

Abstract
Dietary trans fats have been causally linked to atherosclerosis but the mechanism by which they cause the disease remain elusive. Suppressed TGF-β responsiveness in aortic endothelium has been shown to play an important role in the pathogenesis of atherosclerosis in animals with hypercholesterolemia. We investigated the effects of a high trans-fat (TF) diet on TGF-β responsiveness in aortic endothelium and integration of cholesterol in tissues. Here we show that normal mice fed a high TF diet for 24 weeks exhibit atherosclerotic lesions and suppressed TGF-β responsiveness in aortic endothelium. The suppressed TGF-β responsiveness is evidenced by markedly reduced expression of TGF-β type I and II receptors and profoundly decreased levels of P-Smad2, an important TGF-β–response indicator, in aortic endothelium. These mice exhibit greatly increased integration of cholesterol into tissue plasma membranes. These results suggest that dietary trans fats cause atherosclerosis, at least in part, by suppressing TGF-β responsiveness. This effect is presumably mediated by the increased deposition of cholesterol into cellular plasma membranes in vascular tissue, as in hypercholesterolemia.
Keywords: trans fats, cholesterol, aortic endothelium, suppressed TGF-β responsiveness, atherosclerosis
Introduction
Trans fats are composed of trans-fatty acids (TFAs) which are the stereoisomers of the naturally occurring cis-fatty acids (CFAs). They are produced industrially during hydrogenation of unsaturated oils via a process called “hardening”; (1). This process reduces most of the double bonds in unsaturated fatty acid moieties of the oils but isomerizes some of the cis double bonds to a trans configuration, resulting in improved stability, a longer shelf life and the acquisition of desirable tactile, functional and sensory properties (2). Small amounts of naturally occurring TFAs can also be found in various naturally occurring foods and are formed in the rumen of polygastric animals such as cattle, sheep and goats. This is due to the normal bacterial metabolism of unsaturated fatty acids that occurs in the rumen (1). Industrial trans fats are widely used to produce a variety of foods, including margarines, cookies, pastries, salad dressings and cooking oils (2). It has been estimated that trans fats can comprise 4-12% of the dietary fat, which is equivalent to 2-4% of the total calorie intake, in the US (3,4). TFAs are known to be incorporated into plasma lipoproteins and into cell membranes of various tissues, including aortic endothelium (5-11). TFA incorporation into cell membranes is directly proportional to dietary TF levels (8-11). A high concentration of trans fats in the human diet is linked to a variety of disorders and diseases, most notably atherosclerosis (12). The mechanisms by which trans fats cause atherosclerosis and other diseases remain elusive (13).
Transforming growth factor-β (TGF-β) is a family of 25-kDa disulfide-linked dimeric proteins which has three members in mammals (TGF-β1-3) (14). It is a pleiotrophic cytokine and its biological activities include growth regulation, control of chemotaxis and regulation of the synthesis and deposition of extracellular matrix proteins. TGF-β has been implicated in many physiological and pathological processes (14). Accumulating evidence indicates that TGF-β in the circulation protects against atherosclerosis (15-22). Recently, we found that suppressed TGF-β responsiveness in aortic endothelium plays an important role in the pathogenesis of atherosclerosis in hypercholesterolemic animals (23,24). A high concentration of cholesterol in the culture medium suppresses TGF-β responsiveness in cultured cells, including endothelial cells, by causing accumulation of cell-surface TGF-β-TGF-β receptor complexes in lipid rafts/caveolae of the plasma membrane, facilitating rapid degradation of these complexes, and thus attenuating TGF-β signaling and the related responses (23,24). This effect of cholesterol is believed to be mediated by increasing formation of, or stabilization of, lipid rafts/caveolae, presumably via direct integration of cholesterol into plasma membranes of target cells (23,24). Cholesterol is an important structural component of lipid rafts/caveolae. This implies that increased affinity of plasma membranes for cholesterol (due to changes in membrane composition) may result in increased integration of cholesterol into plasma membranes (even in the absence of a high cholesterol level in the medium or plasma) and suppression of responses to TGF-β. Membrane phospholipids containing TFAs have been shown to have increased affinity for cholesterol (4,11). We hypothesized that dietary trans fats may cause atherosclerosis by suppressing TGF-β responses in vascular cells via incorporation of diet-derived TFAs into plasma membrane phospholipids, with resultant increased integration of cholesterol into plasma membranes. In this communication, we provide evidence to support this hypothesis.
Results and Discussion
The C57BL/6 mouse is a standard animal model for studying the pathogenesis of atherosclerosis (20,21). To study the effect of a trans-fat diet on development of atherosclerosis, male mice (5-6 week old; 10 mice per each experimental group) were fed a western diet with trans fats (TF diet) containing 45% of the calories from fat in the form of partially hydrogenated vegetable oil, of which approximately 30% was trans-fat (trans-fat custom diet TD06303, Halen Teklad) (25). Control mice (n = 10) were fed standard rodent chow containing 13.6% of the calories from fat (control diet). After 16 or 24 weeks, mice treated with TF-diet and control diet were sacrificed. Another group of mice (n = 10) fed the TF diet for 16 weeks were fed the control diet for additional 8 weeks, and then sacrificed. Tissues (plasma, hearts, aortas, and livers) were removed from the animals for biochemical, histological and Western blot analyses. After 16 weeks, mice fed the TF diet gained more body weight as compared to those fed the control diet (37.2 ± 3.2 g vs. 28.4 ± 2.4 g in TF diet vs. control diet). Plasma total cholesterol levels were 65% higher in mice fed the TF diet than those fed the control diet (131 ± 13 vs. 80 ± 6 mg/dl, P<0.05). However, plasma triglyceride levels were similar in both groups (59 ± 8 vs. 61 ± 14 mg/dl in control diet vs. TF diet).
Suppressed TGF-β responsiveness in animals caused by a high plasma cholesterol level are characterized by a low ratio of 125I-TGF-β binding to the type II TGF-β receptor (TβR-II) compared to 125I-TGF-β binding to the type I TGF-β receptor (TβR-I) (TβR-II/TβR-I binding) as determined by 125I-TGF-β affinity labeling of the aortic endothelium and by attenuated expression of phosphorylated Smad2 (P-Smad2) in the same tissue (23,24). A low ratio of TβR-II/TβR-I binding indicates that the TGF-β-TGF-β receptor complex accumulates predominantly in lipid rafts/caveolae of the plasma membrane. This mediates rapid degradation of TGF-β-TGF-β receptor complexes following TGF-β binding to the TGF-β receptors, resulting in attenuation of both TGF-β signaling and downstream responses (23,24,26-30). Smad2 is an important signaling molecule directing TGF-β-stimulated cellular responses (31,32). Phosphorylation of Smad2 by TβR-I in the TβR-I-TβR-II heterocomplex stimulated by TGF-β leads to downstream effects such as formation and translocation of trimeric Smad2/3/4 complexes to the nucleus, leading to regulation of expression of TGF-β responsive genes (31,32).
As shown in Fig. 1A, mice fed a TF diet for 24 weeks exhibited a low ratio (0.95) of TβR-II/TβR-I binding as determined by 125I-TGF-β affinity labeling of the aortic endothelium compared to the ratio (2.79) of TβR-II/TβR-I binding found in mice fed the control diet for the same time period (panel a, lane 2 vs. lane 1 and panel b). Interestingly, the low ratio of TβR-II/TβR-I binding could be normalized to that of mice fed the control diet for 24 weeks when mice fed the TF diet for 16 weeks were given the control diet for an additional 8 weeks (the TF→control diet group in panel a, lane 3 vs. the control diet group in panel a, lane 1 and panel b). Mice fed the TF diet for 16 weeks exhibited a ratio (~1.0) of TβR-II/TβR-I binding which was almost identical with that found in mice fed the TF diet for 24 weeks. (data not shown). To further define the functional consequences of the low ratio of TβR-II/TβR-I binding in the mice fed the TF diet, we performed Western blot analysis of TβR-I, TβR-II, P-Smad2 and VCAM-1 in aortic endothelium. P-Smad2 is a TGF-β-response indicator (14,31,32). VCAM-1 is a marker of early lesions of atherosclerosis (23,24,33). As shown in Fig.1B, mice fed a control diet for 24 weeks had a ratio (2:1) of TβR-II protein compared to TβR-I protein (TβR-II/TβR-I protein) in aortic endothelium (panel a, lane 1), which is similar to the ratio of TβR-II/TβR-I binding in the same tissue. Mice fed a TF diet for 24 weeks exhibited markedly reduced expression of TβR-I and TβR-II (to ~10% and ~5%, respectively, of those in mice fed a control diet) (panel a, lane 2 and panel b). The ratio of TβR-II/TβR-I protein in the aortic endothelium of mice fed the TF diet for 24 weeks was estimated to be ~0.9 (panel a, lane 2 and panel b) which is very similar to that of TβR-II/TβR-I binding as determined by 125I-TGF-β affinity labeling. The P-Smad2 protein levels in the aortic endothelium of mice fed the TF diet were too low to be detected (<3% of those found in mice fed the control diet) (panel a, lane 2), in contrast to the significant levels (~10%) of P-Smad2 that were detected in mice fed the control diet for 8 weeks following the 16-week TF diet (panel a, lane 3 and panel b). The relative levels of the total Smad2 protein in mice fed different diets for a total of 24 weeks, which were determined by quantitative Western blot analysis (23,24), were very similar (data not shown). The 24-week TF diet increased VCAM-1 expression by ~4 fold when compared to that in mice fed the control diet (panel a, lane 2 and panel b), corresponding to significant atherosclerotic lesions found in mice fed the TF diet. In mice fed the control diet for an additional 8 weeks after 16 weeks of the TF diet, the aortic endothelium exhibited increased expression of TβR-I and TβR-II protein levels with a TβR-II/TβR-I protein ratio of ~2.2 (panel a, lane 3 and panel b) which was very similar to the ratio (2.38) of TβR-II/TβR-I binding as determined by 125I-TGF-β affinity labeling in the aortic endothelium of these mice. However, the TβR-I and TβR-II protein levels were not completely normalized to control-diet levels in these mice. The TβR-I and TβR-II protein levels were ~10% and ~6% of control-diet levels in TF diet-fed (for 24 weeks) mice and increased to ~35% and ~45 % of control levels respectively, in mice fed a TF diet for 16 weeks followed by a control diet for 8 weeks (panel a, lane 3 and panel b). Interestingly, the VCAM-1 levels were markedly decreased from the 4-fold elevation in TF-fed mice to 1.3-fold above control in mice switched to a control diet following the TF diet (panel b). Switching to the control diet also increased the P-Smad2 level from the undetectable level (<3%) (in mice fed the TF diet for 24 weeks) to ~10% of the level in mice fed a control diet (panel a, lane 3 and panel b) when the P-Smad2 level in mice fed the control diet for 24 weeks was taken as 100%. This ~10% increase appeared to be enough to markedly decrease the VCAM-1 level from 4-fold to 1.3-fold in the aortic endothelium of mice switched to the control diet for 8 weeks.
Fig. 1. 125I-TGF-β affinity labeling (A) and Western blot analysis (B) of the aortic endothelium in mice fed TF and control diets.
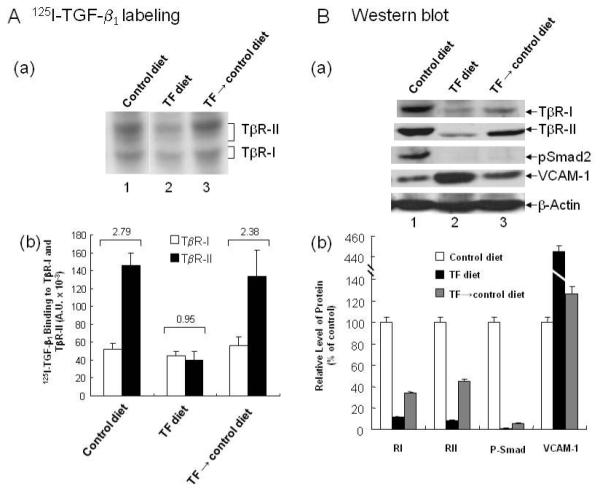
Groups of mice were fed TF and control diets for 24 weeks and were termed the TF and control diet groups, respectively. Another group of mice was fed the TF diet for 16 weeks and then the control diet for additional 8 weeks and was termed the TF→control diet group. At the end of the experiment, the aortic endothelium of these mice was subjected to 125I-TGF-β affinity labeling (A) and Western blot analysis using antibodies to TβR-I, TβR-II, P-Smad2, Smad2, VCAM-1 and β-actin (B). 125I-TGF-β affinity-labeled TβR-I and TβR-II were quantified by a PhosphoImager and expressed as arbitrary units (A.U.). An autoradiogram representative of 125I-TGF-β affinity labeling in the aortic endothelium is shown in panel a (A). The average ratio of 125I-TGF-β binding to TβR-II compared to 125I-TGF-β binding to TβR-I (TβR-II/TβR-II binding) was estimated form 5 mice for each experimental group, as shown in the top of panel a (A). The relative protein levels of TβR-I, TβR-II, P-Smad2, Smad2, VCAM-1 and β-actin were quantified by densitometry (B). The levels in mice fed the control diet were taken as 100%. A representative of a total of 5 animals in each group analyzed is shown (B, panel a). The relative levels of TβR-I, TβR-II, P-Smad2 and VCAM-1 were estimated from the 5 animals in each group; values are mean ± s.d. (B, panel b). The relative levels of Smad2 in mice fed TF, control and TF→control diets for a total of 24 weeks were very similar (data not shown).
To analyze atherosclerotic lesions in mice fed a TF diet and a control diet, we also performed histological analysis after formaldehyde fixation and hematoxylin and eosin (H & E) staining of tissue sections of heart and aorta. As shown in Fig. 2, the heart valves and aorta in mice fed the TF diet exhibited typical early lesions of atherosclerosis whereas mice fed the control diet had normal morphology in these two tissues (Fig. 2B and 2D vs. Fig. 2A and 2C). Foam cells accumulation and fibro-intimal proliferation were found in the heart valves and aorta, respectively, in all of mice fed the TF diet (Fig. 2B and 2D vs. Fig. 2A and 2C). These results also support the hypothesis that the low ratio of TβR-II/TβR-I binding, which reflects the predominant localization of TβR-I and TβR-II in lipid rafts/caveolae microdomains of the plasma membrane, in the aortic endothelium is an excellent indicator of susceptibility to atherosclerosis (23,24,26,27).
Fig. 2. Histological analysis of heart and aorta in mice fed TF and control diets.
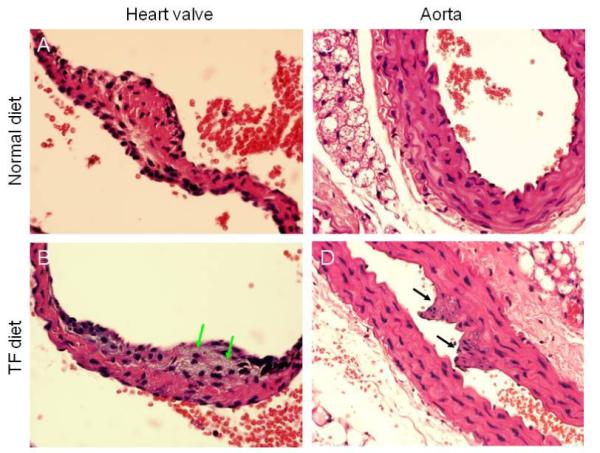
Mice were fed TF and control diets for 24 weeks. Heart valves (A, B) and aorta (C, D) were analyzed following fixation and H & E staining. A representative of a total of 5 mice analyzed is shown. Foam cells and fibro-intimal proliferation were found in the heart valves and aorta as indicated by green and black arrows, respectively.
We hypothesized that tissue membranes derived from mice fed a TF diet would have high affinity for cholesterol, leading to increased integration of cholesterol. As in cells grown in a medium with a high cholesterol level (23,24), this may result in increasing formation of, or stabilization of, lipid rafts/caveolae in plasma membranes, accumulation of TβR-I and TβR-II in lipid rafts/caveolae, lipid raft/caveolae-mediated rapid degradation of the TGF-β receptors following TGF-β binding to these receptors, and subsequent suppressed TGF-β responsiveness in vascular cells and other cell types. To test this hypothesis and investigate the reversibility of the trans-fat effect, we determined the total amount of cholesterol in the livers and hearts of mice fed a TF diet or a control diet for 16 weeks and in the livers of mice fed a control diet and a TF diet for 24 weeks or fed a control diet for 8 weeks following a TF diet for 16 weeks. The reason for analyzing these two organs is that hearts and livers provided enough tissue for quantitative determination of cholesterol. As shown in Fig. 3, there was a ~2.0 fold increase in the cholesterol content in the heart and liver of mice fed a TF diet compared to mice fed a control diet at 16 weeks (Fig. 3A and Fig. 3C, respectively). The increased cholesterol content returned to control values when the mice were given the control diet for additional 8 weeks (Fig. 3B). Since the tissues analyzed were not perfused prior to analysis, the increased cholesterol content detected may be partially due to free cholesterol in blood and extracellular matrix tissue but not membrane-associated cholesterol. To test this possibility, we purified plasma membranes of livers from these mice and determined the cholesterol content of the purified plasma membranes. As shown in Fig. 3D, the TF diet increased plasma-membrane-associated cholesterol by ~ 4-fold compared to the control diet group at 16 weeks. These results suggest that plasma membranes harvested after TF diet feeding possess higher affinity for cholesterol than those derived from a control diet containing mainly cis-fatty acids.
Fig. 3. Cholesterol content in hearts and livers of mice fed TF and control diets.
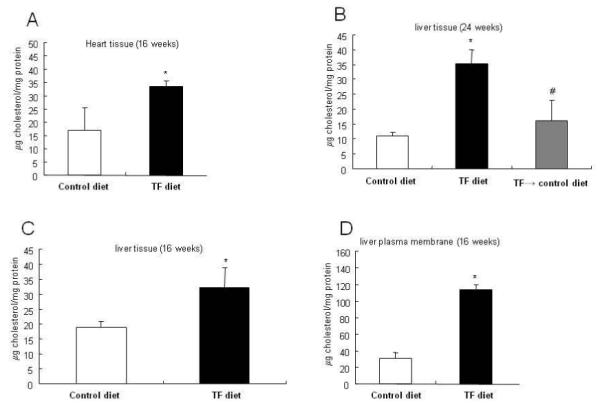
The cholesterol content in hearts and livers of mice fed a TF diet or control diet for 16 or 24 weeks or fed a TF diet for 16 weeks and control diet for additional 8 weeks (TF→control diet) expressed as μg per mg tissue protein is shown. The cholesterol content of purified liver plasma membranes was also determined as per mg membrane protein. The data were estimated from a total of 6-8 mice in each group analyzed; values are mean ± s.d. *Significantly higher than control diet groups (p< 0.05). #Significantly lower than TF diet groups (p< 0.05).
Here we demonstrate that mice fed a TF diet have suppressed TGF-β responsiveness and develop atherosclerotic lesions in the aortic endothelium. Suppressed TGF-β responsiveness in the aortic endothelium is accompanied by a low ratio of TβR-I/TβR-II binding, which indicates that these receptors accumulate predominantly in lipid raft/caveolae, causing lipid raft/caveolae–mediated rapid degradation of TGF-β-TGF-β receptor complexes, and eventual down-regulation of P-Smad2 expression (23,24,26-30). The atherosclerotic lesions in mice fed a TF diet are accompanied by a large increase in the expression of VCAM-1. The VCAM-1 expression caused by the TF diet is actually greater than that found in hypercholesterolemic ApoE-null mice (~4 fold vs ~1.7 fold) (23,24). Normal or wild-type mice fed a TF diet and hypercholesterolemic ApoE-null mice exhibit down-regulation of TβR-II (to ~5% of normal and ~40% of normal, respectively, compared to that in normal mice fed a control diet) (23,24). Interestingly, the levels of TβR-I in normal mice fed a TF diet are also greatly reduced (to ~10% compared to that in normal mice fed a control diet). By contrast, the levels of TβR-I are not significantly altered in hypercholesterolemic ApoE-null mice as compared to those in normal mice fed a control diet (23,24). These results indicate that the TF diet in wild-type mice appears to suppress TGF-β responsiveness more strongly than hypercholesterolemia in ApoE-null mice as evidenced by markedly decreased levels of TβR-I, TβR-II and P-Smad2 in wild-type mice fed a TF diet (compared to those in hypercholesterolemic ApoE-null mice) (23). However, the atherosclerotic lesions in wild-type mice fed a TF diet are less severe than those in ApoE-null mice with hypercholesterolemia as evidenced by high expression of VCAM-1, a marker for early atherosclerotic lesions, and the development of atherosclerotic early lesions in mice fed a TF diet (23,24). This suggests that other factors such as continuous endothelial injury by high plasma LDL and/or cholesterol levels may facilitate the progression of atherosclerosis in hypercholesterolemic animals and that the combination of dietary trans fats and hypercholesterolemia would amplify the effects on progression of atherosclerosis and other related diseases. Lichenstein’s laboratory (34,35) demonstrated that the major risk factor for cardiovascular disease caused by dietary trans fats is LDL-cholesterol levels and total cholesterol/HDL-cholesterol ratios in plasma. Based on these findings, we hypothesize that suppressed TGF-β responsiveness is an important early step in the progression of atherosclerosis caused by hypercholesterolemia or dietary trans fats. This hypothesis is supported by our recent findings that dynasore, a TGF-β mimetic and enhancer, does not alter high plasma cholesterol levels but effectively ameliorates atherosclerosis by antagonizing cholesterol-induced suppression of TGF-β responsiveness in ApoE-null mice with hypercholesterolemia (36).
Based the findings in this communication, we propose a model (Fig. 4) to illustrate how the incorporation of TFAs (e.g., elaidic acid) into phospholipids (possibly at syn-2) in tissues increases integration of cholesterol into the plasma membrane and accumulation of TβR-I and TβR-II in lipid rafts/caveolae. In this model, incorporation of TFAs (which are derived from the TF diet) into phospholipids increases affinity of the plasma membrane for cholesterol (37,38) and formation of, or stabilization of, lipid rafts/caveolae, facilitating accumulation of TβR-I and TβR-II in this plasma membrane microdomain. This is evidenced by a low ratio of TβR-II/TβR-I binding observed in the aortic endothelium of mice fed a TF diet. On the other hand, incorporation of cis-fatty acids (CFAs), which are derived from the normal diet, into phospholipids decreases formation of, or destabilizes, lipid rafts /caveolae, resulting in accumulation of TβR-I and TβR-II in non-lipid raft microdomains. This is evidenced by a high ratio of TβR-II/TβR-I binding found in the aortic endothelium of mice fed a control diet. In the absence of TFAs in the phospholipids of the aortic endothelium in mice fed a normal diet, TβR-I and TβR-II mainly accumulate in non-lipid raft microdomains and have a high ratio of TβR-II/TβR-I binding. The presence of TFAs in the phospholipids of both lipid raft/caveolae and non-lipid raft micrdomains may cause instability of these two microdomains (particularly the lipid raft/caveolae microdomain, because of its high content of TFAs), resulting in rapid turnover and degradation of both the TGF-β receptors. This is supported by the evidence that markedly decreased levels of both types of TβR-I and TβR-II are found in mice fed a TF diet as compared to mice fed a control diet. The TβR-I is relatively stable in mice fed a high cholesterol diet without trans fats (23,24), suggesting that the TGF-β receptors localized to lipid rafts/caveolae (which contain TβR-I > TβR-II) are more stable than those localized to non-lipid raft microdomains (which contain TβR-II > TβR-I) in the aortic endothelium of these hypercholestrolemic ApoE-null mice (23,24).
Fig. 4. A model for the effects of TF and normal diets on TGF-β receptor partitioning between lipid raft/caveolae and non-lipid raft microdomains.
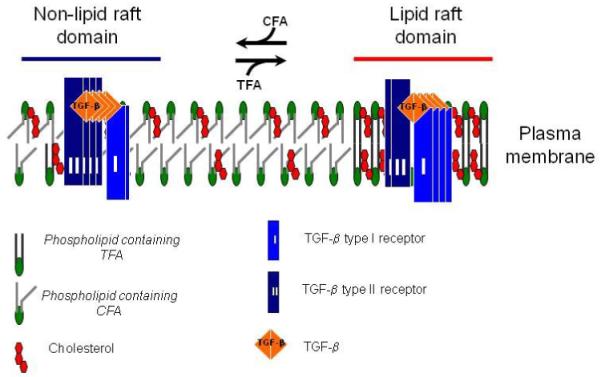
In cells (including vascular cells), two major TβR-I -TβR-II complexes (Complex I and Complex II) present on the cell surface (23,24,29,30). Complex I and Complex II are mainly localized to the non-lipid raft and lipid raft/caveolae microdomains of the plasma membrane, respectively. The numbers of TβR-I and TβR-II molecules (blue and black rectangles, respectively) in Complex I and Complex II shown in the model are arbitrary and intended to indicate that Complex I and Complex II contain TβR-II >TβR-I and TβR-I >TβR-II, respectively. The ratio of TGF-β binding to TβR-II compared to TGF-β binding to TβR-I (TβR-II/TβR-I binding) can be determined by 125I-TGF-β affinity labeling (23,24,29,30). Trans-fatty acid (TFA, derived from the TF diet) incorporation into phospholipids increases formation of, or stabilization of, lipid rafts/caveolae, facilitating accumulation of TβR-I and TβR-II in the microdomain where TGF-β binding to the receptors is incapable of inducing Smad2-dependent signaling but does cause receptor internalization and degradation, and thus attenuated cellular responses. Cis-fatty acid (CFA, derived from the normal diet) incorporation into phospholipids results in increasing formation of, or stabilizing, non-lipid raft microdomains where TGF-β binding to the receptors induces Smad2 signaling which leads to cellular responses. Both lipid raft/caveolae and non-lipid raft microdomains, which contain TFA moieties, in mice fed a TF diet are relatively unstable compared to those in mice fed the normal diet. This may explain the profoundly decreased levels of TβR-I and TβR-II in mice fed a TF diet as compared with those in mice fed the normal diet.
Our demonstration that suppression of TGF-β responsiveness (in aortic endothelium and possibly in other tissues) caused by dietary trans fats is likely to have important implications in other TF-related diseases and disorders. For example, it has been postulated that trans fats are carcinogenic and also contribute to autoimmune disease. These phenomena might be due to suppression of cellular TGF-β responsiveness by dietary trans fats since TGF-β is a well-known tumor suppressor and immunosuppressor (14).
Materials and Methods
Animals
The experimental conditions and physical (body weight, food/water consumption and liver weight), biochemical (plasma triglyceride, cholesterol, glucose, insulin, leptin, resistin, adiponectin and tPA levels, liver function tests and glucose tolerance tests) and histological (liver) analysis for C57BL/6 mice fed the high trans-fat (TF) and normal (control) diets have recently been published (25). Briefly, five-week-old male C57BL/6 mice (Harlan, Indianapolis, IN) were fed a TF diet similar to a western fast food diet with 45% of the calories from fat in the form of partially hydrogenated vegetable oil (trans-fat custom diet TD06303, Harlan Teklad, Madison, WI). Drinking water was provided as gel-water, 2.8% gelatin in the dishes on the cage floor containing 42 g/l high fructose corn syrup (55% fructose and 45% glucose). Control mice were fed standard rodent chow (control diet) containing 13.6% of calories from fat in the form of soybean oil (15% saturated, 23% MUFAs, 61% PUFAs; 2018S, Harlan Teklad). Ten mice were used in each experimental group. After 16 or 24 weeks of ad libitum feeding, mice fasted for 8 hours were sacrificed using ketamine and xylazine anesthesia. To determine if abnormalities induced by this diet were reversible, a group of mice (n=10) treated with the TF diet for 16 weeks were returned to the control diet for another 8 weeks and compared to groups of mice either kept on the TF diet or kept on the control diet for the entire 24 week period. Hearts, aortas, blood and livers were removed for histological (hematoxylin & eosin staining), biochemical and Western blot analyses. The plasma membranes of hearts and livers were prepared according to standard procedures.
Biochemical analyses
Homogenates were prepared by centrifugation following homogenization of livers and hearts removed from mice fed TF and control diets (at 1 g liver or heart tissue/ml phosphate-buffered saline). Plasma membranes of the livers and hearts from mice fed TF and control diets were purified according to standard procedures. Cholesterol contents in tissue homogenates, plasma membranes and serum were analyzed according to standard procedures using a calibrated clinical analyzer (Cobas Mira Plus Chemistry Analyzer (Roche Diagnostics, Indianapolis, IN). Histological analysis of heart and aorta sections were performed as described (23,24). Statistical comparisons among groups were performed by one-way ANOVA followed by post hoc pair-wise multiple-comparison procedures.
125I-TGF-β affinity labeling and Western Blot analysis of aortic endothelium
The endothelium of the aortas from mice fed a TF diet (24 weeks), a control diet (24 weeks), or a TF diet for 16 weeks plus a control diet for additional 8 weeks (TF→control diet) were prepared (5 mice per experimental group) and subjected to 125I-TGF-β affinity labeling and Western blot analyses using antibodies to TβR-I, TβR-II, P-Smad2, Smad2, VCAM-1 and β-actin, as described previously (23,24). The aortic endothelium extracts were characterized by the presence of factor VIII and absence of α-smooth muscle actin (23,24).
Acknowledgement
We thank Frank E. Johnson, M.D. for critical review of the manuscript and Cheng C. Tsai, M.D. for histological analyses and Chris Mabes for typing the manuscript
Footnotes
This work was supported by NIH grants HL 087463 (J.S.H.) and AR 052578 (S.S.H.), and by St. Louis University Liver grant (B.A.N-T).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Emken EA. Nutrition and biochemistry of trans and positional fatty acid isomers in hydrogenated oils. Annu. Rev. Nutr. 1984;4:339–376. doi: 10.1146/annurev.nu.04.070184.002011. [DOI] [PubMed] [Google Scholar]
- (2).Korver O, Katan MB. The elimination of trans fats from spreads: how science helped to turn an industry around. Nutr. Rev. 2006;64:275–279. doi: 10.1111/j.1753-4887.2006.tb00210.x. [DOI] [PubMed] [Google Scholar]
- (3).Allison D, Denke M, Dietschy J, Emken E, Kris-Etherton P, Nicolosi R. Trans fatty acids and cononary heart disease risk. Report of the expert panel on trans fatty acids and coronary heart disease. Am. J. Clin. Nutr. 1995;62:655S–708S. doi: 10.1093/ajcn/62.3.655S. [DOI] [PubMed] [Google Scholar]
- (4).Roach C, Feller SE, Ward JA, Shaikh SR, Zerouga M, Stillwell W. Comparison of cis and trans fatty acid containing phosphatidylcholines on membrane properties. Biochem. 2004;43:6344–6351. doi: 10.1021/bi049917r. [DOI] [PubMed] [Google Scholar]
- (5).Schrock CG, Connor WE. Incorporation of the dietary trans fatty acid (C18:1) into the serum lipids, the serum lipoproteins and adipose tissue. Am. J. Clin. Nutr. 1975;28:1020–1027. doi: 10.1093/ajcn/28.9.1020. [DOI] [PubMed] [Google Scholar]
- (6).Privett OS, Phillips F, Shimasaki H, Nozawa T, Nickell EC. Studies of effects of trans fatty acids in the diet on lipid metabolism in essential fatty acid deficient rats. Am. J. Clin. Nutr. 1971;30:1009–1017. doi: 10.1093/ajcn/30.7.1009. [DOI] [PubMed] [Google Scholar]
- (7).Cook HW. Incorporation, metabolism and positional distribution of trans-unsaturated fatty acids in developing and mature brain. Comparison of elaidate and oleate administered intracerebrally. Biochim. Biophys. Acta. 1978;531:245–256. doi: 10.1016/0005-2760(78)90206-0. [DOI] [PubMed] [Google Scholar]
- (8).Emken EA, Dutton HJ, Rohwedder WK, Rakoff H, Adlof RO. Distribution of deuterium-labeled cis- and trans-12-octadecenoic acids in human plasma and lipoprotein lipids. Lipids. 1980;15:864–871. doi: 10.1007/BF02534378. [DOI] [PubMed] [Google Scholar]
- (9).Blomstrand R, Diczfalusy U, Sisfontes L, Svensson L. Influence of dietary partially hydrogenated vegetable and marine oils on membrane composition and function of liver microsomes and platelets in the rat. Lipids. 1985;20:283–295. doi: 10.1007/BF02534261. [DOI] [PubMed] [Google Scholar]
- (10).Hoy CE, Holmer G. Incorporation of cis- and trans-octadecenoic acids into the membranes of rat liver mitochondria. Lipids. 2005;14:727–733. doi: 10.1007/BF02533898. [DOI] [PubMed] [Google Scholar]
- (11).Niu SL, Mitchell DC, Litman BJ. Trans fatty acid derived phospholipids show increased membrane cholesterol and reduced receptor activation as compared to their cis analogs. Biochem. 2005;44:4458–4465. doi: 10.1021/bi048319+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mozaffarian D, Katan MB, Ascherio A, Stampfer MJ, Willett WC. Trans fatty acids and cardiovascular disease. N. Engl. J. Med. 2006;354:1601–1613. doi: 10.1056/NEJMra054035. [DOI] [PubMed] [Google Scholar]
- (13).Motard-Belanger A, Charest A, Grenier G, Paquin P, Chouinard Y, Lemieux S, Couture P, Lamarche B. Study of the effect of trans fatty acids from ruminants on blood lipids and other risk factors for cardiovascular disease. Am. J. Clin. Nutr. 2008;87:593–599. doi: 10.1093/ajcn/87.3.593. [DOI] [PubMed] [Google Scholar]
- (14).Roberts AB. Molecular and cell biology of TGF-β. Miner. Electrolyte. Metab. 1998;24:111–119. doi: 10.1159/000057358. [DOI] [PubMed] [Google Scholar]
- (15).Metcalfe JC, Grainger DJ. Transforming growth factor-β and the protection from cardiovascular injury hypothesis. Biochem. Soc. Transact. 1995;23:403–406. doi: 10.1042/bst0230403. [DOI] [PubMed] [Google Scholar]
- (16).Grainger DJ, Kempt PR, Metcalfe JC, Liu AC, Lawn RM, Williams NR, Grace AA, Schofield PM, Chauhan A. The serum concentration of active transforming growth factor-β is severely depressed in advanced atherosclerosis. Nat. Med. 1995;1:74–79. doi: 10.1038/nm0195-74. [DOI] [PubMed] [Google Scholar]
- (17).Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, Fradelizi D, Tedgui A. Inhibition of transforming growth factor-β signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Cir. Res. 2001;89:930–934. doi: 10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- (18).Stefoni S, Cianciolo G, Donati A, Silvestri MG, Coli L, De Pascalis A, Ianneelli S. Low TGF-β1 serum levels are a risk factor for atherosclerosis disease in ESRD patients. Kidney Int. 2002;61:324–335. doi: 10.1046/j.1523-1755.2002.00119.x. [DOI] [PubMed] [Google Scholar]
- (19).Grainger DJ. Transforming growth factor β and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler. Thromb. Vasc. Biol. 2004;24:399–404. doi: 10.1161/01.ATV.0000114567.76772.33. [DOI] [PubMed] [Google Scholar]
- (20).Bobik A. Transforming growth factor-βs and vascular disorders. Arterioscler. Thromb. Vasc. Biol. 2006;26:1712–1720. doi: 10.1161/01.ATV.0000225287.20034.2c. [DOI] [PubMed] [Google Scholar]
- (21).Frutkin AD, Ostuka G, Stempien-Otero A, Sesti C, Du L, Jaffe M, Dichek HL, Pennington CJ, Edward DR, Nieves-Cintron M, Minter D, Prusch M, Hu JH, Marie JC, Dichek DA. TGF-β1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2009;29:1251–1257. doi: 10.1161/ATVBAHA.109.186593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-β signaling in T cells accelerates atherosclerosis. J. Clin. Invest. 2003;112:1342–1350. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen C-L, Liu I-H, Fliesler SJ, Han X, Huang SS, Huang JS. Cholesterol suppresses cellular TGF-β responsiveness: implications in atherogenesis. J. Cell Sci. 2007;120:3509–3521. doi: 10.1242/jcs.006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen C-L, Huang SS, Huang JS. Cholesterol modulates cellular TGF-beta responsiveness by altering TGF-β binding to TGF-β receptors. J. Cell. Physiol. 2008;215:223–233. doi: 10.1002/jcp.21303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in sedentary mice fed trans-fats and high fructose syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;295:G987–995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Di Guglielmo GM, Le Roy C, Anne F, Goodfellow AF, Wrana JA. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. 2003. [DOI] [PubMed] [Google Scholar]
- (27).Ito T, Williams JD, Fraser DJ, Phillips AO. Hyaluronan regulates transforming growth factor-β1 receptor compartmentalization. J. Biol. Chem. 2004;279:25326–25332. doi: 10.1074/jbc.M403135200. [DOI] [PubMed] [Google Scholar]
- (28).Huang SS, Huang JS. TGF-β control of cell proliferation (Prospect) J. Cell. Biochem. 2005;96:447–462. doi: 10.1002/jcb.20558. [DOI] [PubMed] [Google Scholar]
- (29).Chen C-L, Huang SS, Huang JS. Cellular heparan sulfate negatively modulates transforming growth factor-β responsiveness. J. Biol. Chem. 2006;281:11506–11514. doi: 10.1074/jbc.M512821200. [DOI] [PubMed] [Google Scholar]
- (30).Chen Y-G. Endocytic regulation of TGF-β signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. 2009. [DOI] [PubMed] [Google Scholar]
- (31).Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- (32).Massague J. TGF-β signal transduction. Ann Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- (33).Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998;18:842–851. doi: 10.1161/01.atv.18.5.842. [DOI] [PubMed] [Google Scholar]
- (34).Vega-López S, Matthan NR, Ausman LM, M. A, Otokozawa S, Schaefer EJ, Lichtenstein AH. Substitution of vegetable oil for a partially-hydrogenated fat favorably alters cardiovascular disease risk factors in moderately hypercholesterolesterolemic postmenopausal women. Atheroscler. 2009;207:208–212. doi: 10.1016/j.atherosclerosis.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lichtenstein AH, Erkkila AT, Lamarche B, Schwab US, Jalbert SM, Ausman LM. Influence of hydrogenated fat and butter on CVD risk factors: remnant-like particles, glucose and insulin, blood pressure and C- reactive protein. Atheroscler. 2003;171:97–107. doi: 10.1016/j.atherosclerosis.2003.07.005. [DOI] [PubMed] [Google Scholar]
- (36).Chen C-L, Hou W-H, Liu I-H, Huang SS, Huang JS. Inhibitors of clarthrin-dependent endocytosis inhibitors enhance signaling and responses. J. Cell Sci. 2009;122:1863–1871. doi: 10.1242/jcs.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Katz AM. Should trans fatty acids be viewed as membrane-active drugs? Atheroscler. 2006;(Suppl. 7):41–42. doi: 10.1016/j.atherosclerosissup.2006.04.009. [DOI] [PubMed] [Google Scholar]
- (38).Ibrahim A, Natrajan S, Ghafoorunissa R. Dietary trans-fatty acids alter adipocyte plasma membrane fatty acid composition and insulin sensitivity in rats. Metab. Clin. Exp. 2005;54:240–246. doi: 10.1016/j.metabol.2004.08.019. [DOI] [PubMed] [Google Scholar]