

Abstract
Calciphylaxis or calcific uremic arteriolopathy is an infrequent complication of end stage kidney disease. It is characterized by arteriolar medial calcification, thrombotic cutaneous ischemia, tissue necrosis often leading to ulceration, secondary infection and increased mortality rates. Current, multimodality treatment involves local wound care, well-controlled calcium, phosphate and parathyroid hormone levels and combination therapy with sodium thiosulfate and hyperbaric oxygen therapy. This combination therapy may be changing the historically poor prognosis of calcific uremic arteriolopathy reported in the literature. Peritoneal dialysis is considered a risk factor based on limited publications, however this remains to be proven. Clinical presentation, diagnosis, pathogenesis and treatment of calcific uremic arteriolopathy in these patients are no different from other patients manifesting with this condition.
1. Introduction
Calcific uremic arteriolopathy (CUA), commonly referred to as calciphylaxis, is an uncommon but increasingly recognized disorder associated with high morbidity and mortality. Although the disorder was first reported in 1898 [1], the term calciphylaxis was coined by Selye in 1962 as a condition of systemic hypersensitivity induced by a sensitizing agent that resulted in metastatic calcification in various organs analogous to anaphylaxis [2].
It is characterized by tender plaques or nodules with violaceous discoloration associated with severe pain that is often refractory to standard analgesia [3]. Historically, CUA has been associated with significant morbidity including debilitating pain, surgical resections, and amputations. More than 50% die within one year of diagnosis, usually due to superimposed sepsis [4]. Approximately 1–4% patients with chronic kidney disease (CKD) manifest CUA, and a recent report estimates a prevalence of 4% in patients with end-stage kidney disease (ESKD) [5]. The pathogenesis of CUA is poorly understood; however, many putative risk factors such as female gender, hyperphosphatemia, hypercalcemia, high calcium phosphorous product [6], use of calcium containing phosphate binders, and hypercoagulable states are reported [6, 7]. Nonuremic CUA is also reported, mostly in the context of primary hyperparathyroidism, malignancy, alcoholic liver disease, or connective tissue diseases [8]. Mineral abnormalities that are postulated in CUA are often absent in these patients suggesting that various factors play a role in its pathogenesis. Peritoneal dialysis (PD) is shown to be a risk factor for CUA in one prospective cohort [9] but has not been subsequently substantiated adequately. Further, the exact mechanism of presumed increased incidence of CUA in PD patients is not well described and in one report was attributed to the use of calcium containing phosphate binders [10]. The introduction of early aggressive therapy with sodium thiosulfate (STS) and hyperbaric oxygen therapy (HBO), in addition to local wound care, calcium, phosphate, and parathyroid hormone (PTH) control, is hoped to improve the very poor outcome of this debilitating condition. The aim of this paper is to study the clinical features, pathogenesis and management of CUA in PD patients.
2. Clinical Presentation
CUA is now a well-described entity in the nephrology literature in both hemodialysis (HD) and PD patients. A spectrum of disease has been described (Table 1), most likely representing a continuum dependent on both the underlying severity and the duration of disease.
Table 1.
CUA cases in PD population described within the literature.
| Patient numbers, patient characteristics | PD duration | Clinical presentation | CCa2+ (mmol/L) | PO4− (mmol/L) | PTH (ng/L) | Treatment | Outcome | Reference |
|---|---|---|---|---|---|---|---|---|
| n = 54 most female diabetics | > 2 months | 73% subcutaneous plaques 95% lower legs (95% bilateral) 5% abdomina l wall | n/a | n/a | n/a | Parathyroidectomy (n = 6) | 5 improve | |
| [11] | ||||||||
| Change to HD (n = 13) | 11 improve | |||||||
| Steroid therapy (n = 19) | 3 die within 3 month8 improve 8 no change | |||||||
| n = 169 female HTN, IHD, renal calculi, osteoarthritis, graves thyroid disease, osteoporosis | 3 months | Unilateral right calf violaceous lesion 1.5 inch, progressing to bilateral indurated lesions calf and thighs | 2.22 | 2.10 | 109 | D/C caltrate, calcitriol start sevelamer prednisolone STS (IV) | Resistant to all but STS, with improved pain in 2 weeks, and reduced lesion size in 8 weeks | [12] |
| n = 126 female 2 failed transplant not on calcium salts | 4 years | Violaceous lesions progressing in 6 weeks to ulcerations of left proximal arm and bilateral thigh | 2.4 | 1.77 | 89 | Prior parathyroidectomy, initially STS (IV) 2 months STS (IP) 4 months | STS(IV) response but D/C due to N&V. STS(IP) no response and died from sepsis | [13] |
| n = 153 female Moroccan obese, IDDM, IHD, CVA | 3 years | Redness right breast progressed to ulcerate Further progressed to bilateral breast and fingertip ulceration. Further progression to thigh | 2.7 | 2.0 | 1376 | Parathyroidectomy mastectomy STS—although very late | Poor response to all. Poor wound healing. Died from sepsis | [14] |
| n = 117 male Afro-American Wegener's granulomatosis (receiving cyclophosphamide and prednisolone) previous warfarin (right atrial thrombus) | 3 years | Erythema and swelling bilateral metatarsals, progressed to necrotic feet and fingers, with scintigraphy evidence of widespread disease | 2.42 | 2.68 | 321 | D/C caltrate Sevelamer preexisting Aluminium commenced STS (IV) 3 months HBO Change to HD | Pain relief with STS but lesions progress. HBO did not halt Proceed to bilateral BKA. Change to HD 5x/week and disease healed | [15] |
PD: peritoneal dialysis, CCa2+: corrected calcium, PO4−: phosphate, PTH: parathyroid hormone, n: number, HD: hemodialysis, HTN: hypertension, IHD: ischemic heart disease, D/C: discontinue, STS: sodium thiosulphate, IV: intravenous, IP: intraperitoneal, N&V:, nausea and vomiting, IDDM: insulin-dependent diabetes mellitus, CVA: cerebrovascular accident, HBO: hyperbaric oxygen therapy, and BKA: below knee amputation.
Early lesions present with localized erythema, skin induration, and associated pain and tenderness, as documented in the cases identified by Fine and Zacharias [10]. Untreated, these lesions tend to progress to become painful plaques with violaceous discoloration and formation of a central eschar. Progression leads to central ulceration (Figure 1) with an expanding border of necrosis (Figure 2) invariably associated with intense pain requiring, and often resistant to, high doses of opioids. Severity of disease has been proposed to correlate with high mortality rate and likely treatment failure.
Figure 1.
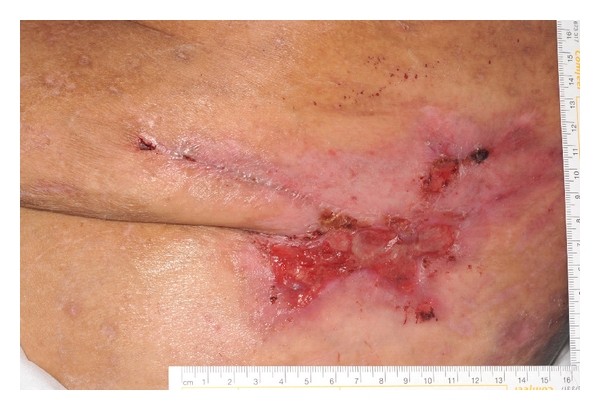
CUA lesion on the lateral abdominal wall with early ulceration, surrounding erythema and induration.
Figure 2.
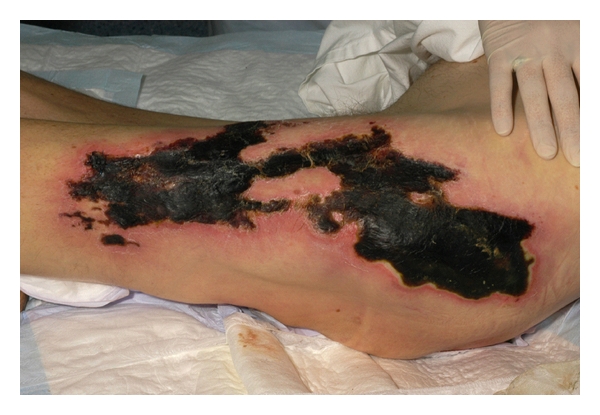
Large CUA lesion of the proximal lower limb with necrotic ulceration.
The distribution of the lesions predominantly involves the lower limbs and the abdomen. Upper extremity [13, 14], breast [13], penis, vulva [16], and cardiac and pulmonary [17] involvement have also been documented. Some authors suggest a distal distribution, in comparison with more proximal, portends a better prognosis [16]. However, this has not been proven.
Radiological investigations support but not confirm the diagnosis. Plain radiographic films of involved soft tissue may demonstrate areas of calcification representative of small vessel calcification. Standard mammography technique of involved soft tissue increases the sensitivity of imaging and is more sensitive than high-resolution-computed tomography [18]. The characteristic mesh-like pattern of arterioles demonstrated in CUA using a mammographic technique was not present in a long-term HD patient when CUA was absent [18].
Histological diagnosis remains the gold standard although it is often avoided for fear of poor wound healing and exacerbating associated ulceration [9]. Histopathologic features are small and medium vessel involvement with circumferential wall calcification involving both the medial and intimal layers. There may be associated intimal hyperplasia with partial obliteration of the vessel lumen. Acute or chronic panniculitis with a relative absence of inflammatory cells is a frequent feature [16]. Fibrin thrombi are often noted and are in close proximity to epidermal and dermal necrosis. Necrosis can extend into the subcutaneous tissue.
3. A Local Cohort of Peritoneal Dialysis Patients with CUA
A retrospective chart analysis from 2007 to 2010 identified 5 PD patients treated for CUA within our centre (Table 2). This represents an annual incidence of 0.97%, with one patient excluded due to referral from outside our local treatment population. An additional 4 HD patients in our centre were identified, not included here, with an annual incidence of 0.75%. Patient characteristics were similar to those documented in the literature (Table 1). Three of five were male, all with hypertension and two with ischemic heart disease. Only one had diabetes and none treated with therapeutic anticoagulation with either heparin or warfarin. Hyperphosphatemia was universal, while two of the five also had hypercalcemia at presentation. Time-averaged phosphate and calcium levels were not available. The average calcium-phosphate product was 5.46 mmol2/L2. There was a wide range in parathyroid hormone levels (PTH). Of note is a 28-years old female with ESKD secondary to lupus nephritis with suppressed PTH and intact parathyroid glands. Similar presentations have been noted previously with underlying SLE and normal or suppressed PTH [19, 20].
Table 2.
CUA cases within our local PD population.
| Patient characteristics | PD duration | Clinical presentation | CCa2+ (mmol/L) | PO4− (mmol/L) | PTH (ng/L) | Treatment | Response to treatment |
|---|---|---|---|---|---|---|---|
| 79 yo female ESRD (? cause) HTN, IHD, PVD, dyslipidemia, depression | 9 years CAPD | Painful bilateral foot ulceration | 2.64 | 2.16 | 598 | D/C calcium carbonate and calcitriol Started cinacalcet and aluminium OHSTS (IV) 3 monthsIntensive PD—change to HD | With STS and change to HD lesions resolvedDied 7/12 after resolution of lesions. Withdrawal of HD for functional decline |
| 67 yo male ESKD (FSGS) IHD, HTN, dyslipidemia, OSA, BPH, GORD | 7 months CAPD | Painful red violaceous ulceration right calf: Multiple indurated lesions on thighs and bilateral calf | 2.42 | 2.61 | 1386 | Started cinacalcet. Parathyroidectomy and STS (IV) 6 months commenced at same time. Caltrate and calcitriol not stopped due to hungry bone | Complete resolution of lesions including ulceration. Died 18 months later with ischemic CCF not for intervention |
| 75 yo male ESKD (DM/ obstructive) DM, IHD, dyslipidemia, CVA, GORD, HTN, gout | 7 months CAPD | Ulcerations right anterior lower leg, initially presumed DM ulcers | 2.73 | 1.73 | 132 | D/C calcium carbonate. Started sevelamer, cinacalcet, aluminium OHHBO + pamidronate + Intensive PD STS (IV) 5 weeksPD changed to HD while on STS | No response to change in phosphate binders. Pain relief gained while on STS but no wound healing. Ulcers began to heal off STS after HD initiated. Died secondary to sepsis |
| 74 yo male ESKD (ADPKD) PVD, HTN, gout, ex-smoker | 3 years CAPD | Painful ulceration of left shin, superimposed infection | 2.45 | 2.28 | 38 | Prior Parathyroidectomy. STS (IP) 12 weeks + HBO for 5 days. PD changed to HD after 2nd PD peritonitis | Complete response was documented. STS complicated by 2 episodes PD peritonitis and was D/C |
| 28 yo female ESKD (SLE) AIHA, Epilepsy, HTN | 27 months CAPD | 4 painful ulcerating lesions left lower leg medial and posterior | 2.42 | 1.90 | 8.54 | Started sevelamer. STS (IP) 3 months −25 g was reduced to 12.5 g for nausea. HBO 30 treatments | Complete response, with durability to 2 years. Remaining on PD |
PD peritoneal dialysis, CCa2+ corrected calcium, PO4− phosphate, PTH parathyroid hormone, ESRD (aetiology), HD haemodialysis, HTN hypertension, IHD ischemic heart disease, PVD peripheral vascular disease, CAPD continuous abdominal PD, D/C discontinue, STS sodium thiosulphate, IV intravenous, IP intraperitoneal, OSA obstructive sleep apnoea, AIHA autoimmune hemolytic anemia, DM diabetes mellitus, CVA cerebrovascular accident, and HBO hyperbaric oxygen therapy.
All our patients presented with ulcerative lesions. The margins of ulceration were biopsied and examined by a pathologist who was blinded to clinical severity (Figure 3.) There was no association between the size of involved vessel, maximal degree of luminal narrowing, associated changes including: ulceration, epidermal necrosis, panniculitis, fat necrosis, thrombosis, calcification (septal, lobular, or both), and outcome. Despite patients presenting with ulcerative lesions, outcomes were better than expected from the literature. We suggest the better outcomes are due to a multimodality approach with STS and HBO.
Figure 3.
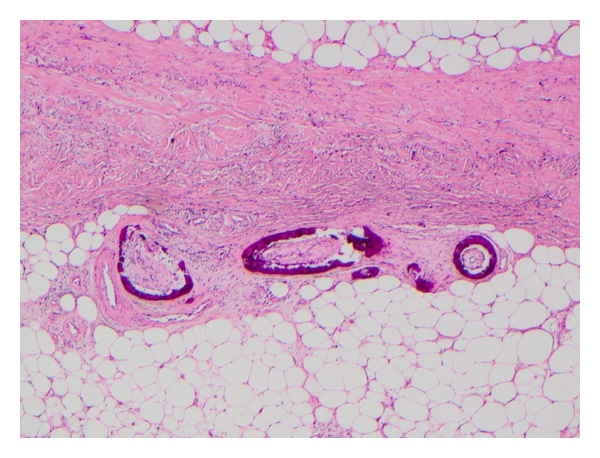
Characteristic biopsy specimen of CUA demonstrating circumferential calcification of small blood vessels with narrowing of the vascular lumen.
4. Risk Factors
Although many authors have proposed potential risk factors in CUA, its low frequency has restricted analysis design to retrospective case control studies with only one prospective study reported in the PD population [11]. Females are disproportionately represented with Zacharias documenting 75% of cases being female, compared with 53% females in the background PD population [10]. Female predominance has also been observed in HD populations [7]. Diabetes is another potential risk factor in patients with a frequency of 67% in CUA, compared to 29% in PD patients without CUA [9]. This association is not confirmed in the HD population [7].
Fine reported a link between PD and increased risk of CUA with a reported incidence of approximately 4% during the late 1990s, but with a declining incidence during the last decade [9]. However, the literature reports a similar incidence of 4.1% within a HD population [21]. Single-centre studies report the proportion of PD suffering CUA to be higher than the HD population [9]. Supporting the notion of dialysis modality being a contributing factor is the reported therapeutic response in CUA with a change from PD to HD. Reasons for this remain unclear although the constant high levels of phosphate in PD, in comparison with the fluctuations seen in HD, may contribute.
Use of calcium containing phosphate binders during the months preceding CUA onset was suggested to contribute to the incidence in PD patients [10] with the observation of decreasing incidence following the introduction of non-calcium containing phosphate binders [9]. It must be noted that these observations were not controlled for hyperphosphatemia. Hyperphosphatemia was identified as the only significant risk factor in a HD population although the small sample size of nineteen limited the power of this study [7].
Retrospective case-controlled cohort analyses have identified numerous other potential risk factors, including time on dialysis [7], obesity [22], and warfarin use [23], in the development of CUA.
5. Pathogenesis
The molecular biology of intimal and medial vascular calcification is a subject currently attracting considerable interest in the literature and is particularly pertinent to the ESKD population where widespread large vessel, that is, arterial, calcification is common [24]. Small and medium arteriolar vessel calcification, as seen in CUA, is much less common. In the absence of data in CUA, the molecular biology is presumed to be similar [3, 24]. Identifying the stimulus for the development of CUA, and how it selects patients, remains elusive. However, it is unlikely to be a single injury but rather the sum of a variable combination of multiple processes. Pathophysiological mechanisms of established CUA remain largely theoretical. The emergence of possible treatment modalities has sparked interest in research into identification of these mechanisms. Hans Selye [2] hypothesized a “two-hit” mechanism for CUA in a rat model, whereby sensitization with either vitamin D or intact endogenous parathyroid hormone would prime the animal for the development of “calciphylaxis” when injected with either iron preparation or egg white compound.
Disordered mineral metabolism of the calcium, phosphate, and parathyroid axis is almost universally assumed to play a role in CUA. An association between osteoporosis or osteopenia and vascular calcification has been well documented [24], although this is restricted to large vessel calcification. Available literature does not confirm a causative relationship between disordered mineral metabolism and CUA, possibly due to heterogeneity and lack of well powered prospective studies.
Hyperphosphatemia induced vascular smooth muscle cell (vSMC) mineralization and transition from contractile smooth muscle to an osteochondrogenic phenotype expressing osteopontin, Cbfa-1/Runx2, alkaline phosphatase, and osteocalcin in in vitro studies (Figure 4) [24]. Similar expression markers have been identified in human CUA biopsy samples [25]. Long-term exposure to hypercalcemia at levels similar to that seen in ESKD-induced mineralization of cultured human smooth muscle cells [26]. Hypercalcemia induced increased expression of the type III sodium dependent phosphate cotransporter, Pit-1 [26]. In the presence of both hyperphosphatemia and hypercalcemia mineralization was dramatically enhanced.
Figure 4.
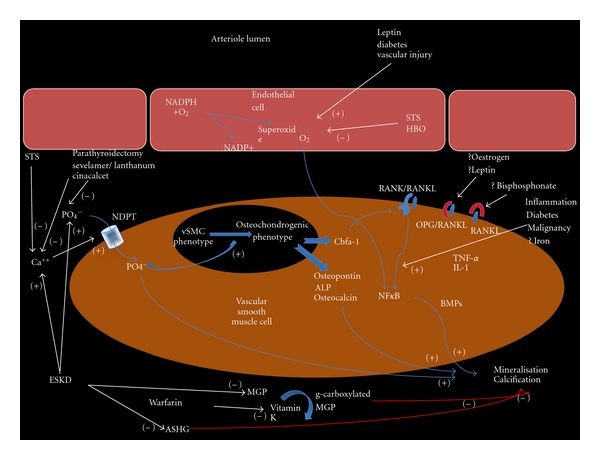
Schematic representation for the pathogenic mechanisms of CUA development and the potential sites of action for therapeutic intervention, where (+) indicates augmentation and (−) indicated inhibition; STS, sodium thiosulfate; HBO, hyperbaric oxygen; PO4−, phosphate; Ca++, calcium; NDPT, sodium-dependent phosphate cotransporter (Pit-1); vSMC, vascular smooth muscle cell; Cbfa-1, core-binding factor alpha 1; NFκB, nuclear factor κ−B; RANK(L), receptor activator of NFκB (ligand); OPG, osteoprotegerin; TNF-α, tumour necrosis factor alpha; IL-1 interleukin 1; BMP, bone morphogenic protein; ESKD, end-stage kidney disease; MGP, matrix G1a protein; ASHG, α-2 Heremans-Schmid glycoprotein.
The final common pathway of the varying mechanisms is the expression of nuclear factor κ-B (NFκB), a nuclear transcription factor, through the complex interaction of receptor activator of NFκB (RANK), its ligand (RANKL) and antagonist to the ligand, osteoprotegerin (OPG) [27]. Increased expression of NFκB promotes extraosseous mineralization and decreases osseous mineralization. OPG under-expression results in osteoporosis and vascular calcification, while increased expression results in an osteopetrosis phenotype [27]. The therapeutic mechanism of bisphosphonate therapy in CUA may be explained by its inhibition of the RANK/RANKL interaction [27]. This pathogenic mechanism does not, however, explain the higher incidence in the female and obese populations, where oestrogen, in the former, and leptin, in the later, increase OPG levels resulting in an expected protective effect.
Other proteins have been implicated in vascular calcification in the ESKD population. Fetuin-A or alpha-Heremans-Schmid glycoprotein (ASHG) has been demonstrated to inhibit ectopic calcification in knockout mice [28]. ASHG levels have been demonstrated to be low within both HD [29] and PD populations. Interestingly, low ASHG levels were also shown to be a predictor of mortality and cardiovascular death [28]. A second protein, matrix Gla protein (MGP) has been shown to regulate medial calcification, which develops spontaneously in knock-out mice [30]. MGP requires vitamin K as an activating cofactor, thereby providing a mechanism of involvement for warfarin, by depleting vitamin K. Supporting the role of these two proteins is the observation of low MGP and ASHG levels in a HD population, with a negative correlation to time-averaged phosphate levels [31].
Iron deposition may also be involved in the development of CUA. Farah identified iron deposition in all the analyzed biopsy specimens of their series of CUA [32]. Eleven had iron deposited within the affected vessel wall and iron around the vessel wall only in the twelfth [32]. No iron deposits in areas unaffected by CUA were seen in any biopsies. Whether iron deposition is a cause, a consequence, or an unrelated association remains to be determined. Calcific narrowing of small and medium vessels provides the milieu for additional processes that may ultimately culminate in the development of CUA [27]. Associated thrombosis can develop secondary to low blood flow rates through narrowed arteriolar lumens [27]. Ahmed proposes a second mechanism, whereby vSMCs sloughed into the lumen aggregate to form thrombus [33]. Local small and medium vessel luminal thrombus in addition to vessel lumen narrowing, promotes ischemia and secondary necrosis. Systemic procoagulant states may also increase the risk of CUA development through enhanced thrombus development.
Local and systemic inflammatory mediators are thought to potentiate the development of CUA. Chronic dialysis and diabetes are known to be associated with increased levels of proinflammatory mediators, such as tumor necrosis factor alpha, Interleukin-1 and Interleukin-6 which can result in endothelial disruption promoting local thrombosis and necrosis in addition to potentiation of NFκB stimulating further vessel calcification [3, 27]. Such mechanisms may explain the possible increased risk of CUA in diabetic populations and with local minor trauma such as with insulin and erythropoietin injections.
6. Treatment of CUA
6.1. General Measures
6.1.1. Local Wound Care
There is some evidence to support surgical debridement of the wounds [27, 34]. The opposing point of view is that debridement can increase the extent of the nonhealing ulcer surface. The use of antibiotics is limited to obvious bacterial infection rather than colonization.
6.1.2. Nutrition
CUA is most often painful and associated with depression which both impact the nutritional state. Necrotic tissues reduce appetite, possibly through the release of cytokines. Adequate analgesia with nonsteroidal anti-inflammatory drugs and opioids without significantly affecting quality of life is important, as is supportive psychotherapy.
6.1.3. Cessation of Warfarin
Warfarin inhibits matrix GLA metalloproteinases, thus promoting vascular calcification. Based on this molecular mechanism, stopping warfarin with vitamin K reversal has become a common practice despite scant clinical evidence [23]. Substitution with low molecular weight heparin (LMWH) is a reasonable long-term management strategy, where anticoagulation is mandated. Therapeutic bridging from warfarin to LMWH with monitoring of Factor Xa levels is performed in those with mandated therapeutic anticoagulation. Conditions such as atrial fibrillation necessitate and individualized decision, recognizing traditional risk benefit ratios demonstrated in the general public cannot be generalized to the dialysis population [35]. Management is determined by the balance of risk of stroke or thrombosis, ability to administer and monitor LMWH, patient preferences and risk of LMWH, including the cumulative risk of precipitating new CUA lesions through endothelial damage from subcutaneous injections [3].
6.2. Vitamin K Administration
Regular administration of Vitamin K has not been investigated. Through the γ–carboxylation of MGP, Vitamin K administration may have a therapeutic benefit [36] although likely small.
6.3. Lowering the Calcium Phosphorus Product
6.3.1. Substitution with Non-Calcium Containing Phosphate Binders Like Sevelamer and Lanthanum Carbonate May Be Beneficial [27, 37]
Change of dialysis prescription to: intensified PD, extended hours of HD or daily HD has been shown to be beneficial in some patients [38]. Changing to HD from PD has been trialed in the past with dramatic improvement in some patients and not in others [11].
6.3.2. Reduction of Elevated PTH Levels
The relationship of CUA with hyperparathyroidism is best seen in the nonuremic population [8]. There are also reports of ulcer healing, pain relief and survival benefit in patients on dialysis with CUA and hyperparathyroidism who undergo parathyroidectomy [39]. Medical treatment of hyperparathyroidism with cinacalcet, a relatively recent option, was reported to be beneficial [40]. Our local practice is surgical parathyroidectomy in the presence of uncontrolled hyperparathyroidism, that is, hormone levels greater than seven to nine times the upper limit of reference range (CARI guidelines [41]). Cinacalcet is used in those who are not surgical candidates.
6.4. Emerging Treatment Options
6.4.1. Sodium Thiosulfate (STS)
STS acts as a chelating agent for calcium and iron and also as a potent antioxidant resulting in decreased vasospasm and pain along with solubilization and decalcification of the deposits [42, 43]. STS can be given orally [40], intravenously (IV) [12] or intraperitoneally (IP) [13]. There is no consensus on the best dosing schedule and STS is given at varying regimes, including 7.5 g/week orally [40], 12.5 g [44] to 25 g IV or IP three times a week. Intravenous STS can cause abdominal cramping, nausea, vomiting, and diarrhea if infused rapidly [45]. Acidosis and hypocalcemia have been reported [46]. We used STS in all our PD patients with CUA. In three patients, we administered STS IV and converted them to HD. However we left the last two patients on PD and administered 12.5 g of STS in 2 litres of the long dwell dialysate, three times a week. One patient developed bacterial peritonitis after one week of therapy, which was treated according to the International Society of Peritonitis Dialysis guidelines and continued on IP STS and PD. Two months later, he presented again with culture negative peritonitis raising the possibility of chemical peritonitis. He also developed tremors and we stopped IP STS due to concern over STS causing these side effects. The second patient received STS IP for 3 months uneventfully and there was a complete healing of skin lesions. The optimum dose and duration of treatment with STS is yet to be determined.
6.4.2. Hyperbaric Oxygenation Therapy (HBO)
This modality is increasingly used for its benefits of better tissue oxygenation, improved angiogenesis, and enhanced phagocytic activity and bactericidal action in tissues with CUA and hypoxemia driven injury [47]. Ulcers heal in 40% to 75% of treated patients [48, 49]. Reported side effects are barotrauma, reversible myopia and neurological complications from oxygen toxicity [50]. Logistical constraints may limit its use as HBO may not be available in all centers, necessitating transfer of patients to a unit with such facilities.
6.4.3. Combination Therapy
Aggressive therapy with a combination of all these modalities, including HBO and STS, in early lesions is likely to promote healing through multiple pathways [47]. Treatment using this approach in our PD population resulted in healing of skin lesions of all cases despite aggressive presentations.
6.4.4. Bisphosphonate Therapy
Bisphosphonates have an anti-inflammatory action and inhibit osteoprotegerin mediated calcification [51]. Despite bisphosphonates current product information contraindication in ESKD, case reports have document benefit with their use in CUA [52, 53]. Data to guide safe dosing of bisphosphonate therapy in ESKD is absent, hence routine use in this population cannot be recommended.
Due to increasing success with combination therapy of STS and HBO, our unit reserves bisphosphonate use for last line therapy. The decision to use bisphosphonate should include the bone metabolism status; however, the high mortality in CUA often supervenes a concern of later development of adynamic bone disease. The emergence of more accurate markers of bone turnover such as C-telopeptide and N-terminal propeptide of collagen type I may better guide our use in the future but would require investigation prior to routine use.
7. Conclusions
The clinical features of CUA in PD patients, the diagnosis, and possible pathogenetic mechanisms are comparable to other patient groups with CUA. The risk association of PD with CUA requires confirmation. Aggressive therapy with combination STS and HBO, in addition to general measures, may reduce morbidity and mortality in PD patients with CUA. Treatment of CUA in PD patients does not differ from other patient groups except for the intraperitoneal route of administering STS.
Further prospective studies are needed to clearly identify the risk factors, to describe the pathophysiology, in particular the differences from atherosclerosis seen in large arteries, and to define and standardize therapy. Adequately powered randomized studies will be difficult to conduct due to the low incidence of disease and the high associated mortality causing hesitancy and ethical consideration in randomization to a control group. Recently, international CUA registries have been established to promote knowledge of CUA and progress therapeutic strategies:
Germany: Calciphylaxie Register, International Collaborative Calciphylaxis Network (http://www.calciphylaxie.de/),
USA: Calciphylaxis Registry, KU Medical Center, University of Kansas (http://www2.kumc.edu/calciphylaxisregistry/),
UK: UK Calciphylaxis Registry, International Collaborative Calciphylaxis Network (http://www.calciphylaxis.org.uk/).
References
- 1.Bryant JH, White WH. A case of calcification of the arteries and obliterative endarterities associated with hydronephrosis in a child age six months. Guys Hospital Report. 1898;55: 1719 [Google Scholar]
- 2.Selye H. Calciphylaxis. Chicago, Ill, USA: University of Chicago Press; 1962. [Google Scholar]
- 3.Rogers NM, Teubner DJO, Coates PTH. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Seminars in Dialysis. 2007;20(2):150–157. doi: 10.1111/j.1525-139X.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- 4.Weenig RH, Sewell LD, Davis MDP, McCarthy JT, Pittelkow MR. Calciphylaxis: natural history, risk factor analysis, and outcome. Journal of the American Academy of Dermatology. 2007;56(4):569–579. doi: 10.1016/j.jaad.2006.08.065. [DOI] [PubMed] [Google Scholar]
- 5.Wilmer WA, Magro CM. Calciphylaxis: rmerging concepts in prevention, diagnosis, and treatment. Seminars in Dialysis. 2002;15(3):172–186. doi: 10.1046/j.1525-139x.2002.00052.x. [DOI] [PubMed] [Google Scholar]
- 6.Russell R, Brookshire MA, Zekonis M, Moe SM. Distal calcific uremic arteriolopathy in a hemodialysis patient responds to lowering of Ca × P product and aggressive wound care. Clinical Nephrology. 2002;58(3):238–243. doi: 10.5414/cnp58238. [DOI] [PubMed] [Google Scholar]
- 7.Mazhar AR, Johnson RJ, Gillen D, et al. Risk factors and mortality associated with calciphylaxis in end-stage renal disease. Kidney International. 2001;60(1):324–332. doi: 10.1046/j.1523-1755.2001.00803.x. [DOI] [PubMed] [Google Scholar]
- 8.Mehta RL, Scott G, Sloand JA, Francis CW. Skin necrosis associated with acquired protein C deficiency in patients with renal failure and calciphylaxis. American Journal of Medicine. 1990;88(3):252–257. doi: 10.1016/0002-9343(90)90150-c. [DOI] [PubMed] [Google Scholar]
- 9.Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clinical Journal of the American Society of Nephrology. 2008;3(4):1139–1143. doi: 10.2215/CJN.00530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fine A, Zacharias J. Calciphylaxis is usually nonulcerating: risk factors, outcomes, and therapy. Kidney International. 2002;61(6):2210–2217. doi: 10.1046/j.1523-1755.2002.00375.x. [DOI] [PubMed] [Google Scholar]
- 11.Zacharias JM, Fontaine B, Fine A. Calcium use increases risk of calciphylaxis: a case-control study. Peritoneal Dialysis International. 1999;19(3):248–252. [PubMed] [Google Scholar]
- 12.Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Peritoneal Dialysis International. 2008;28(3):268–270. [PubMed] [Google Scholar]
- 13.Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. American Journal of Kidney Diseases. 2004;43(6):1104–1108. doi: 10.1053/j.ajkd.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Mataic D, Bastani B. Intraperitoneal sodium thiosulfate for the treatment of calciphylaxis. Renal Failure. 2006;28(4):361–363. doi: 10.1080/08860220600583781. [DOI] [PubMed] [Google Scholar]
- 15.Thang OHD, Jaspars EH, ter Wee PM. Necrotizing mastitis caused by calciphylaxis. Nephrology Dialysis Transplantation. 2006;21(7):2020–2021. doi: 10.1093/ndt/gfl135. [DOI] [PubMed] [Google Scholar]
- 16.Amin N, Gonzalez E, Lieber M, Salusky IB, Zaritsky JJ. Successful treatment of calcific uremic arteriolopathy in a pediatric dialysis patient. Pediatric Nephrology. 2010;25(2):357–362. doi: 10.1007/s00467-009-1313-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daudén E, Oñate MJ. Calciphylaxis. Dermatologic Clinics. 2008;26(4):557–568. doi: 10.1016/j.det.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Rogers NM, Coates PTH. Calcific uraemic arteriolopathy: an update. Current Opinion in Nephrology and Hypertension. 2008;17(6):629–634. doi: 10.1097/MNH.0b013e32830f4566. [DOI] [PubMed] [Google Scholar]
- 19.Bleibel W, Hazar B, Herman R. A case report comparing various radiological tests in the diagnosis of calcific uremic arteriolopathy. American Journal of Kidney Diseases. 2006;48(4):659–661. doi: 10.1053/j.ajkd.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 20.Sakr SH, Russell EB, Jasin HE. Systematic lupus erythematosus and calciphylaxis. Journal of Rheumatology. 2004;31(9):1851–1853. [PubMed] [Google Scholar]
- 21.Igaki N, Moriguchi R, Hirota Y, et al. Calciphylaxis in a patient with end-stage renal disease secondary to systemic lupus erythematosus associated with acral gangrene and mesenteric ischemia. Internal Medicine. 2001;40(12):1232–1237. doi: 10.2169/internalmedicine.40.1232. [DOI] [PubMed] [Google Scholar]
- 22.Angelis M, Wong LL, Myers SA, Wong LM. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122(6):1083–1090. doi: 10.1016/s0039-6060(97)90212-9. [DOI] [PubMed] [Google Scholar]
- 23.Bleyer AJ, Choi M, Igwemezie B, De La Torre E, White WL. A case control study of proximal calciphylaxis. American Journal of Kidney Diseases. 1998;32(3):376–383. doi: 10.1053/ajkd.1998.v32.pm9740152. [DOI] [PubMed] [Google Scholar]
- 24.Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. American Journal of Kidney Diseases. 1998;32(3):384–391. doi: 10.1053/ajkd.1998.v32.pm9740153. [DOI] [PubMed] [Google Scholar]
- 25.Giachelli CM, Speer MY, Li X, Rajachar RM, Yang H. Regulation of vascular calcification: roles of phosphate and osteopontin. Circulation Research. 2005;96(7):717–722. doi: 10.1161/01.RES.0000161997.24797.c0. [DOI] [PubMed] [Google Scholar]
- 26.Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circulation research. 2000;87(7):E10–E17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 27.Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney International. 2004;66(6):2293–2299. doi: 10.1111/j.1523-1755.2004.66015.x. [DOI] [PubMed] [Google Scholar]
- 28.Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor κ-B. Journal of the American Academy of Dermatology. 2008;58(3):458–471. doi: 10.1016/j.jaad.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Wang AY-M. Vascular and other tissue calcification in peritoneal dialysis patients. Peritoneal Dialysis International. 2009;29(supplement 2):S9–S14. [PubMed] [Google Scholar]
- 30.Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361(9360):827–833. doi: 10.1016/S0140-6736(03)12710-9. [DOI] [PubMed] [Google Scholar]
- 31.Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protien. Nature. 1997;386(6620):78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 32.Hermans MMH, Vermeer C, Kooman JP, et al. Undercarboxylated matrix GLA protein levels are decreased in dialysis patients and related to parameters of calcium-phosphate metabolism and aortic augmentation index. Blood Purification. 2008;25(5-6):395–401. doi: 10.1159/000108629. [DOI] [PubMed] [Google Scholar]
- 33.Farah M, Crawford RI, Levin A, Chan Yan C. Calciphylaxis in the current era: emerging ‘ironic’ features? Nephrology Dialysis Transplantation. 2011;26(1):191–195. doi: 10.1093/ndt/gfq407. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed S, Oneill KD, Hood AF, Evan AP, Moe SM. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. American Journal of Kidney Diseases. 2001;37(6):1267–1276. doi: 10.1053/ajkd.2001.24533. [DOI] [PubMed] [Google Scholar]
- 35.Kang AS, McCarthy JT, Rowland C, et al. Is calciphylaxis best treated surgically or medically? Surgery. 2000;128(6):967–972. doi: 10.1067/msy.2000.110429. [DOI] [PubMed] [Google Scholar]
- 36.Chan KE, Lazarus JM, Thadhani R, Hakim RM. Anticoagulant and antiplatelet usage associates with mortality among hemodialysis patients. Journal of the American Society of Nephrology. 2009;20(4):872–881. doi: 10.1681/ASN.2008080824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy R. Potential treatment of calciphylaxis with vitamin K2: comment on the article by Jacobs-Kosmin and DeHoratius. Arthritis Care and Research. 2007;57(8):1575–1576. doi: 10.1002/art.23107. [DOI] [PubMed] [Google Scholar]
- 38.Chan MR, Yevzlin AS, Hinshaw M, Jaffery JB. Calciphylaxis responsive to lanthanum carbonate (FOSRENOL) therapy. Wisconsin Medical Journal. 2008;107(7):335–338. [PubMed] [Google Scholar]
- 39.Llach F. The evolving pattern of calciphylaxis: therapeutic considerations. Nephrology Dialysis Transplantation. 2001;16(3):448–451. doi: 10.1093/ndt/16.3.448. [DOI] [PubMed] [Google Scholar]
- 40.Duffy A, Schurr M, Warner T, Chen H. Long-term outcomes in patients with calciphylaxis from hyperparathyroidism. Annals of Surgical Oncology. 2006;13(1):96–102. doi: 10.1245/ASO.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 41.Velasco N, MacGregor MS, Innes A, MacKay IG. Successful treatment of calciphylaxis with cinacalcet—an alternative to parathyroidectomy? Nephrology Dialysis Transplantation. 2006;21(7):1999–2004. doi: 10.1093/ndt/gfl114. [DOI] [PubMed] [Google Scholar]
- 42.Elder G. The CARI guidelines. Biochemical and haematological targets: Parathyroid hormone. 2006, http://www.cari.org.au/
- 43.Musso CG, Enz P, Vidal F, et al. Oral sodium thiosulfate solution as a secondary preventive treatment for calciphylaxis in dialysis patients. Saudi Journal of Kidney Diseases and Transplantation. 2008;19(5):820–821. [PubMed] [Google Scholar]
- 44.Hayden MR, Goldsmith DJA. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Seminars in Dialysis. 2010;23(3):258–262. doi: 10.1111/j.1525-139X.2010.00738.x. [DOI] [PubMed] [Google Scholar]
- 45.Brucculeri M, Cheigh J, Bauer G, Serur D. Long-term intravenous sodium thiosulfate in the treatment of a patient with calciphylaxis. Seminars in Dialysis. 2005;18(5):431–434. doi: 10.1111/j.1525-139X.2005.00082.x. [DOI] [PubMed] [Google Scholar]
- 46.Courtney MJ, Pannu N. Calcific uremic arteriolopathy should be treated conservatively. Seminars in Dialysis. 2010;23(1):34–37. doi: 10.1111/j.1525-139X.2009.00655.x. [DOI] [PubMed] [Google Scholar]
- 47.Subramaniam K, Wallace H, Sinniah R, Saker B. Complete resolution of recurrent calciphylaxis with long-term intravenous sodium thiosulfate. Australasian Journal of Dermatology. 2008;49(1):30–34. doi: 10.1111/j.1440-0960.2007.00416.x. [DOI] [PubMed] [Google Scholar]
- 48.Rogers NM, Coates PTH. Calcific uremic arteriolopathy—the argument for hyperbaric oxygen and sodium thiosulfate. Seminars in Dialysis. 2010;23(1):38–42. doi: 10.1111/j.1525-139X.2009.00656.x. [DOI] [PubMed] [Google Scholar]
- 49.Basile C, Montanaro A, Masi M, Pati G, De Maio P, Gismondi A. Hyperbaric oxygen therapy for calcific uremic arteriolopathy: a case series. Journal of Nephrology. 2002;15(6):676–680. [PubMed] [Google Scholar]
- 50.Podymow T, Wherrett C, Burns KD. Hyperbaric oxygen in the treatment of calciphylaxis: a case series. Nephrology Dialysis Transplantation. 2001;16(11):2176–2180. doi: 10.1093/ndt/16.11.2176. [DOI] [PubMed] [Google Scholar]
- 51.Leach RM, Rees PJ, Wilmshurst P. ABC of oxygen: hyperbaric oxygen therapy. British Medical Journal. 1998;317(7166):1140–1143. doi: 10.1136/bmj.317.7166.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. American Journal of Kidney Diseases. 1998;32(3):384–391. doi: 10.1053/ajkd.1998.v32.pm9740153. [DOI] [PubMed] [Google Scholar]
- 53.Monney P, Nguyen QV, Perroud H, Descombes E. Rapid improvement of calciphylaxis after intravenous pamidronate therapy in a patient with chronic renal failure. Nephrology Dialysis Transplantation. 2004;19(8):2130–2132. doi: 10.1093/ndt/gfh305. [DOI] [PubMed] [Google Scholar]