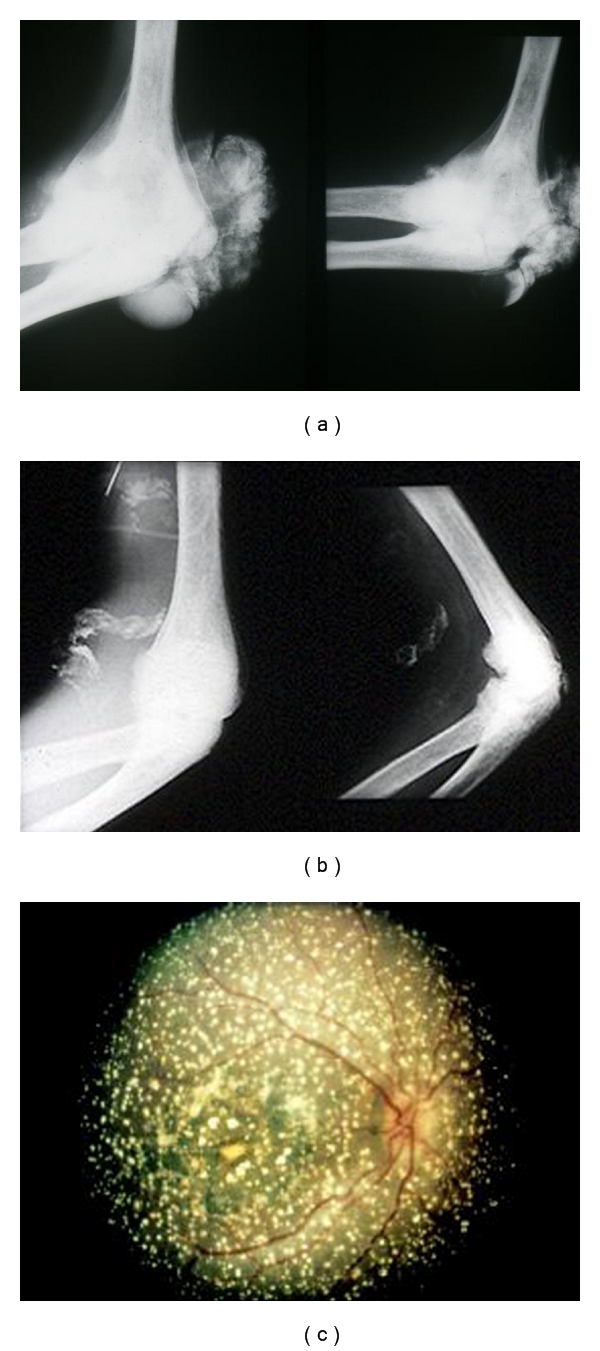
Systemic involvement (“oxalosis”) in PH1. Calcium oxalate deposition in the bones and joints (a), the vessels (b), and the retina (c).
5. Diagnosis
5.1. PH1
The median age at initial symptoms of PH1 is 4 to 7 years in Europe and 13 years in Japan, ranging from birth to the sixth decade [13, 27, 28]. PH1 has variable presentation. The infantile form often presents as a life-threatening condition because of rapid progression to ESRD due to both early oxalate load and immature GFR; one half of the patients experience ESRD at the time of diagnosis and 80% develop ESRD by the age of 3 years [29]. Other presentations include recurrent urolithiasis and progressive renal failure in childhood or adolescence, late-onset form with occasional stone passage and sometimes ESRD as the first symptom in adulthood, diagnosis made after recurrence posttransplantation and presymptomatic individuals with a family history of PH1 [30]. Sonographic examination may show either cortical or medullary nephrocalcinosis [31]. Because of the rarity of disease, physicians are likely to encounter few or no PH patients during their practicing lifetime. The diagnosis of PH is, therefore, often delayed for years [12, 27, 32]. At time of diagnosis, a high proportion of patients (10–40%) had already reached ESRD [11–13, 27, 32, 33]. Overall, ESRD is reached by the age of 24 to 33 years in half of the patients with PH1 [27, 33]. The combination of both clinical and sonographic findings, that is, the association of stone passage, nephrocalcinosis, and renal impairment, is a strong argument for clinical diagnosis of PH1. Family history may bring additional information.
Crystalluria and infrared spectroscopy are of major interest for identification (qualitative) and quantitative analysis of crystals and stones (Figure 4), showing CaOx monohydrate crystals (type Ic whewellite) [34]. In patients with normal or significant residual GFR, concomitant hyperoxaluria (urine oxalate >1 mmol/1.73 m2 per day, reference value <0.5) and hyperglycoluria (urine glycolate >0.5 mmol/1.73 m2, reference value <0.5) are indicative of PH1, but some patients do not present with hyperglycoluria. The measurement of oxalate in a timed 24-hour urine collection corrected for body surface area is preferred for the diagnosis of PH [35]. Random urine oxalate/creatinine ratios can be useful to estimate oxalate excretion, particularly in infants, or patients with inability to provide complete 24-hour collection [36, 37]. Reference age-related values for oxalate and glycolate excretion have been established (Table 1). Urinary oxalate excretion may be falsely low in patients with decreased GFR because of oxalate retention and systemic deposition as calcium oxalate. In PH patients with GFR <30 mL/min per 1.73 m2, Pox is usually >20–30 μmol/L, and in those with ESRD, Pox is >50 μmol/L. In ESRD patients, plasma oxalate (± glycolate)/creatinine ratio and oxalate (± glycolate) measurement in dialysate might also be helpful for screening [38].
Figure 4.
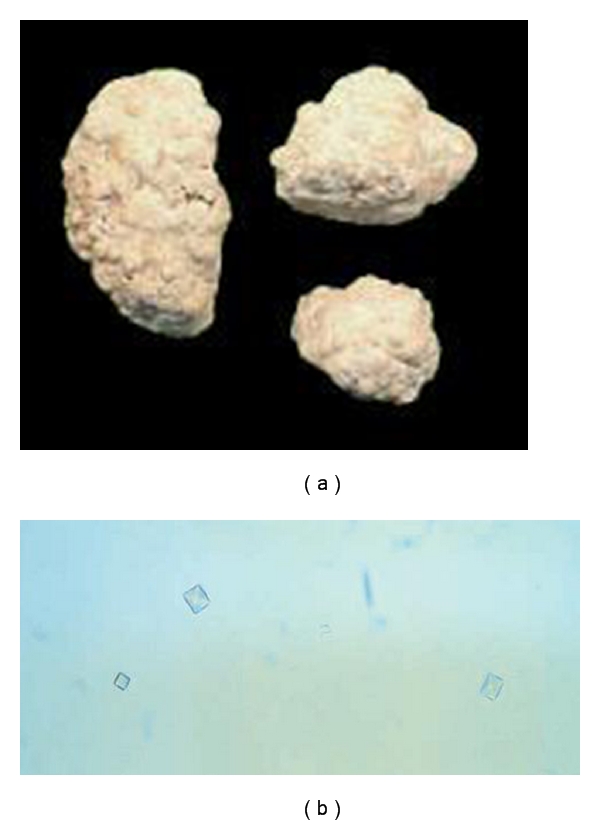
Calcium oxalate stones (a) and urine microscopic examination showing calcium oxalate monohydrate crystals (b) in PH1.
Table 1.
Reference age-related values for urinary oxalate, glycolate, and L-glycerate excretion (adapted from [8]).
| Units | Normal values | ||
|---|---|---|---|
| Oxalate | Glycolate | L-Glycerate | |
| mmol/1.73 m² per day | <0.5 | <0.5 | |
| μmol/l | <5 | ||
| mmol/mol creatinine | |||
| 0–6 months | <325–360 | <363–425 | 14–205 |
| 7–24 months | <132–174 | <245–293 | 14–205 |
| 2–5 years | <98–101 | <191–229 | 14–205 |
| 5–14 years | <70–82 | <166–186 | 23–138 |
| >16 years | <40 | <99–125 | <138 |
Until recently, liver biopsy to measure AGT catalytic activity has been essential for definitive diagnosis of PH1. As an alternative approach, genetic analysis of AGXT gene allows to detect mutations in most of suspected patients, thereby, supplanting the need for liver biopsy as a first step [39]. In the presence of atypical presentation or in patients with no mutation identified, however, a definitive diagnosis requires AGT activity measurement in liver tissue. The AGXT gene is located on chromosome 2 (2p37.3). It is noteworthy that among more than 150 mutations responsible for PH1 found throughout the 11 exons of the AGXT gene [40], many, corresponding to almost 50% of the patients, cosegregate with a so-called minor allele, the most prominent feature of which is a proline to leucine change in position 11 [41]. The frequency of this minor allele is highest in the Caucasian population, and it acts in synergy with some of the mutations, in particular, the common p.Gly170Arg change [41, 42]. If most of the mutations in PH1 are “private” mutations, some mutations occur more commonly [39]. The most frequent mutation, p.Gly170Arg, is found in 20 to 40% of patients and is associated with significant residual catalytic AGT activity in liver biopsies [43]. Some mutations are found among specific ethnic groups, the most obvious example being the p.Ile244Thr mutation which is found in many patients of North African/Spanish origin [44]. Prenatal diagnosis can be performed from DNA obtained from crude chorionic villi or amniocytes, on the basis of a restricted analysis of exons including mutation(s) identified in the index case. Such a procedure allows the identification of fetuses affected or not with PH1. Independently of the diagnostic value, mutation analysis may bring information on pyridoxine responsiveness, on complex enzyme phenotype, and sometimes on clinical prognosis [27, 40]. Although PH1 usually shows marked inter- and intrafamilial heterogeneity [45–47], genotype-phenotype correlations have been described for some mutations [27, 48].
5.2. Other Types of PH
Symptoms onset in PH2 typically occurs in childhood. Although patients with PH2 are also at risk for ESRD and systemic oxalosis, they appear to form less stones, have a less pronounced nephrocalcinosis, and a lower incidence of ESRD over time than PH1 patients [49, 50]. In the presence of hyperoxaluria without hyperglycoluria, a diagnosis of PH2 should be considered, especially when AGT activity is normal. The diagnosis is based on increased urinary excretion of L-glycerate (reference values in Table 1), but the definitive diagnosis may require the measurement of GRHPR activity in a liver biopsy [51, 52] as some PH2 patients have normal level of L-glycerate in urine [53]. Genotyping can provide reliable diagnosis for PH2. The GRPHR gene is located on chromosome 9 (9q11) [5] and more than 15 mutations have been identified so far.
A few patients referred to as non-1 non-2 PH have been described. These patients have a phenotype similar to PH1 or 2 but no AGT or GRHPR deficiency. Recently, a third type of PH, PH3, caused by mutations in the gene DHDPSL has been characterized among non-1 non-2 PH patients [6].
In this paper, we propose an algorithm for the diagnosis and the conservative treatment of PH as illustrated in the Figure 5.
Figure 5.
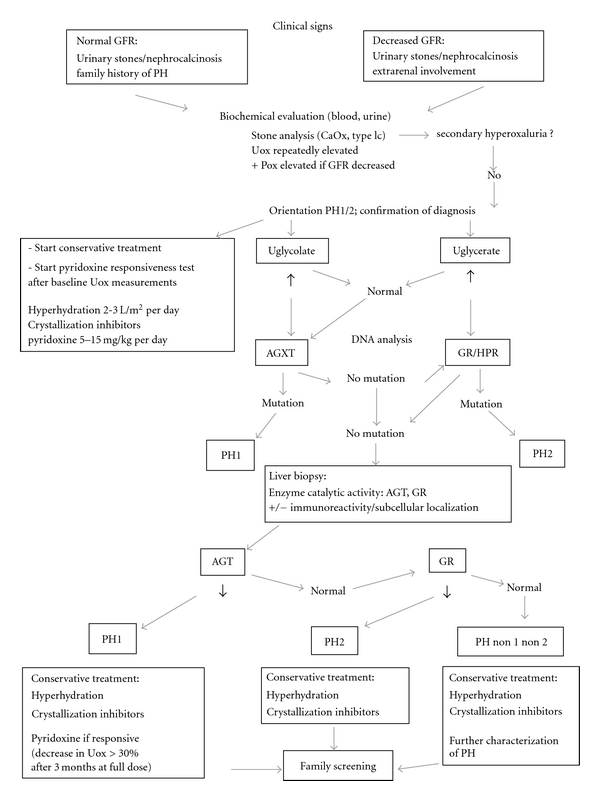
Proposed algorithm for the diagnosis and conservative treatment of primary hyperoxalurias. Abbreviations: PH: primary hyperoxaluria; GFR: glomerular filtration rate; Uox: urinary oxalate; Pox: plasma oxalate; CaOx: calcium oxalate; AGT: alanine:glyoxylate amino transferase; AGXT: AGT gene; GR/HPR: glyoxylate reductase/hydroxypyruvate reductase.
6. Treatment
6.1. Conservative Measures
Conservative measures should be initiated as soon as basal urinary oxalate measures have been completed and while renal function persists. Once ESRD has been reached, pyridoxine is the only stone treatment that should be pursued, if the patient is responsive. These measures apply to all types of PH with the exception of pyridoxine which is specific to some PH1 patients.
6.1.1. High Fluid Intake
The positive effect of high fluid intake has been proven in epidemiological and prospective intervention studies in stone formers [54]. In PH, the fluid intake recommended is >2 L/m2 per day (up to 3 L/m2 per day), distributed throughout the day and night. In small children and infants, a feeding or gastrostomy tube may be required. Special care should be taken in situations of fluid losses (diarrhea, vomiting, and fever) or limited oral hydration (surgery) and intravenous fluid intake instituted if necessary.
6.1.2. Diet
Oral calcium can bind oxalate in the bowels, and calcium restriction has been shown to result in higher oxalate intestinal absorption [55, 56]. Calcium intake should thus remain normal. A restriction in oxalate intake is of limited usefulness in PH patients as the main source of oxalate is endogenous. Excessive intake of vitamin C and vitamin D are to be avoided.
6.1.3. Inhibition of Calcium Oxalate Crystallization
Alkali citrate can reduce the urinary calcium oxalate saturation by forming complexes with calcium thus decreasing stone growth or nephrocalcinosis [57]. Potassium, or sodium, citrate at a dose of 100–150 mg/kg body weight per day (0.3–0.5 mmol/kg) is recommended. Pyrophosphate ions may decrease calcium oxalate crystallization although orthophosphate has never been evaluated in isolation of other treatments [58]. Moderate doses of elemental phosphorus 20–30 mg/kg body weight per day may be administered. There is as yet no evidence that probiotics, such as Oxalobacter formigenes despite its ability to metabolize oxalate [59], can significantly decrease urinary oxalate excretion in PH patients.
6.1.4. Pyridoxine
The cofactor of AGT is pyridoxal phosphate, one of the B6 vitamins. The administration of pyridoxine hydrochloride has been shown to be associated with a decrease in urinary oxalate in about 10–30% of patients with PH [60, 61]. The metabolic basis of pyridoxine responsiveness is unclear. All PH1 patients should be tested for pyridoxine responsiveness and treated until liver transplantation is performed, if responsive, even while undergoing hemodialysis. Studies have determined that a subset of patients with PH1, carrying 1 or 2 copies of AGT mutations (p.Gly170Arg or p.Phe152Ile), may be responsive to pharmacological doses of pyridoxine, though this list is not limitative [48, 62]. Responsiveness has been defined as a minimum 30% decrease in urinary oxalate excretion after a test period of a minimum 3 months at maximum dose [61]. The starting dose recommended is 5 mg/kg per day and can be increased to 10 mg/kg per day. No sensory neurotoxicity has been described in PH patients, and a dose of 500 mg per day is thought to be below toxic levels. Absorption of pyridoxine may vary between patients, and assessing plasma levels of pyridoxal phosphate may be useful, though therapeutic levels are not defined.
Added monitoring of the tolerance and efficacy of these measures may require evaluation of stone formation, and nephrocalcinosis by ultrasounds or X-ray, urinary pH and volume, urinary citrate, magnesium and oxalate excretion and supersaturation, and crystalline volume in the urines. In summary, an aggressive supportive management should be started as soon as the diagnosis of PH has been suspected and may improve renal survival provided compliance is optimal [63].
6.2. Urology
The treatment of stones should avoid open and percutaneous surgery because further renal lesions will alter GFR. The use of extracorporeal shock wave lithotripsy may be an available option in selected patients, but the presence of nephrocalcinosis may be responsible for parenchymal damage. In patients with repeated renal colic, a ureteral JJ stent may be helpful for pain control and protection of renal damage. Bilateral nephrectomy may be proposed in patients on renal replacement therapy in order to limit the risk of infection, obstruction, and passage of stones.
6.3. Dialysis
Conventional dialysis is inadequate for patients who have reached ESRD, because it cannot overcome the ongoing oxalate production [64]. This results in continuing tissue deposition of oxalate and risk of organ damage. The removal of oxalate by hemodialysis exceeds that by peritoneal dialysis [65]. Better results may be obtained by combining daily high-flux hemodialysis and peritoneal dialysis or by long daily hemodialysis sessions [66, 67].
In summary, there are limited indications for dialysis in patients with PH1 [30]: (1) if diagnosis of PH1 has not been established, (2) in small children with infantile oxalosis waiting for organ Tx, (3) in preparation for kidney Tx, before or after liver Tx, in order to deplete oxalate from the body, (4) following isolated kidney or combined liver-kidney Tx with any delay in achieving optimal renal function, as a temporary adjunct in the case of high oxalate burden, or with transient loss of transplant function, (5) very exceptionally in older patients if the only alternative is no dialysis, (6) in developing countries; hemodialysis (or even less satisfactory, peritoneal dialysis) may be indicated as only a preference to absolute withdrawal of all therapy.
7. Organ Transplantation
Ideally, organ transplantation should be planned prior to the onset of ESRD and systemic oxalosis. The different options for organ Tx for PH1 according to residual GFR are summarized in Table 2 [68].
Table 2.
Suggestions for organ transplantation strategies in PH1 patients according to residual GFR (ml/min per 1.73 m²), systemic involvement, and local facilities (from [68]).
| Tx strategy | Combined simultaneous liver + kidney | Liver first Kidney as a second step | Isolated kidney | Isolated liver |
|---|---|---|---|---|
| HD strategy | Peroperative ± postoperative according to Pox and GFR | Standard HD following liver Tx aiming at Pox <20 μmol/L | Pre- and peroperative | Sometimes peroperative |
| 40 < GFR < 60 | No | No | No | Optional |
| 20 < GFR < 40 | Yes | No | (1) In developing countries (2) In very selected patients |
No |
| GFR < 20 | Yes | Yes | No | No |
| Infantile form (ESRD <2 years) |
Yes | Yes (emergency) | No | No |
Abbreviations: ESRD end-stage renal disease; GFR glomerular filtration rate; HD hemodialysis; Pox plasma oxalate; Tx transplantation.
7.1. Kidney Transplantation
Kidney Tx allows significant removal of soluble Pox [69]. However, because the biochemical defect is in the liver, overproduction of oxalate and subsequent deposition in tissues continues unabated. The high rate of urinary oxalate excretion originates from both ongoing oxalate production from the native liver and oxalate deposits in tissues leading to high risk for disease recurrence. Isolated kidney Tx for PH1 is no longer recommended, because frequent recurrence leads to poor graft survival and patient quality of life. In Europe, the European Dialysis Transplantation Association (EDTA) registry reported poor results of isolated kidney Tx two decades ago with a 3-year graft survival of 17 to 23% according to donor source [70]. A United States Renal Data System (USRDS) analysis of 190 adults transplanted in the US between 1988 and 1998 showed superior death-censored kidney graft survival in combined liver-kidney Tx than in isolated kidney Tx, but no difference in patient survival [71]. More recently, data from 58 PH1 patients showed a death-censored kidney graft survival at 3 years of 95% with combined liver-kidney Tx versus 56% with isolated kidney Tx [72]. Proven pyridoxine responsive patients may benefit from isolated kidney Tx, provided pyridoxine is maintained, but the clinical evidence for this is still sparse. Isolated kidney Tx may be also regarded as a temporary solution in some developing countries before managing the patient in a specialized center for further combined liver-kidney procedure.
Isolated kidney Tx might be the treatment of choice in patients with PH2. Indeed, PH2 patients with kidney Tx alone appear to have a more favorable course than PH1 patients, and the benefit of liver Tx in such patients is still unclear.
7.2. Liver Transplantation
Since the liver is the only organ responsible for glyoxylate detoxification by AGT, the excessive production of oxalate will continue as long as the native liver is left in place. Therefore, PH1 can be cured only when the deficient host liver has been removed. Liver Tx is a form of gene therapy as well as enzyme replacement therapy as it will supply the missing enzyme in the correct organ (liver), cell (hepatocyte), and intracellular compartment (peroxisome).
In Europe, combined liver-kidney Tx has been the preferred approach in the past 25 years. A European PH1 Transplant registry reported 127 liver Tx including more than 100 liver-kidney Tx in 117 patients between 1984 and 2004 [73]. Results were encouraging with patient survival rates of 86, 80, and 69% at 1 year, 5 years, and 10 years, respectively. There were 13 kidney graft failures. Comparable results have been reported from the USRDS, with a patient survival above 80% at 5 years and a death-censored graft survival of 76% at 8 years post Tx [71]. Such a strategy can be successfully proposed to infants with PH1 [74]. Worldwide experience indicates that once perioperative mortality is avoided, combined liver-kidney Tx seems to be an acceptable treatment for PH1 [75–77]. The strategy of combined liver-kidney Tx may be influenced by the stage of the disease (Table 2). Simultaneous liver and kidney Tx is logical in patients with a GFR between 15 and 40 mL/min per 1.73 m², because, at this level, oxalate retention increases rapidly. A sequential procedure (first liver Tx, then dialysis until sufficient oxalate has been cleared from the body, followed by kidney Tx) may be proposed to ESRD patients, mainly infants with a long waiting time [78].
Preemptive isolated liver Tx might be the first option in selected patients before advanced chronic renal failure has occurred, that is, at a GFR between 60 and 40 mL/min/1.73 m². In the Hamburg experience, 4 pediatric recipients with a GFR between 27 and 98 mL/min per 1.73 m² received a preemptive liver transplant, and 3 of them still have significant residual renal function after a median followup of ~12 years [79]. Another group reported good results at 5 years in 4 PH1 children who received a preemptive liver Tx with a mean pretransplant GFR of 81 mL/min/1.73 m² [80]. Such a strategy has a strong rationale but raises ethical controversies especially when the GFR is superior to 60 mL/min per 1.73 m². Indeed PH1 is the only peroxisomal disease without psychomotor delay due to cerebral involvement, and the conservative management of PH1 patients has significantly improved during the last 10 years; this may influence the role of preemptive liver Tx in such patients.
The choice of donor source may be based either on immunological bases (i.e., using the same donor for both organs) or on biochemical rationale (i.e., using a two-step procedure according to oxalate body store). Indeed most publication currently report on the use of cadaver donors but a living related donor may be considered under certain conditions [78].
7.3. Posttransplantation Reversal of Renal and Extrarenal Involvement
After combined liver-kidney Tx, urinary glycolate immediately returns to normal and Pox returns to normal before urine oxalate does [81]. Indeed, urinary oxalate can remain elevated for as long as several years post-Tx, due to the slow resolubilization of systemic CaOx deposition [72]. Therefore, recurrent nephrocalcinosis or renal calculi are still a risk and may jeopardize graft function. Thus, independent of the Tx strategy, the kidney must be protected against the damage that can be induced by heavy oxalate load suddenly released from tissues. Forced fluid intake (3–5 L per 1.73 m² per day) supported by the use of crystallization inhibitors is the most important approach. Pox, crystalluria, and CaOx saturation are helpful tools in renal management after combined liver-kidney Tx. The benefit of daily high-efficiency post-transplant hemodialysis is still debated and should be limited to patients with significant systemic involvement (Table 2). It will provide a rapid drop in Pox and, therefore, reduce the exposure of the transplanted organs to high Pox, but it may also increase the risk of CaOx supersaturation due to reduction in urine volume in the case of inappropriate fluid removal. However, posttransplant hemodialysis is mandatory in patients with acute tubular necrosis or delayed graft function.
8. Conclusion and Future Prospects
Patients with hyperoxaluria should be referred for diagnosis and management to reference centers with interest and experience in the conditions and access to the appropriate biochemical and molecular biological facilities. Major advances in biochemistry, enzymology, genetics, and management have been achieved during recent years. Improved knowledge of the disease, early and accurate diagnosis, before renal failure occurs, and aggressive supportive treatment are of critical importance for the prognosis. In (pre)ESRD patients, the greatest experience has been obtained with one-step combined liver-kidney transplantation. New insights into potential therapies including the restoration of defective enzymatic activity through the use of chemical chaperones and hepatocyte cell transplantation, or recombinant gene therapy for enzyme replacement [82–84], provide hope for curative treatments of primary hyperoxalurias in the future.
References
- 1.Danpure CJ, Jennings PR. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Letters. 1986;201(1):20–24. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 2.Purdue PE, Lumb MJ, Fox M, et al. Characterization and chromosomal mapping of a genomic clone encoding human alanine: glyoxylate aminotransferase. Genomics. 1991;10(1):34–42. doi: 10.1016/0888-7543(91)90481-s. [DOI] [PubMed] [Google Scholar]
- 3.Mistry J, Danpure CJ, Chalmers RA. Hepatic D-glycerate dehydrogenase and glyoxylate reductase deficiency in primary hyperoxaluria type 2. Biochemical Society Transactions. 1988;16(4):626–627. [Google Scholar]
- 4.Rumsby G, Cregeen DP. Identification and expression of a cDNA for human hydroxypyruvate/glyoxylate reductase. Biochimica et Biophysica Acta. 1999;1446(3):383–388. doi: 10.1016/s0167-4781(99)00105-0. [DOI] [PubMed] [Google Scholar]
- 5.Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Human Molecular Genetics. 1999;8(11):2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 6.Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. American Journal of Human Genetics. 2010;87(3):392–399. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danpure CJ. Primary hyperoxaluria. In: Scriver CR, Beaudet A, Sly WS, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 2. New York, NY, USA: McGraw-Hill; 2001. pp. 3323–3367. [Google Scholar]
- 8.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney International. 2009;75(12):1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Roe SM, Hou Y, et al. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. Journal of Molecular Biology. 2003;331(3):643–652. doi: 10.1016/s0022-2836(03)00791-5. [DOI] [PubMed] [Google Scholar]
- 10.Booth MPS, Conners R, Rumsby G, Brady RL. Structural basis of substrate specificity in human glyoxylate reductase/hydroxypyruvate reductase. Journal of Molecular Biology. 2006;360(1):178–189. doi: 10.1016/j.jmb.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Nephrology Dialysis Transplantation. 1995;10(8):3–7. doi: 10.1093/ndt/10.supp8.3. [DOI] [PubMed] [Google Scholar]
- 12.Kopp N, Leumann E. Changing pattern of primary hyperoxaluria in Switzerland. Nephrology Dialysis Transplantation. 1995;10(12):2224–2227. doi: 10.1093/ndt/10.12.2224. [DOI] [PubMed] [Google Scholar]
- 13.van Woerden CS, Groothoff JW, Wanders RJA, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrology Dialysis Transplantation. 2003;18(2):273–279. doi: 10.1093/ndt/18.2.273. [DOI] [PubMed] [Google Scholar]
- 14.Lorenzo V, Alvarez A, Torres A, Torregrosa V, Hernández D, Salido E. Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: single-center experience. Kidney International. 2006;70(6):1115–1119. doi: 10.1038/sj.ki.5001758. [DOI] [PubMed] [Google Scholar]
- 15.North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Annual Report. Rockville, Md, USA: The EMMES Corporation; 2010. [Google Scholar]
- 16.Lewis M, Shaw J, Reid C, Evans J, Webb N, Verrier-Jones K. Demography and management of childhood established renal failure in the UK (chapter 13) Nephrology, Dialysis, Transplantation. 2007;22:vii165–175. doi: 10.1093/ndt/gfm336. [DOI] [PubMed] [Google Scholar]
- 17.Hattori S, Yosioka K, Honda M, Ito H. The 1998 report of Japanese National Registry data on pediatric end-stage renal disease patients. Pediatric Nephrology. 2002;17(6):456–461. doi: 10.1007/s00467-002-0848-8. [DOI] [PubMed] [Google Scholar]
- 18.Al-Eisa AA, Samhan M, Naseef M. End-stage renal disease in Kuwaiti children: an 8-year experience. Transplantation Proceedings. 2004;36(6):1788–1791. doi: 10.1016/j.transproceed.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatric Nephrology. 1996;10(4):479–482. doi: 10.1007/s004670050143. [DOI] [PubMed] [Google Scholar]
- 20.Marangella M, Vitale C, Petrarulo M, et al. Bony content of oxalate in patients with primary hyperoxaluria or oxalosis-unrelated renal failure. Kidney International. 1995;48(1):182–187. doi: 10.1038/ki.1995.283. [DOI] [PubMed] [Google Scholar]
- 21.Toussaint C, Vienne A, De Pauw L, et al. Combined liver-kidney transplantation in primary hyperoxaluria type 1: bone histopathology and oxalate body content. Transplantation. 1995;59(12):1700–1704. doi: 10.1097/00007890-199506270-00010. [DOI] [PubMed] [Google Scholar]
- 22.Day DL, Scheinman JI, Mahan J. Radiological aspects of primary hyperoxaluria. American Journal of Roentgenology. 1986;146(2):395–401. doi: 10.2214/ajr.146.2.395. [DOI] [PubMed] [Google Scholar]
- 23.Ring E, Wendler H, Ratschek M, Zobel G. Bone disease of primary hyperoxaluria in infancy. Pediatric Radiology. 1989;20(1-2):131–133. doi: 10.1007/BF02010661. [DOI] [PubMed] [Google Scholar]
- 24.Fisher D, Hiller N, Drukker A. Oxalosis of bone: report of four cases and a new radiological staging. Pediatric Radiology. 1995;25(4):293–295. doi: 10.1007/BF02011105. [DOI] [PubMed] [Google Scholar]
- 25.Behnke B, Kemper MJ, Kruse HP, Müller-Wiefel DE. Bone mineral density in children with primary hyperoxaluria type I. Nephrology Dialysis Transplantation. 2001;16(11):2236–2239. doi: 10.1093/ndt/16.11.2236. [DOI] [PubMed] [Google Scholar]
- 26.Bacchetta J, Fargue S, Boutroy S, et al. Bone metabolism in oxalosis: a single-center study using new imaging techniques and biomarkers. Pediatric Nephrology. 2010;25(6):1081–1089. doi: 10.1007/s00467-010-1453-x. [DOI] [PubMed] [Google Scholar]
- 27.Harambat J, Fargue S, Acquaviva C, et al. Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney International. 2010;77(5):443–449. doi: 10.1038/ki.2009.435. [DOI] [PubMed] [Google Scholar]
- 28.Takayama T, Nagata M, Ichiyama A, Ozono S. Primary hyperoxaluria type 1 in Japan. American Journal of Nephrology. 2005;25(3):297–302. doi: 10.1159/000086361. [DOI] [PubMed] [Google Scholar]
- 29.Cochat P, Koch Nogueira PC, Ayman Mabmoud M, Jamieson NV, Scheinman JI, Rolland MO. Primary hyperoxaluria in infants: medical, ethical, and economic issues. Journal of Pediatrics. 1999;135(6):746–750. doi: 10.1016/s0022-3476(99)70095-8. [DOI] [PubMed] [Google Scholar]
- 30.Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: still challenging! Pediatric Nephrology. 2006;21(8):1075–1081. doi: 10.1007/s00467-006-0124-4. [DOI] [PubMed] [Google Scholar]
- 31.Diallo O, Janssens F, Hall M, Avni EF. Type I primary hyperoxaluria in pediatric patients: renal sonographic patterns. American Journal of Roentgenology. 2004;183(6):1767–1770. doi: 10.2214/ajr.183.6.01831767. [DOI] [PubMed] [Google Scholar]
- 32.Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatric Nephrology. 2003;18(10):986–991. doi: 10.1007/s00467-003-1234-x. [DOI] [PubMed] [Google Scholar]
- 33.Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. American Journal of Nephrology. 2005;25(3):290–296. doi: 10.1159/000086360. [DOI] [PubMed] [Google Scholar]
- 34.Daudona M, Jungers P. Clinical value of crystalluria and quantitative morphoconstitutional analysis of urinary calculi. Nephron - Physiology. 2004;98(2):p31–p36. doi: 10.1159/000080261. [DOI] [PubMed] [Google Scholar]
- 35.Gibbs DA, Watts RWE. The variation of urinary oxalate excretion with age. The Journal of Laboratory and Clinical Medicine. 1969;73(6):901–908. [PubMed] [Google Scholar]
- 36.Leumann EP, Dietl A, Matasovic A. Urinary oxalate and glycolate excretion in healthy infants and children. Pediatric Nephrology. 1990;4(5):493–497. doi: 10.1007/BF00869828. [DOI] [PubMed] [Google Scholar]
- 37.Hoppe B, Leumann E, Milliner D. Urolithiasis in childhood. In: Geary D, Schäfer F, editors. Comprehensive Pediatric Nephrology. New York, NY, USA: Elsevier/WB Saunders; 2008. pp. 499–525. [Google Scholar]
- 38.Wong PN, Law ELK, Tong GMW, Mak SK, Lo KY, Wong AKM. Diagnosis of primary hyperoxaluria type 1 by determination of peritoneal dialysate glycolic acid using standard organic-acids analysis method. Peritoneal Dialysis International. 2003;23(2):S210–S213. [PubMed] [Google Scholar]
- 39.Rumsby G, Williams E, Coulter-Mackie M. Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney International. 2004;66(3):959–963. doi: 10.1111/j.1523-1755.2004.00842.x. [DOI] [PubMed] [Google Scholar]
- 40.Williams EL, Acquaviva C, Amoroso A, et al. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Human Mutation. 2009;30(6):910–917. doi: 10.1002/humu.21021. [DOI] [PubMed] [Google Scholar]
- 41.Purdue PE, Takada Y, Danpure CJ. Identification of mutations associated with peroxisome-to-mitochondrian mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria Type 1. Journal of Cell Biology. 1990;111(6):2341–2351. doi: 10.1083/jcb.111.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lumb MJ, Danpure CJ. Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. Journal of Biological Chemistry. 2000;275(46):36415–36422. doi: 10.1074/jbc.M006693200. [DOI] [PubMed] [Google Scholar]
- 43.Danpure CJ. Primary hyperoxaluria: from gene defects to designer drugs? Nephrology Dialysis Transplantation. 2005;20(8):1525–1529. doi: 10.1093/ndt/gfh923. [DOI] [PubMed] [Google Scholar]
- 44.Santana A, Salido E, Torres A, Shapiro LJ. Primary hyperoxaluria type 1 in the Canary Islands: a conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(12):7277–7282. doi: 10.1073/pnas.1131968100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoppe B, Danpure CJ, Rumsby G, et al. A vertical (pseudodominant) pattern of inheritance in the autosomal recessive disease primary hyperoxaluria type 1: lack of relationship between genotype, enzymic phenotype, and disease severity. American Journal of Kidney Diseases. 1997;29(1):36–44. doi: 10.1016/s0272-6386(97)90006-8. [DOI] [PubMed] [Google Scholar]
- 46.Amoroso A, Pirulli D, Florian F, et al. AGXT gene mutations and their influence on clinical heterogeneity of type 1 primary hyperoxaluria. Journal of the American Society of Nephrology. 2001;12(10):2072–2079. doi: 10.1681/ASN.V12102072. [DOI] [PubMed] [Google Scholar]
- 47.Frishberg Y, Rinat C, Shalata A, et al. Intra-familial clinical heterogeneity: absence of genotype-phenotype correlation in primary hyperoxaluria type 1 in Israel. American Journal of Nephrology. 2005;25(3):269–275. doi: 10.1159/000086357. [DOI] [PubMed] [Google Scholar]
- 48.Van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJA, Waterham HR. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney International. 2004;66(2):746–752. doi: 10.1111/j.1523-1755.2004.00796.x. [DOI] [PubMed] [Google Scholar]
- 49.Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney International. 2001;59(1):31–36. doi: 10.1046/j.1523-1755.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 50.Johnson SA, Rumsby G, Cregen D, Hulton SA. Primary hyperoxaluria type 2 in children. Pediatric Nephrology. 2002;17(8):597–601. doi: 10.1007/s00467-002-0858-6. [DOI] [PubMed] [Google Scholar]
- 51.Giafi CF, Rumsby G. Kinetic analysis and tissue distribution of human D-glycerate dehydrogenase/glyoxylate reductase and its relevance to the diagnosis of primary hyperoxaluria type 2. Annals of Clinical Biochemistry. 1998;35(1):104–109. doi: 10.1177/000456329803500114. [DOI] [PubMed] [Google Scholar]
- 52.Marangella M, Petrarulo M, Cosseddu D, et al. Detection of primary hyperoxaluria type 2 (L-glyceric aciduria) in patients with maintained renal function or end-stage renal failure. Nephrology Dialysis Transplantation. 1995;10(8):1381–1385. [PubMed] [Google Scholar]
- 53.Rumsby G, Sharma A, Cregeen DP, Solomon LR. Primary hyperoxaluria type 2 without L-glycericaciduria: is the disease under-diagnosed? Nephrology Dialysis Transplantation. 2001;16(8):1697–1699. doi: 10.1093/ndt/16.8.1697. [DOI] [PubMed] [Google Scholar]
- 54.Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. Journal of Urology. 1996;155(3):839–843. [PubMed] [Google Scholar]
- 55.Von Unruh GE, Voss S, Sauerbruch T, Hesse A. Dependence of oxalate absorption on the daily calcium intake. Journal of the American Society of Nephrology. 2004;15(6):1567–1573. doi: 10.1097/01.asn.0000127864.26968.7f. [DOI] [PubMed] [Google Scholar]
- 56.Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney International. 2001;59(1):270–276. doi: 10.1046/j.1523-1755.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- 57.Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatric Nephrology. 1993;7(2):207–211. doi: 10.1007/BF00864405. [DOI] [PubMed] [Google Scholar]
- 58.Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. New England Journal of Medicine. 1994;331(23):1553–1558. doi: 10.1056/NEJM199412083312304. [DOI] [PubMed] [Google Scholar]
- 59.Hoppe B, Beck B, Gatter N, et al. Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney International. 2006;70(7):1305–1311. doi: 10.1038/sj.ki.5001707. [DOI] [PubMed] [Google Scholar]
- 60.Watts RWE, Veall N, Purkiss P. The effect of pyridoxine on oxalate dynamics in three cases of primary hyperoxaluria (with glycollic aciduria) Clinical Science. 1985;69(1):87–90. doi: 10.1042/cs0690087. [DOI] [PubMed] [Google Scholar]
- 61.Leumann E, Hoppe B. The primary hyperoxalurias. Journal of the American Society of Nephrology. 2001;12(9):1986–1993. doi: 10.1681/ASN.V1291986. [DOI] [PubMed] [Google Scholar]
- 62.Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney International. 2005;67(5):1704–1709. doi: 10.1111/j.1523-1755.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- 63.Fargue S, Harambat J, Gagnadoux MF, et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney International. 2009;76(7):767–773. doi: 10.1038/ki.2009.237. [DOI] [PubMed] [Google Scholar]
- 64.Marangella M, Petrarulo M, Cosseddu D, Vitale C, Linari F. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. American Journal of Kidney Diseases. 1992;19(6):546–553. doi: 10.1016/s0272-6386(12)80833-x. [DOI] [PubMed] [Google Scholar]
- 65.Hoppe B, Graf D, Offner G, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatric Nephrology. 1996;10(4):488–492. doi: 10.1007/s004670050145. [DOI] [PubMed] [Google Scholar]
- 66.Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney International. 2006;70(9):1642–1648. doi: 10.1038/sj.ki.5001806. [DOI] [PubMed] [Google Scholar]
- 67.Díaz C, Catalinas FG, De Alvaro F, Torre A, Sánchez C, Costero O. Long daily hemodialysis sessions correct systemic complications of oxalosis prior to combined liver-kidney transplantation: case report. Therapeutic Apheresis and Dialysis. 2004;8(1):52–55. doi: 10.1111/j.1526-0968.2004.00106.x. [DOI] [PubMed] [Google Scholar]
- 68.Cochat P, Fargue S, Harambat J. Primary hyperoxaluria type 1: strategy for organ transplantation. Current Opinion in Organ Transplantation. 2010;15(5):590–593. doi: 10.1097/MOT.0b013e32833e35f5. [DOI] [PubMed] [Google Scholar]
- 69.Scheinman JI, Najarian JS, Mauer SM. Successful strategies for renal transplantation in primary oxalosis. Kidney International. 1984;25(5):804–811. doi: 10.1038/ki.1984.93. [DOI] [PubMed] [Google Scholar]
- 70.Broyer M, Brunner FP, Brynger H, et al. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrology Dialysis Transplantation. 1990;5(5):332–336. doi: 10.1093/ndt/5.5.332. [DOI] [PubMed] [Google Scholar]
- 71.Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU. Renal allograft survival in patients with oxalosis. Transplantation. 2002;74(5):707–710. doi: 10.1097/00007890-200209150-00020. [DOI] [PubMed] [Google Scholar]
- 72.Bergstralh EJ, Monico CG, Lieske JC, et al. Transplantation outcomes in primary hyperoxaluria. American Journal of Transplantation. 2010;10(11):2493–2501. doi: 10.1111/j.1600-6143.2010.03271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jamieson NV. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. American Journal of Nephrology. 2005;25(3):282–289. doi: 10.1159/000086359. [DOI] [PubMed] [Google Scholar]
- 74.Millan MT, Berquist WE, So SK, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation. 2003;76(10):1458–1463. doi: 10.1097/01.TP.0000084203.76110.AC. [DOI] [PubMed] [Google Scholar]
- 75.Gagnadoux MF, Lacaille F, Niaudet P, et al. Long term results of liver-kidney transplantation in children with primary hyperoxaluria. Pediatric Nephrology. 2001;16(12):946–950. doi: 10.1007/s004670100001. [DOI] [PubMed] [Google Scholar]
- 76.Ellis SR, Hulton SA, McKiernan PJ, De Goyet JVD, Kelly DA. Combined liver-kidney transplantation for primary hyperoxaluria type 1 in young children. Nephrology Dialysis Transplantation. 2001;16(2):348–354. doi: 10.1093/ndt/16.2.348. [DOI] [PubMed] [Google Scholar]
- 77.Shapiro R, Weismann I, Mandel H, et al. Primary hyperoxaluria type 1: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation. 2001;72(3):428–432. doi: 10.1097/00007890-200108150-00012. [DOI] [PubMed] [Google Scholar]
- 78.Malla I, Lysy PA, Godefroid N, et al. Two-step transplantation for primary hyperoxaluria: cadaveric liver followed by living donor related kidney transplantation. Pediatric Transplantation. 2009;13(6):782–784. doi: 10.1111/j.1399-3046.2008.01049.x. [DOI] [PubMed] [Google Scholar]
- 79.Brinkert F, Ganschow R, Helmke K, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87(9):1415–1421. doi: 10.1097/TP.0b013e3181a27939. [DOI] [PubMed] [Google Scholar]
- 80.Perera MTPR, Sharif K, Lloyd C, et al. Pre-emptive liver transplantation for primary hyperoxaluria (PH-I) arrests long-term renal function deterioration. Nephrology Dialysis Transplantation. 2011;26(1):354–359. doi: 10.1093/ndt/gfq353. [DOI] [PubMed] [Google Scholar]
- 81.Inoue Y, Masuyama H, Ikawa H, Mitsubuchi H, Kuhara T. Monitoring method for pre- and post-liver transplantation in patients with primary hyperoxaluria type I. Journal of Chromatography B. 2003;792(1):89–97. doi: 10.1016/s1570-0232(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 82.Koul S, Johnson T, Pramanik S, Koul H. Cellular transfection to deliver alanine-glyoxylate aminotransferase to hepatocytes: a rational gene therapy for Primary Hyperoxaluria-1 (PH-1) American Journal of Nephrology. 2005;25(2):176–182. doi: 10.1159/000085410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang J, Salido EC, Guha C, et al. Correction of hyperoxaluria by liver repopulation with hepatocytes in a mouse model of primary hyperoxaluria type-1. Transplantation. 2008;85(9):1253–1260. doi: 10.1097/TP.0b013e31816de49e. [DOI] [PubMed] [Google Scholar]
- 84.Salido E, Rodriguez-Pena M, Santana A, Beattie SG, Petry H, Torres A. Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Molecular Therapy. 2011;19:870–875. doi: 10.1038/mt.2010.270. [DOI] [PMC free article] [PubMed] [Google Scholar]