

Abstract
Introduction:
Secondary hyperparathyroidism (SHPT) is an insidious disease that develops early in the course of chronic kidney disease (CKD) and increases in severity as the glomerular filtration rate deteriorates. Recent studies have identified fibroblast growth factor-23 (FGF23) as a new protein with phosphaturic activity. It is mainly secreted by osteoblasts and is now considered the most important factor for regulation of phosphorus homeostasis. It is not yet proven if there is any direct relation between parathyroid hormone (PTH) and FGF23. The present study aims to evaluate the relation between serum FGF23, phosphorus, and PTH in end-stage renal disease in patients with SHPT on regular hemodialysis.
Materials and Methods:
Forty-six consecutive CKD adult patients (case group) and 20 healthy adults (control group) were included in the study. All patients had SHPT and were on regular hemodialysis. Both groups were subjected to full medical history, clinical examination and biochemical studies. Serum phosphorus, calcium, ferritin, hemoglobin level, blood urea, creatinine, PTH, and FGF23 were analyzed.
Results:
Levels of FGF23 were significantly higher in the case group in comparison with those in the control group, viz., 4-fold, and positively correlated with PTH. Phosphorus levels in the case group were significantly high in spite of the increasing levels of FGF23. Both PTH and FGF23 were positively correlated with phosphorus and negatively with hemoglobin levels.
Conclusion:
SHPT and FGF23 may have a partial role in the development of anemia in patients with CKD. FGF23 could be a central factor in the pathogenesis of SHPT. Its role in controlling hyperphosphatemia in CKD is vague.
Keywords: Chronic kidney disease, fibroblast growth factor–23, hyperparathyroidism
INTRODUCTION
Secondary hyperparathyroidism (SHPT) is a major complication of chronic kidney disease (CKD), resulting from disturbances in the regulation of parathyroid hormone (PTH), calcium, phosphorus, and vitamin D.[1] Although hyperphosphatemia appears to be particularly important in the development of SHPT, the complication often occurs early in stage 3 of kidney failure, before the development of hyperphosphatemia.[2] The systemic balance of phosphate is maintained mainly by threeorgans, viz., intestine, kidney and bone. Several factors, including PTH and vitamin D, play a criticalrole in this system.[3] Fibroblast growth factor–23 (FGF23), a recently identified phosphatonin, is also implicated. It is predominately expressed in osteoblasts in bone.[4,5] Its level increases in response to phosphate load by yetunknown mechanisms. It promotes phosphaturia andsuppressesrenal 1,25D production.[6] Although, a significant role of FGF23 inphosphate homeostasis in physiological conditions is described in many studies with the kidney functioning normally, few studies are available on end-stage renal disease.
One of the most commonly cited morbidity associated with end-stage kidney disease is renal osteodystrophy, secondary to the alterations in mineral metabolism.[7] To understand the interrelatedseries of events leading to that, it is first necessary tostudy the mineral metabolism and its physiological process. The present study aims to evaluate the relation between serum FGF23, phosphorus, and PTH in end-stage renal disease patients with SHPT on regular hemodialysis.
MATERIALS AND METHODS
A case-control study was performed. All patients who had SHPT and were on regular hemodialysis in nephrology unit in Suez Canal University Hospital, from January to July 2010, were included in the study (case group, comprising 46 patients). Twenty healthy adults (with normal kidney function) matched for age and gender were considered as the control group. Both groups were subjected to full medical history, clinical examination and biochemical studies. Serum phosphorus, calcium, ferritin, albumin, hemoglobin level, blood urea, and creatinine were assessed by standard protocols at the Department of Clinical Pathology in Suez Canal University Hospital. PTH levels were analyzed using the Immulite 2000 Intact PTH assay. Intact FGF23 concentrations were measured using Human FGF-23 (C-Term) ELISA kit according to manufacturer's (Immutopics, Inc. San Clemente, CA 92673) protocol. Serum samples were collected from each individual at a single time point and kept at –70°C until analysis.[8]
Ethical consideration
Before the commencement of the study, informed consent was obtained from all individuals selected for the study. The aim and the value of the work were explained to them in a simplified manner. There was no chance of any harm being inflicted on them; on the contrary, all would have the benefits of follow-up and the results of the study. The study was approved by the ethics committee of the Faculty of Medicine, Suez Canal University.
Statistical aspects
The data were coded and organized. The final study results were stated using the SPSS program version 14. Results were presented through tables and figures. The Student t test, correlation coefficient, and Chi-square tests were used to evaluate the results. Chi-square test was used for qualitative variables, while independent t test was used for quantitative variables. Correlation analysis was performed using Pearson's test.
RESULTS
All the patients (46 patients) of the case group were on hemodialysis for an average duration of 4.9 years. The age of the patients ranged from 20 to 52 years (34.7+11.8) years. Twenty-seven (58.7%) of them were males. Forty-one (89.8%) patients were on regular vitamin D and calcium supplements. Thirty-eight (82.6%) patients received regular parenteral iron. None of the patients received erythropoietin. The control group consisted of 20 healthy adults with normal kidney function; their mean age was 30.3±7.9 years. Half of them were males. Laboratory characteristics of both case and control groups are presented in Table 1. There are significant differences in all parameters except serum calcium. Table 2 shows that FGF23 has a significant positive correlation with serum phosphorus, urea, creatinine, ferritin, hemoglobin, and PTH levels (P< 0.01). Table 3 shows that PTH has a significant positive correlation with serum phosphorus level (P< 0.01). In addition, PTH level is negatively correlated with hemoglobin level and calcium level. The scatter plots show positive correlation between FGF23 and serum phosphorus levels (r= +0.9; P< 0.001) and between FGF23 and serum PTH levels (r= +0.6; P< 0.01) [Figure 1 and Figure 2].
Table 1.
Laboratory characteristics of the studied patients in comparison to control
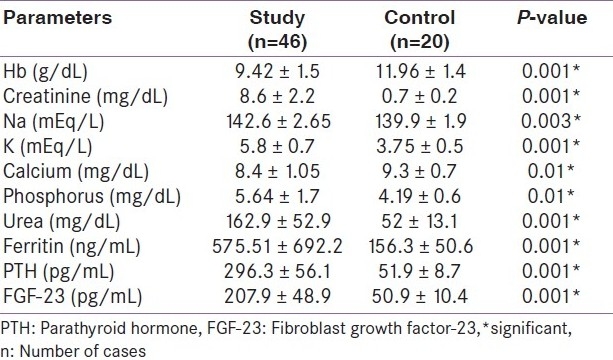
Table 2.
Correlation between FGF-23 and various parameters
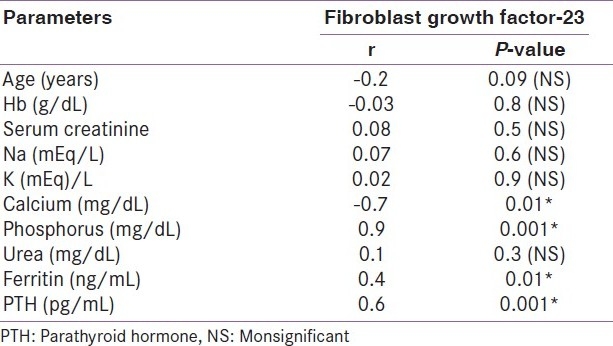
Table 3.
Correlation between parathyroid hormone level and other parameters
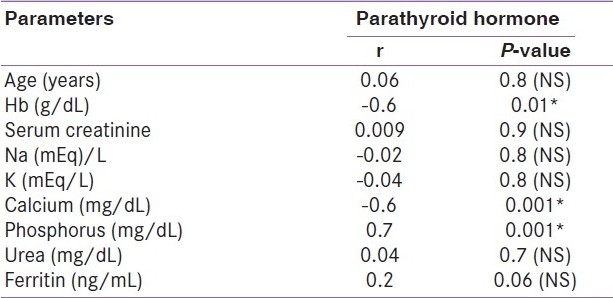
Figure 1.
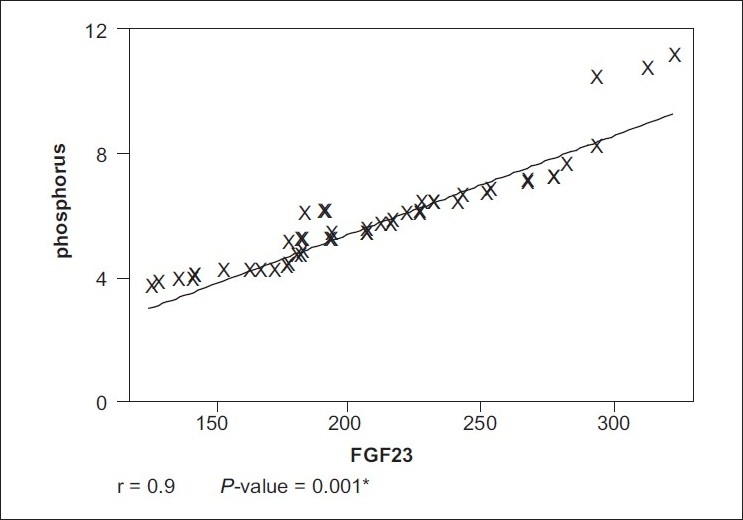
Correlation between FGF-23 and serum phosphorus level
Figure 2.
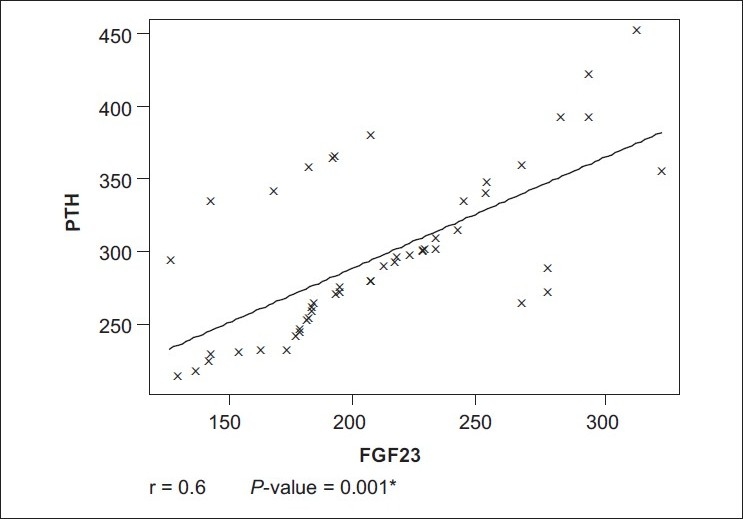
Correlation between FGF-23 and parathyroid hormone
DISCUSSION
SHPT is an insidious disease that develops early in the course of CKD and increases in severity as the glomerular filtration rate deteriorates. Recent studies have identifiedFGF23 as a new protein with phosphaturicactivity.[9,10] It is not yet proven if there is any direct relationbetweenPTH and FGF23.
In our study, levels of both PTH and serum phosphorus in the case group, were significantly higher in comparison with those in the control group. As the number of functioning nephrons decreases, the failing kidneys are unable to excrete phosphorus and there is a progressive increase in serum phosphorus levels. The ongoing inability to excrete phosphorus leads to continual over-stimulation of the parathyroid glands, tissue hyperplasia, and over-secretion of PTH.[1,11] The development of SHPT in our patients was most likely a sequela of this series of classic events.
Hemoglobin levels were significantly low in the case group and negatively correlated with both PTH and FGF23 levels, which indicates that SHPT may have a partial role in the development of anemia in patients with CKD. High PTH levels may contribute to anemia by directly inhibiting the production of red blood cells and increasing their fragility. SHPT can also cause marrow fibrosis, further decreasing the production of red blood cells.[12] It is worth noting, that a recent prospective study has shown that FGF23 mediates the development of hypophosphatemia after I.V iron poly-maltose administration.[13] In our study, most of the patients received parenteral iron, and further correlation was observed between ferritin and FGF23 blood levels, which warrants further studies to evaluate the mechanism whereby parenteral iron therapy influences FGF23 metabolism and to identify additional systemic factors regulating FGF23 production, in order to enhance our understanding of phosphate homeostasis. In addition, the possible role of FGF23 in the development of anemia is explicable.
The degree of FGF23 elevation, seen in the current data, correlates positively with thedegree of hyperphosphatemia. In experimental studies, phosphate loading in mice increasesFGF23 levels.[5,6] However, the data in humans are variable. Previous studies on dietary phosphate as a regulator of FGF23 showed conflicting results.[14–17] Small sample size, and differences in magnitude or duration of altered phosphate intake may explain the contradictory results. Although FGF23 inhibits phosphate re-absorption in the proximaltubule, leading to hypophosphatemia,[2] phosphate levels were significantly high in our case group in spite of the concomitant 4-fold increases in FGF23 levels. The possible explanation is that the kidney, which is the principal target of FGF23,is no longer responsive to FGF23 in CKD. Klothoproduction by the kidney is reduced in end-stage renal disease. It is an essentialco-factor for FGF23 activation.[11] Another explanation is that, in early stage of CKD, serum FGF23 is elevated to maintain normal serum phosphate levels by promoting urinary phosphate excretion; but in patients at the advanced stage, overt phosphate loading may overcome such compensation for decreased glomerular filtration rate (GFR) despite markedly elevated FGF23 levels. On the other hand, the precise mechanism and site of definitely increased FGF23 levels havenot been evaluated in the present study. It is possible thatboth increased production and decreased metabolism or clearanceof FGF23 contributes to elevated levels of FGF23 in CKD.[18] Althoughno studies have yet documented increased production of FGF23,the correlation of FGF23 levels with increasing blood urea and creatinine in our study and therapid decrease in FGF23 levels in some patients after renaltransplantation in other studies,[19] suggest that FGF23 is cleared by the kidney. Previous in vivo studies have shown a stimulatory effect of 1,25(OH)2vit D3 on circulating FGF23 levels in rodents and in humans.[20–22] It is an interesting point as most of our studied patients received high doses of active vitamin D, which may be implicated in the pathophysiology of the observed rises in FGF23. However, the function ofhigh FGF23 levels in the control of phosphate levels in CKD is still unexplained, and more studies are necessary.
Data with regard to the role of PTH in FGF23 regulation are conflicting. However, there is growing evidence that PTH may stimulate FGF23 expression and secretion by bone tissue. In the setting of primary hyperparathyroidism, elevated FGF23 concentrations have been observed by several groups.[23–25] On the other hand, in the study by Hiroyuki et al., no difference in serum FGF23 levels was found between healthy controls and primary hyperparathyroidism patients with normal renal function; also, there were no significant relations detected between serum FGF23 levels and the levels of PTH.[26] As several agents influence the release of PTH and FGF23, the presence of altered calcium or magnesium levels, subsequent to administration of variable supplements to the patients, confounds the interpretation and conflicts of any association between FGF23 and circulating levels of PTH. A strongpositive correlation between elevated FGF23 levels and the severity ofhyperparathyroidism in CKD group (case group) was observed in our study. Although the mechanism of this finding is unclear, it is possible that chronic phosphate retention as reflected by elevated FGF23 levels may have contributed to further stimulation of PTH secretion, progression of parathyroid hyperplasia, and parathyroid cell proliferation. Another possibility is that high levels of FGF23 at baseline may be a consequence of prolonged active vitamin D administration for severe hyperparathyroidismin our patients, as mentioned above. Therefore, FGF23 might indirectly contribute to the development of SHPT associated with renal insufficiency. Furtherstudies are required to document the effects of FGF23 on PTH production and secretion and on parathyroid cellproliferation and to assess the role of FGF23 estimation in predicting the future development of refractory SHPTin CKD.
CONCLUSION
The precise role of extremely elevated FGF23 in the control of hyperphosphatemia; and their direct effect on parathyroid function and development of anemia in CKD patients on hemodialysis still remain unclear. Their positive correlation with PTH may suggest that FGF23 is a central factor in the early pathogenesis of SHPT.Using FGF23 as a biomarker for assessing phosphate retention or as a predictor of morbidity and future development of refractory SHPT in CKD needs more studies.
It is imperative to note the several limitations of this study design. First, the relatively small sample size. Second, current data were obtained in the setting of parathyroid disorders. These results may not be extrapolated to normal physiology.
Acknowledgments
The authors wish to thank the members of the dialysis unit in the Suez Canal University Hospital for their participation and cooperation. We are also indebted to professor Adel Morshedy, the chairman of the Clinical Epidemiology Unit in the faculty of medicine for his valuable guide and great help in revising the manuscript.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, et al. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44:250–6. doi: 10.1053/j.ajkd.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 2.Gutie´rrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–15. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 3.Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, et al. Vitamin D receptor independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289:1088–95. doi: 10.1152/ajprenal.00474.2004. [DOI] [PubMed] [Google Scholar]
- 4.Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by Phex. J Biol Chem. 2003;278:37419–26. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 5.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 6.Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543–9. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 7.Moe S, Drueke T, Cunningham J, Goodman W, Martin K, Olgaard K, et al. Definition, evaluation and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69:1945–53. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 8.Immutopics. Inc Human FGF-23 (C-Term) ELISA Kit: 2nd generation Enzyme-Linked Immuno Sorbent Assay (ELISA) for the determination of human fibroblast growth factor 23 Levels in plasma or cell culture media. [last accessed on 2011 Feb 10]. Available from: http://www.immutopicsintl.com .
- 9.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–8. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 10.Pierre K, Jean-Jacques B, Rafik K. Fibroblast growth factor 23 and its role in phosphate homeostasis. Eur J Endocrinol. 2010;162:1–10. doi: 10.1530/EJE-09-0597. [DOI] [PubMed] [Google Scholar]
- 11.Nabeshima Y. Clinical discovery of alpha-Klotho and FGF-23 unveiled new insight into calcium and phosphate homeostasis. Calcium. 2008;18:923–34. [PubMed] [Google Scholar]
- 12.Rao DS, Shih MS, Mohini R. Effect of serum parathyroid hormone and bone marrow fibrosis on the response to erythropoietin in uremia. N Engl J Med. 1993;328:171–5. doi: 10.1056/NEJM199301213280304. [DOI] [PubMed] [Google Scholar]
- 13.Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metabol. 2009;94:2332–7. doi: 10.1210/jc.2008-2396. [DOI] [PubMed] [Google Scholar]
- 14.Burnett SA, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–96. doi: 10.1359/jbmr.060507. [DOI] [PubMed] [Google Scholar]
- 15.Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006;70:2141–7. doi: 10.1038/sj.ki.5002000. [DOI] [PubMed] [Google Scholar]
- 16.Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, et al. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. 2004;65:1943–6. doi: 10.1111/j.1523-1755.2004.00604.x. [DOI] [PubMed] [Google Scholar]
- 17.Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18:1227–34. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- 18.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–9. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 19.Pande S, Ritter CS, Rothstein M, Wiesen K, Vassiliadis J, Kumar R, et al. FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol. 2006;104:23–32. doi: 10.1159/000093277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito H, Maeda A, Ohtomo SI, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543–9. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Tang W, Jianping Z, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–15. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 22.Collins MT, Lindsay JR, Jain A, Kelly MH, Cutler CM, Weinstein LS, et al. Fibroblast growth factor-23 is regulated by 1,25-dihydroxyvitamin D. J Bone Miner Res. 2005;20:1944–50. doi: 10.1359/JBMR.050718. [DOI] [PubMed] [Google Scholar]
- 23.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–8. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi K, Imanishi Y, Miyauchi A, Onoda N, Kawata T, Tahara H, et al. Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol. 2006;154:93–9. doi: 10.1530/eje.1.02053. [DOI] [PubMed] [Google Scholar]
- 25.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-O M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–8. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiroyuki Y, Takeyoshi Y, Masashi M, Takashi S, Junichiro J, Takashi S, et al. Fibroblast growth factor (FGF)-23 in patients with primary hyperparathyroidism. Eur J Endocrinol. 2004;151:55–60. doi: 10.1530/eje.0.1510055. [DOI] [PubMed] [Google Scholar]