

Abstract
Glutathione S-transferase P is abundantly expressed in some mammalian tissues, particularly those associated with malignancies. While the enzyme can catalyze thioether bond formation between some electrophilic chemicals and GSH, novel non-detoxification functions are now ascribed to it. This review summarizes recent material that implicates GSTP in mediating S-glutathionylation of specific clusters of target proteins and in reactions that define a negative regulatory role in some kinase pathways through ligand or protein:protein interactions. It is becoming apparent that GSTP participates in the maintenance of cellular redox homeostasis through a number of convergent and divergent mechanisms. Moreover, drug platforms that have GSTP as a target have produced some interesting preclinical and clinical candidates.
1. Introduction
Since the 1970s much attention has focused around those properties of glutathione S-tranferases (GST) that facilitate the catalysis of thioether bonds between glutathione (GSH) and electrophilic centers on small molecules. Human GSTs can be divided into two distinct super families, membrane bound microsomal and cytosolic. Microsomal GST contain three isoforms designated mGST 1, 2, and 3 encoded by a single gene located on chromosome 12 (MGST1) and are involved in the endogenous metabolism of leukotrienes and prostaglandins [1]. All cytosolic GST have genetic polymorphisms in human populations. They are divided into 6 classes that in humans are found on six different chromosomes but share ~30% sequence identity: Alpha (chromosome 6), Mu (chromosome 1), Omega (chromosome 10), Pi (chromosome 11), Theta (chromosome 22), and Zeta (chromosome 14) [2]. There are indications of both structural as well as functional redundancies between isozyme family members. Multiple alleles sharing >50% sequence identity exist within each class [3]. Promoter regions vary between classes and can contain one or more of the following response elements:-antioxidant-response element; xenobiotic response element; GSTP enhancer 1; glucocorticoid-response element; Barbie box element [4]. Promoters may also contain putative binding sites for transcription factors such as, AP-1, MAF, Nrf1, Jun, Fos, and NF-kappa B, the occurrence of which is species specific. Cytosolic GSTs have catalytic activity as homo- or hetero-dimeric proteins, allowing the formation of greater numbers of enzymes from a limited number of genes [4]; however, dimerization is usually limited to subunits within the same class. The subunits range in size from 24 to 29 kDa [3]. Each subunit contains an active site with two sub-sites: a highly conserved G site for GSH binding and an H site for hydrophobic substrates. Although <10% of the protein is strictly conserved, all GST isozymes have two domains and similar topologies.
Because GSTP is a predominant protein in many tumors the majority of this review focuses upon its increasingly pleiotropic role in the cancer phenotype. Some structural properties of GSTP are quite critical in facilitating and promoting GSH dependent reactions. Its N-terminal domain 1 (essentially residues 1–80, as in all other GSTs) adopts a topology similar to that of the thioredoxin fold [5], consisting of four β-sheets with three flanking α-helices (Figure 1 panel A). This is a structure common to several proteins from a thioredoxin fold super-family, all of which bind cysteine or GSH with high affinity. Examples of these include, DsbA (the bacterial enzyme equivalent to protein disulfide isomerase; [6]), glutaredoxin [7], glutathione peroxidases [8], and peroxiredoxins [9]. This fold consists of distinct N- and C-terminal motifs which have a βαβ and ββα arrangement respectively and which are linked by an α-helix (α-2). Domain 1 is highly conserved in all GST isozymes and provides a binding domain primarily involved in binding GSH. Glutathione occupies a site on domain 1 (referred to as the G-site [10]) which is situated in a cleft formed between the intra-subunit domains. The cleft extends from a segment (residues 8–10) connecting strand β1 to helix α1, to about Ser63 at the N-terminal end of helix α3. One end of the cleft opens out to bulk solvent, while the other, near Ser63, is adjacent to the cavity at the center of the dimer interface. Side chains lining the G-site for GSTP include: Tyr7, Gly12, Arg13, Trp38, Lys42, Gln49, Pro51, Gln62, Ser63 and Glu95 [11]. Domain 1 is connected to domain 2 by a short linker sequence (Figure 1 panels B and C). C-terminal domain 2 (essentially residues 87–210) begins at the C-terminus of the linker sequence and in the case of the GSTP and GSTM family members consists of five α-helices [11, 12] and in the case of the GSTA members, six α-helices. The C- terminal domain together with a loop from the N-terminal domain forms the substrate-binding site (H-site). The H-site is proposed to be hydrophobic and must be adjacent to the G-site, and should also permit proper orientation of the bound reactants. Several possible locations for this site were suggested [11]. One was a hydrophobic region in the cleft adjacent to the G-site that could accommodate small molecules. This region is coated by the side chains of: Phe8, Pro9, Val1O, Met35, Tyr1O6, Pro200 and Gly203 (Figure 1 panel C). Different amino acids in the H site of isozymes can account for substrate specificities. Within the C-terminus an additional α helix is present in the Alpha and Theta classes while the Mu class has an extra loop [13], differences that are proximal to the H site and create a more constricted active site. While GSTs are ubiquitously expressed, their tissue (and even cellular within the same tissue) distribution in mammals is variable and complex [14, 15].
Figure 1.
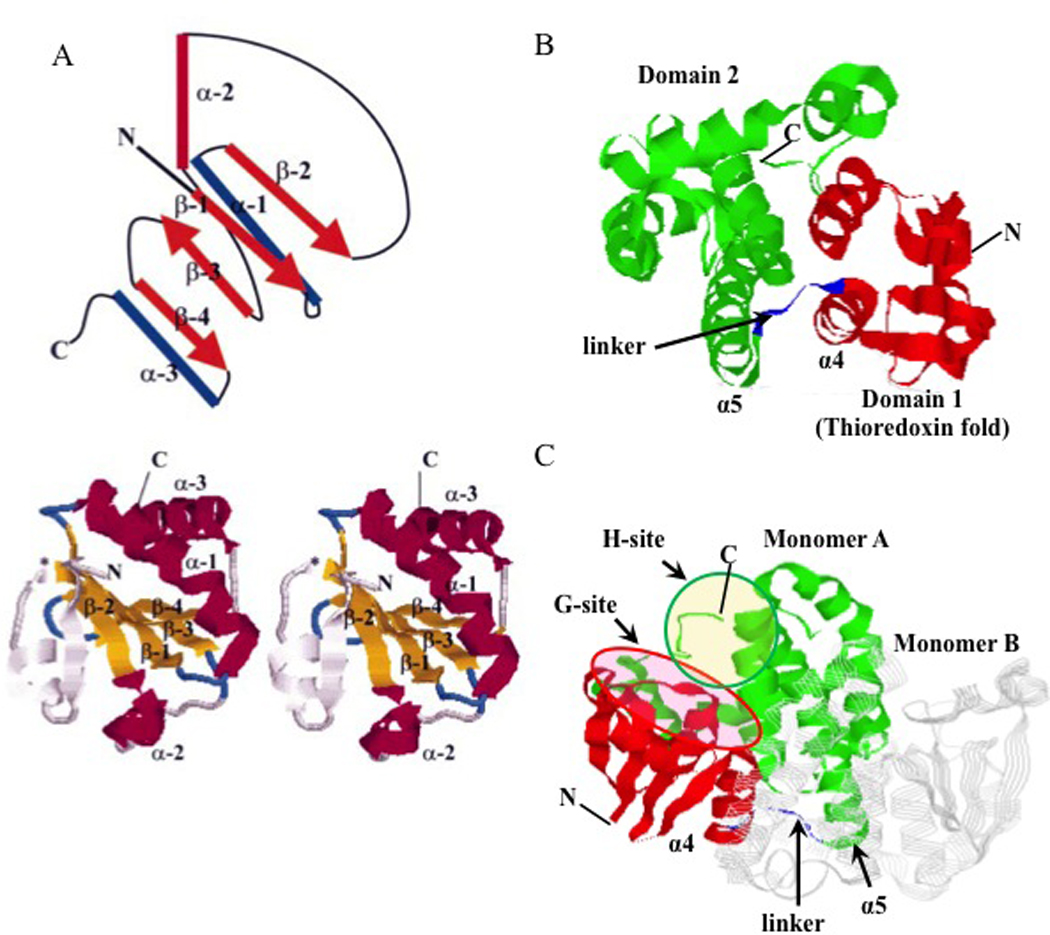
Panel A: schematic diagram representing the thioredoxin fold is shown above a RasMol depiction of the thioredoxin dimer [134]. In the diagram, α-helices are shown as cylinders, while β-sheets are shown as orange arrows. The four β-sheets are essentially co-planar, with one helix (α-2) shown in red above this plane and the other two α-helices (α-1 and α-3) shown in blue below the plane. In this case, the cis-Pro loop links α-2 to β-3. In GSTs, domain 2 is connected to the C-terminus by a short linker peptide. In thioredoxin itself, β-sheets are colored yellow, while α-helices are magenta. The thioredoxin fold has an extra β-sheet and α-helix at the N-terminus (residues 1±21) ending at the point denoted by the asterisk * where the fold begins proper. These additional N-terminal features are colored grey. Domains 1 (red) and 2 (green) of GSTP (5GSS, PDB) together with α4 and α5 helices are presented in panel B. The characteristic GSH-binding site (G-site, purple oval) and electrophilic substrate-binding site (H-site, orange oval) are presented in panel C. Monomer B of a GSTP homodimer is presented in strands (grey). Structures in panels B and C are generated using RasMol (version 2.7.5). Adapted from references [17, 134].
The homodimeric structure is common for proteins containing thioredoxin folds and structural interactions at the inter-subunit interface are crucial for complex assembly and stability. The domains at the inter-subunit surface of the GSTP homodimer are dominated by hydrophobic interactions between residues from domain 1 of one subunit and domain 2 of the other. Aromatic residues play major roles in these interactions. Tyr-49 in GSTP can act as a `key' extending from the loop preceding β-3 that fits into a hydrophobic `lock' provided by helices α-4 and α-5 of the other subunit. The interface is approximately 25 Å to 35 Å and at a height of ~ 25 Å, diverges to create a V-shaped crevice that is solvent-accessible [16]. In addition, the active site cleft in GSTP is shallow, while in other GST isozymes it is larger and more open [17]. Thus, based on structural similarities of C-terminal domains of GSTM and GSTP, similar homodimerization/ monomerization properties can be predicted for these enzymes. Using similar logic, these should be different from the GSTA isozymes.
An early name for one of the GST family was ligandin [18] premised upon their capacity to bind to a number of hydrophobic compounds without their catalytic processing [19] - for example heme and bilirubin. Ironically, there is now a renewed reconsideration of the ligand binding properties of GSTP with particular emphasis on protein interactions. In oncology, interest in the GST family of proteins has been fueled by the fact that high levels of GSTP (the most ubiquitous and prevalent GST in non-hepatic tissues) are found in many tumors (in particular ovarian, non-small cell lung, breast, colon, pancreas and lymphomas) and in a wide range of drug resistant cell lines and tumors [20]. When compared to normal tissues or wild type cell lines, these enhanced expression ratios have not always been readily explained. In two of the earliest reports of increased GST expression in drug resistance, one was in response to chlorambucil [21], where evidence of catalytic formation of the thioether conjugate of this alkylating agent was subsequently documented [22] and could provide a cause:effect relationship for selection of GST over-expression. However, an MCF7 human breast carcinoma cell line resistant to adriamycin was found to have approximately 50-fold more GSTP than the albeit low expressing wild-type line [23]. This correlation was not explicable by GST catalysis, since GSH conjugates of adriamycin do not occur under physiological conditions. Since these reports, tacit (and sometimes unjustified) assumptions have linked GST-mediated detoxification processes with many acquired drug resistant phenotypes. The importance of GST in kinase regulation and a role for GSTP in the forward reaction of S-glutathionylation has provided a maturing approach in understanding alterations in GSTP expression patterns. In this regard, some tumors or drug resistant cells may depend upon this protein. Because of the proliferative nature of tumor cells, kinase pathways are frequently aberrantly regulated, and consequently, there could be a homeostatic attempt to compensate by enhancing expression of GSTP to counterbalance increased kinase activity. Addiction to over-expressed proteins has been identified as a characteristic of the transformed phenotype. In addition, there is a literature of growing abundance delineating the importance of S-glutathionylation in regulating protein structure and function. As one example, phosphatases such as PTP1B [24] and cdc25 [25] are regulated by S-glutathionylation of specific cysteine residues. Because the kinase/phosphatase cycle effects multiple pathways critical to uncontrolled cell growth it would not be unreasonable to speculate that a relative abundance of GSTP may reflect roles unrelated to its capacity to enact catalytic detoxification. In context, the absence of external electrophilic stress implies that GSTP can be such a prevalent protein only as a consequence of the influence of specific tumor related endogenous factors. Over a protracted time period, selective pressures could produce convergent evolution and the emergence of properties of GSTP unrelated to small molecule detoxification.
While it appears that cells are quite capable of adapting to high expression levels of GSTP, how cells might adapt to the absence of GSTP would also provide an insight into the importance of this protein. Mice null for GSTP1-1 and GSTP2-2 are viable, fertile, with essentially normal development and life expectancy [26]. The animals are more susceptible to carcinogen induced skin papillomas [27]. Mouse embryo fibroblast (MEF) cells isolated from wild type differed from GSTP null animals in a number of characteristics relevant to signaling and growth pathways [28]. The doubling time for wild type cells was 33.6 h versus 26.2 h for GSTP null. Both early passage and immortalized MEF cells from GSTP null animals had elevated activities of extracellular regulated kinases (ERK1/ERK2). Knockout animals had constitutively elevated c-jun NH2-terminal kinase (JNK) activities compared to wild type correlating with altered regulation of genes downstream of JNK control [29]. In general, ablation of GSTP influences the capacity of stress kinases to regulate gene expression impacting on cell proliferation pathways. The lack of lethality of the deletion suggests functional redundancy and implies that other GST, or other redox proteins, may compensate for the absence of GSTP. This is supported by the data suggesting general redundancy of function among and within the GST protein cluster.
Although high levels of GSTP frequently accompany the malignant phenotype, exceptions do exist. Hypermethylation of the GSTP regulatory region is a common somatic alteration identified in human prostate cancer [30]. This alteration results in the loss of GSTP expression and is proposed to occur during pathogenesis of the disease. A methyl-CpG binding domain (MBD) protein has been identified that mediates hyper-methylation of the GSTP regulatory region [30]. GST expression (and/or activity) of specific isoforms is lost in some individuals with allelic variation and it has been speculated that reduced GSTP may alter the capacity to detoxify possible carcinogens causing malignant transformation and disease progression in the prostate. While this could be true, absence of GSTP would also alter the regulation of kinase dependent proliferation pathways and/or protein S-glutathionylation patterns. As discussed in the pertinent sections, pharmacological suppression of GSTP, while not as efficient as genetic ablation, also causes changes in these same pathways and can influence cell proliferation (particularly in the bone marrow) in a therapeutic setting.
GSTP and cell redox homeostasis
Since most cancer drugs are not good substrates for GSTP, the question of why acquired drug resistant cells have such high levels of this isozyme seems perplexing. Moreover, even without drug selection GSTP can be one of the more prevalent cytosolic proteins in cancer cells. These observations would seem to indicate that GSTP has a diversity of functions in cancer cells, some of which are likely unrelated to the detoxification of chemicals or drugs. General considerations of cellular redox homeostasis begin to shed light on the apparent conundrum.
The balance between oxidation and reduction reactions determines cellular redox homeostasis and plays an essential role in numerous signaling cascades including those associated with proliferation, inflammatory responses, apoptosis and senescence. Reactive oxygen and nitrogen species (ROS; RNS) are invariable components of aerobic metabolism and are key contributors to cellular redox. The sensing of redox changes is most actively mediated through cysteine residues in various proteins. While there is some debate as to the precise definition of what constitutes redox “sensing” versus redox “signaling” [31] there is little doubt that cysteine residues at various oxidation states are at the center of the process. There appears to be a correlation between organism cysteine content and degree of biological complexity, i.e. evolutionary development [32]. Analysis by Jones and colleagues suggested that there are slightly more than 200,000 cysteines encoded by the human genome [33] a number suggestive of sparing usage, but evolutionary importance. An estimate of current literature suggests that more than 150 proteins have cysteine residues that are susceptible to the post-translational modification of S-glutathionylation (i.e. the addition of GSH), but in context, since susceptible protein targets for S-glutathionylation are still appearing, this number will likely increase. Selective modification can alter the structure and/or function of a number of proteins including, enzymes, receptors, structural proteins, transcription factors and transport proteins and may also alter a variety of protein-protein interactions. This process is discussed in more detail in the next section. However, within the cancer field, it is generally accepted that phosphorylation is perhaps the most critical protein modification that influences cell signaling pathways. As a consequence, it is worth reflecting that there are some parallels between S-glutathionylation and phosphorylation. Of note, some microorganisms deprived of a source of phosphorus will substitute sulfur, creating thiolipid instead of phospholipid membranes [34]. A coalescence of sulfur and phosphorus biochemistry may also be represented by S-glutathionylation of phosphatases [24] critical in maintaining the cyclical nature of kinome regulation (phosphorylation/ dephosphorylation). Many proteins that are S-glutathionylated are also involved in growth regulatory pathways, including many kinases [35]. Thus, the S-glutathionylation cycle may provide an extra level of control of the phosphorylation cascades controlled by kinases or phosphatases.
GSTP and S-glutathionylation
S-glutathionylation Cycle: General Considerations
S-glutathionylation generally occurs when a cysteine in an essentially basic environment within the protein (e.g. three dimensionally surrounded by Arg, His or Lys residues) forms a disulfide bond with GS− (Figure 2). S-glutathionylation can occur either in response to endogenous (physiological) oxidative (ROS) or nitrosative stress (RNS) mediated signaling events, or from exposure to external environmental drug treatments. A wide range of chemicals can induce S-glutathionylation, but literature examples tend to be dominated by hydrogen peroxide, glutathione disulfide, diamide and various nitric oxide donors [35, 36]. In particular, our group has utilized a GST activated diazeniumdiolate prodrug PABA/NO (O2- [2,4-dinitro-5- (N methyl-N-4-carboxyphenylamino) phenyl] 1-N, N-dimethylamino) diazen-1-ium-1, 2-diolate) [37] that releases NO and has been shown to cause limited amounts of nitrosylation, but impressive levels of S-glutathionylation [38]. In this instance, and probably for the many NO donors, nitrosylated cysteines can have short half-lives and can be quite rapidly converted to their S-glutathionylated derivative. Spectroscopic studies indicate that at physiological pH GST effectively lower the pKa of the cysteine thiol of glutathione, resulting in formation of the nucleophilic thiolate anion (GS−) at the active site [39]. The ability of GST to bind and activate GSH is important in numerous reactions where activated GSH can act as a thiol donor [40]. The immediate use or delivery of an activated GSH may determine a particular catalytic or carrier GST function. Cysteines on the surfaces of globular proteins are exposed to GSH and GSSG and are prone to spontaneous S-glutathionylation [41]. They are also influenced by reducing/deglutathionylating enzymes such as thioredoxin (Trx), [42] glutaredoxin (Grx) [43], and sulfiredoxin (Srx) [44]. Although an appreciation of the importance S-glutathionylation has been evident since the 1990s, the identification of specific protein substrates has been made easier by the more recent advances in proteomic technologies. Relative to the proteome the actual number of S-glutathionylated proteins is not proportionally large. By analyzing those clusters of proteins sensitive to the modification some general patterns do emerge. For example, Table 1 summarizes those categories of proteins so far described as being susceptible to S-glutathionylation and the impact that the post-translational modification has upon their functions. These proteins include, enzymes with catalytically important cysteines (in particular those involved with protein folding and stability, nitric oxide regulation, redox homeostasis); cytoskeletal proteins; signaling proteins—particularly kinases and phosphatases; transcription factors; ras proteins; heat shock proteins; ion channels, calcium pumps and binding proteins (involved in calcium homeostasis); energy metabolism and glycolysis.
Figure 2.
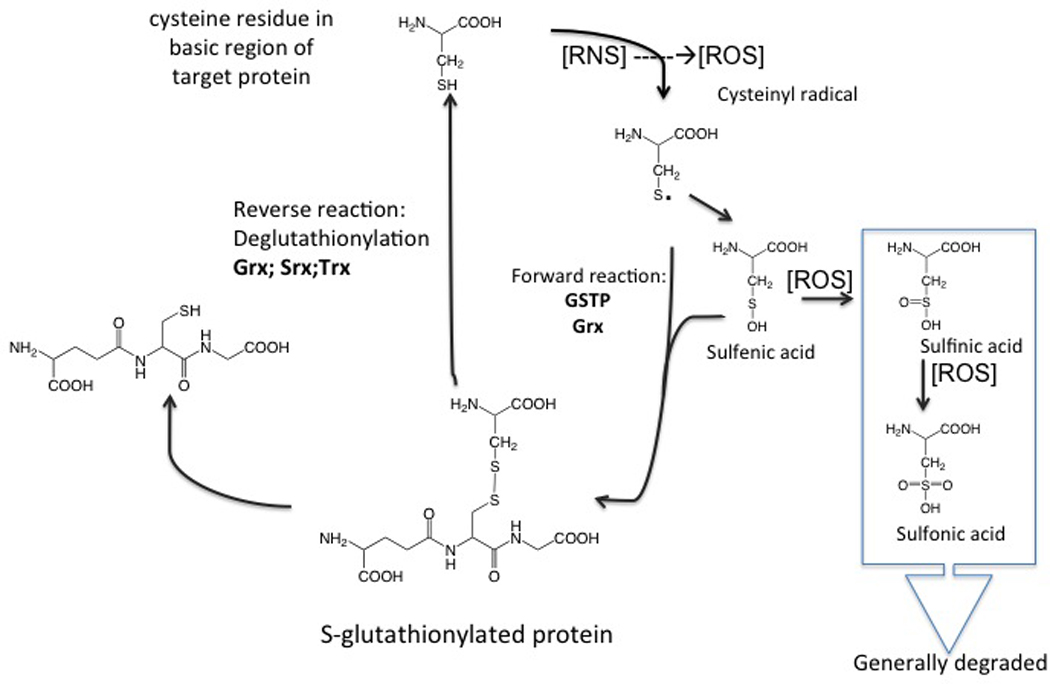
Schematic representation of the S-glutathionylation cycle.
Table 1.
Impact of S-glutathionylation on those proteins (categorized in clusters) reported sensitive to the post-translational modification.
| Protein | Reported impact of S- glutathionylation |
References |
|---|---|---|
| Enzymes with thiols in active center. | ||
| Carbonic anhydrase III | Inhibition | [135, 136] |
| Tyrosine hydrolase | Inhibition | [137] |
| α-Ketoglutarate dehydrogenase | Inhibition | [135] |
| Aldose reductase | Inhibition | [138] |
| Creatine kinase | Inhibition | [139, 140] |
| GAPDH | Inhibition | [141–143] |
| HIV-1 protease | Inhibition | [144] |
| Peroxiredoxin I | Protection | [145] |
| Peroxiredoxin VI | Reactivation | [55] |
| Inosine 5’-monophosphate dehydrogenase 2 |
Inhibition | [145] |
| Protein disulfide isomerase | Inhibition | [72] |
| Elonase 1α | Inhibition | [145] |
| Phosphoglycerate kinase | Inhibition | [145] |
| Aldolase | Inhibition | [146] |
| Phosphorylase kinase delta | Inhibition | [145] |
| 6-Phosphogluconolactonase | Inhibition | [145] |
| Triosephosphate isomerase | Inhibition | [145] |
| Adenylate kinase 2 | Inhibition | [145] |
| dUTP pyrophosphatase | Inhibition | [145] |
| Peptidylprolyl isomerase (cyclophilin A) | Inhibition | [145] |
| Cytochrome c oxidase | Inhibition | [145] |
| Ubiquitin-conjugating enzyme E2N | Inhibition | [145] |
| Thioredoxin 1 | Inhibition | [147] |
| Glutathione S-transferase P | Inhibition | [47] |
| Endothelial nitric oxide synthase (eNOS) | Inhibition | [64, 71] |
| Cytoskeletal proteins | ||
| Vimentin | Inhibition of filament formation | [145] |
| G-Actin | Inhibition of polymerization | [148, 149] |
| Tropomyosin | Inhibition | [146] |
| Transgelin, SM22 homolog calponin-like | Inhibition | [145] |
| Cofilin | Inhibition | [145] |
| Myosin | Inhibition | [145] |
| Profilin | Inhibition | [145] |
| Βeta-Tubulin | Inhibition | [146, 150] |
| Annexin II | Inhibition of binding | [151] |
| Spectrin | Inhibition | [152] |
| Signaling proteins. | ||
| Protein kinase A | Inhibition | [153, 154] |
| Protein kinase C | Inhibition | [153, 154] |
| ERK | Inhibition | [153, 154] |
| T cell p59fyn kinase | Activation | [155] |
| PTP1B | Inhibition | [38, 156] |
| MEKK1 | Inhibition | [157] |
| PTEN | Inhibition | [158] |
| Protein kinase G | Inhibition | [159] |
| c-Abl | Inhibition | [160] |
| p53 | Inhibition | [161] |
| Caspase 3 | Inhibition | [162] |
| Transcription factors. | ||
| c-Jun | Inhibition | [163–165] |
| NF-κB subunits 65 and 50 | Inhibition | [163–165] |
| IKK β-subunit | Inhibition | [166] |
| Pax-8 | Inhibition | [167] |
| OxyR | Inhibition | [168] |
| Ras proteins. | ||
| P21ras | Activation | [169, 170] |
| Heat shock proteins. | ||
| HSP60 | Inhibition | [145] |
| HSP70 | Inhibition | [145] |
| Ion channels, Ca2+pumps and Ca2+–binding proteins. | ||
| RyR1 | Activation | [171] |
| CFTR | Inhibition | [172] |
| SERCA | Activation | [173, 174] |
| S100 A1, S100 A4, S100 B | Activation | [175–177] |
| Energy metabolism, glycolysis. | ||
| Complex I | Inhibition | [178] |
| NADP+-dependent isocitrate dehydrogenase |
Inhibition | [179] |
| Cytochrome oxidase | Inhibition | [145] |
| ATPase | Inhibition | [38] |
| NADH ubiquinone reductase | Inhibition | [180] |
| Carbonic anhydrase III | Inhibition | [162] |
| Catechol-O-methyltransferase | Inhibition | [181] |
| Pyruvate dehydrogenase | Inhibition | [182] |
Partly because of the sensitivity of detection issues, there is still debate over whether S-glutathionylation is primarily a response to external stress or whether it has an important role in physiological processes in "unstressed" cells. Although this cannot yet be answered definitively, it is clear that cytoskeletal restructuring during cell growth is impacted by S-glutathionylation, particularly of actin. However, mitochondria produce significant ROS as byproducts of metabolism, and a number of mitochondrial proteins are quite susceptible to S-glutathionylation. It seems probable that maintenance or stabilization of proteins in a glutathionylated state may be a process that could have important structural/functional consequences. Since a number of proteins have GSH recognition motifs (e.g. sulfiredoxin, glutaredoxin, thioredoxin), perhaps they may be candidates for such a process. The importance of S-glutathionylation as a post-translational modification may best be exemplified by discussion of specific examples where the structure and function of the protein is influenced by the change.
Actin
The most readily detected S-glutathionylated protein is actin and growth factor stimulation of cells can produce extensive actin S-glutathionylation and alter the ratios of the soluble:polymerized protein. This may influence cellular architecture and membrane ruffling with consequential changes in a number of cytoskeletal functions, including intracellular trafficking. S-glutathionylation of actin influences cell adhesion and protein–protein interactions as well as cell–cell interactions [45] and the modified protein has a weaker affinity for tropomyosin [46]. S-glutathionylation of actin provides a primary example where the post-translational modification results from a physiological response consequential to cell growth - rather than a stress response to an exogenous insult.
GSTP
The role of GSTP in regulating the forward reaction of protein S-glutathionylation under oxidative and/or nitrosative stress has recently been described [47]. This function is based on the catalytic activity of enzyme and is influenced by auto-S-glutathionylation of GSTP itself on two critical residues, Cys47 and Cys101 (Figure 3), each of which can affect catalytic activity and binding to target proteins. Specific S-glutathionylation also causes GSTP oligomerization, a process with probable consequences to other components of cellular stress response. Within the GSTP monomer, S-glutathionylation causes a change in secondary structure by decreasing α-helical content (Figure 3) with resultant impact on tertiary and quaternary structure. GSTP has two tryptophan residues (Trp28 and Trp38). Trp38 is ~3.2 Å from the sulfhydryl of Cys47, where S-glutathionylation causes a quenching of tryptophan fluorescence [48] (Figure 3). In addition, the tryptophan fluorescence of S-glutathionylated GSTP has a shift of emission maximum (~4nm) to a shorter wavelength, suggesting a less polar environment around this residue. Thus, there are changes in GSTP tertiary and quaternary structure that imply that self- S-glutathionylation will alter its capacity to interact with any ligand binding partner proteins. One such example is the GSTP:JNK regulation of kinase signaling (4, 5). By immunoprecipitating JNK1/2 (in HEK293 cells) it was demonstrated that Cys47 and Cys101 in GSTP are critical for these protein-protein interactions [47]. S-glutathionylation of Cys47 and/or Cys101 interferes with complex formation with other proteins and with respect to the JNK:GSTP interaction, the result may be an attenuation of cellular signaling events mediated by JNK.
Figure 3.
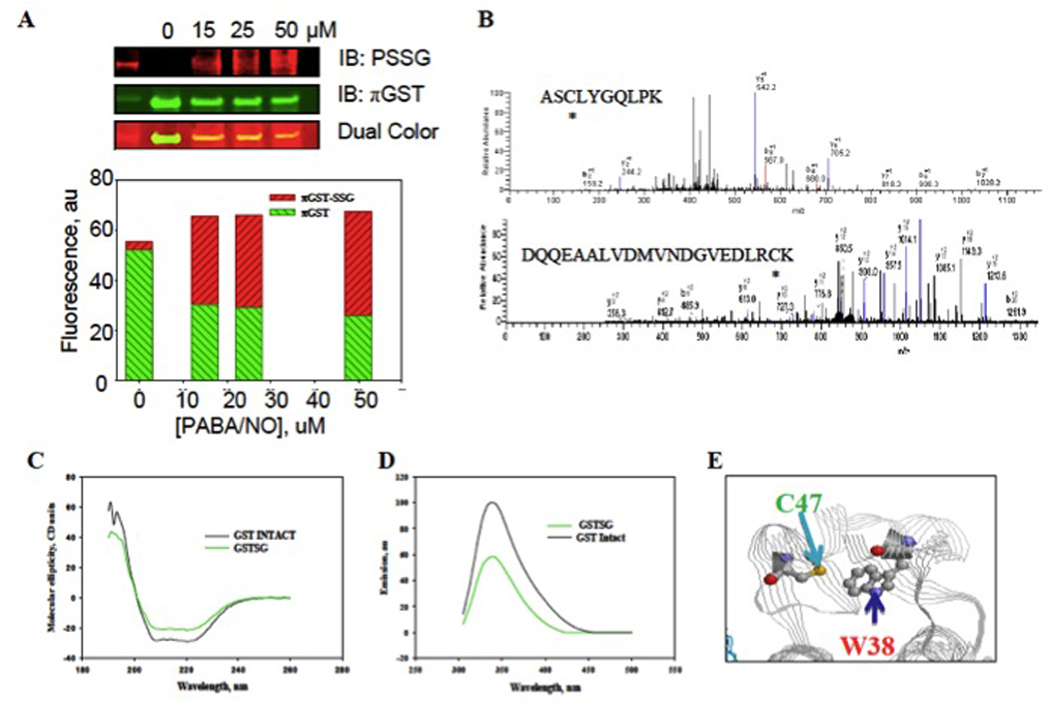
Auto-regulation of GSTP occurs through S-glutathionylation of the Cys47 and Cys101 residues. HEK293-WT cells were treated with 50 uM PABA/NO for 1h (A). The proteins were separated by non-reducing SDS-PAGE and S-glutathionylation (PSSG, red: commercially available fluorescently labeled antibodies to S-glutathionylated cysteine residues) and GSTP (green) evaluated by immunoblots. MALDI-MS analysis of GSTP treated with 50 uM PABA/NO showed that peptides containing Cys47 and Cys101 are S-glutathionylated (B). S-glutathionylation of Cys47 and Cys101 on GSTP alters structure. Spectroscopic analysis of native (black) and S-glutathionylated (green) GSTP in vitro was performed using CD (C) and tryptophanyl fluorescence (D). The relative position of GSTP’s Cys47 and Trp38 are depicted (E). Adapted from [47].
Peroxiredoxin VI
There are further indications that GSTP can store and deliver reducing equivalents (GS−) to regions of target proteins that may not be readily accessible (i.e. buried hydrophobic regions of, for example, a globular protein). Delivery may occur through heterodimerization of a GSH-loaded GSTP with the target protein, resulting in S-glutathionylation of specific “hidden” cysteine residues in the latter [49]. Considerations for this to occur will include the affinity of the binding interface of the GSTP monomer for that of the target protein, as well as the proximity of GS− (bound to the G-site of GSTP) to target the cysteine of the partner protein [50]. Thus, GSTP serves to overcome an accessibility barrier for delivery of the hydrophilic GSH (GS−) into a hydrophobic domain of the protein. This S-glutathionylation would result in a redox-mediated modification of the structure/function of the target protein.
The capacity for homodimeric GSTP (or in some cases GSTM) to dissociate into monomers and to form heterodimers with other monomeric proteins is crucial for their capacity to deliver reducing equivalents [51]. The cytosolic GSTs are catalytically active as dimers, with the dimer interface providing a non-catalytic site for ligand binding. Some studies indicate that mammalian GSTP and GSTM exist as monomers when they interact with ASK1, JNK, or peroxiredoxin VI (PrdxVI) [52–55]. It has been reported that heterodimers can be formed between GSTM and GSTP proteins in vitro without the need for denaturants, an observation that might reflect some promiscuity in the subunit dimerization of these two enzymes [51]. In addition, the monomers of cytosolic GST isozymes have also been demonstrated in non-mammalian species [56]. The majority of the published structural data provides convincing evidence that specificity of its C-terminus structure can facilitate dissociation of GSTP homodimers into monomers. On the other hand, the thioredoxin fold in the N-terminus domain of GSTP can facilitate heterodimerization of its monomers with other (especially thioredoxin fold containing) proteins.
There is a specific example where GSTP-mediated delivery of reducing equivalents (GS−) occurs in the globular domain of PrdxVI. GSH-loaded GSTP activates PrdxVI through heterodimerization, subsequent to S-glutathionylation of its catalytic Cys47 residue ([57]; Figure 4). Chromatographic purification and N-terminal sequencing showed the presence of equimolar amounts of the two proteins in this complex [57] and a schematic representation of the heterodimer is shown in Figure 4. Proof that GSH loading in GSTP is critical for the formation of this complex was provided by using mutants of the catalytically active tyrosine residue in GSTP (Y7F), which compromises GSH binding [47]. The Y7F mutant does not form a complex with PrdxVI. The peroxidase activity (towards both H2O2 and phospholipid hydroperoxide) of PrdxVI in the PrdxVI-GSTP complex was high indicating the importance of both delivery of the reducing equivalent (GS−) and the subsequent S-glutathionylating reduction/activation step for PrdxVI. Rapid (minutes) S-glutathionylation of PrdxVI is detectable by immunostaining [50]. Two regions of GSTP: 41–85 and 115–124 are critical for the protein:protein (GSTP-PrdxVI) interactions [49] and perhaps unsurprisingly these domains are found in the N-terminus, which includes the catalytic Cys47. Those component domains responsible for the surface interactions are shown in Figure 5. As this field advances, it will be interesting to understand how commonly GSTP provides reducing equivalents to acceptor cysteines in biologically inaccessible sites. As outlined in Table 1, families or clusters of proteins that are potential targets of the GSTP-mediated delivery of reducing equivalents may be quite limited. As a consequence, the general relevance of this process in human disease pathologies may be significant.
Figure 4.
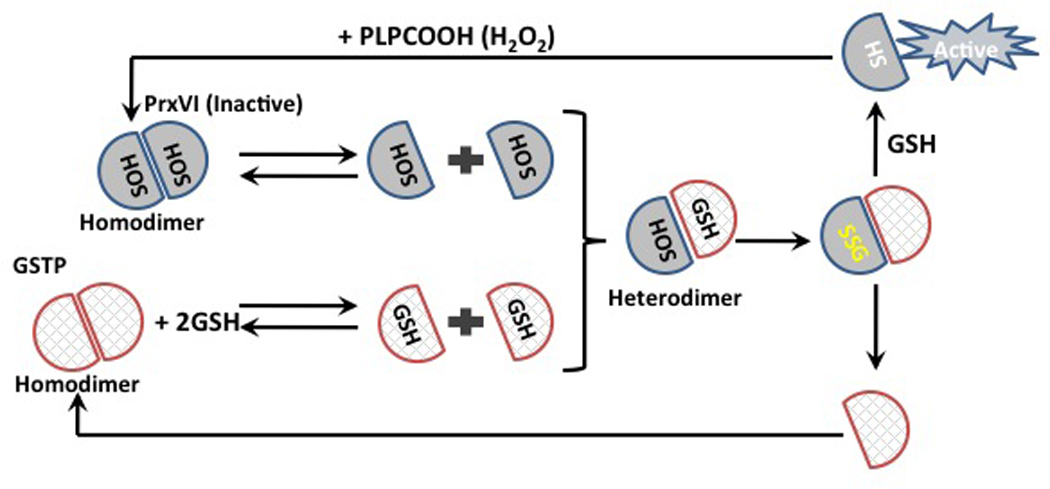
A role for GSTP in the catalytic cycle of PrdxVI. The catalytic Cys47 of PrdxVI (grey hemisphere) becomes oxidized to a sulfenic acid (SOH) by peroxide and forms “head-to-tail” homodimers. Under normal physiological conditions PrdxVI exists in equilibrium between homodimers and monomers as well as GSH-loaded GSTP (cross hatched hemisphere). Heterodimerization of PrdxVI with the GSH-loaded GSTP results in S-glutathionylation of catalytic Cys47 (SSG). This alters PrdxVI structure resulting in dissociation of the hetero-dimer and consequential opening up of the milieu of the protein to cytosolic GSH and subsequent spontaneous reduction of Cys47 and reactivation of PrdxVI.
Figure 5.
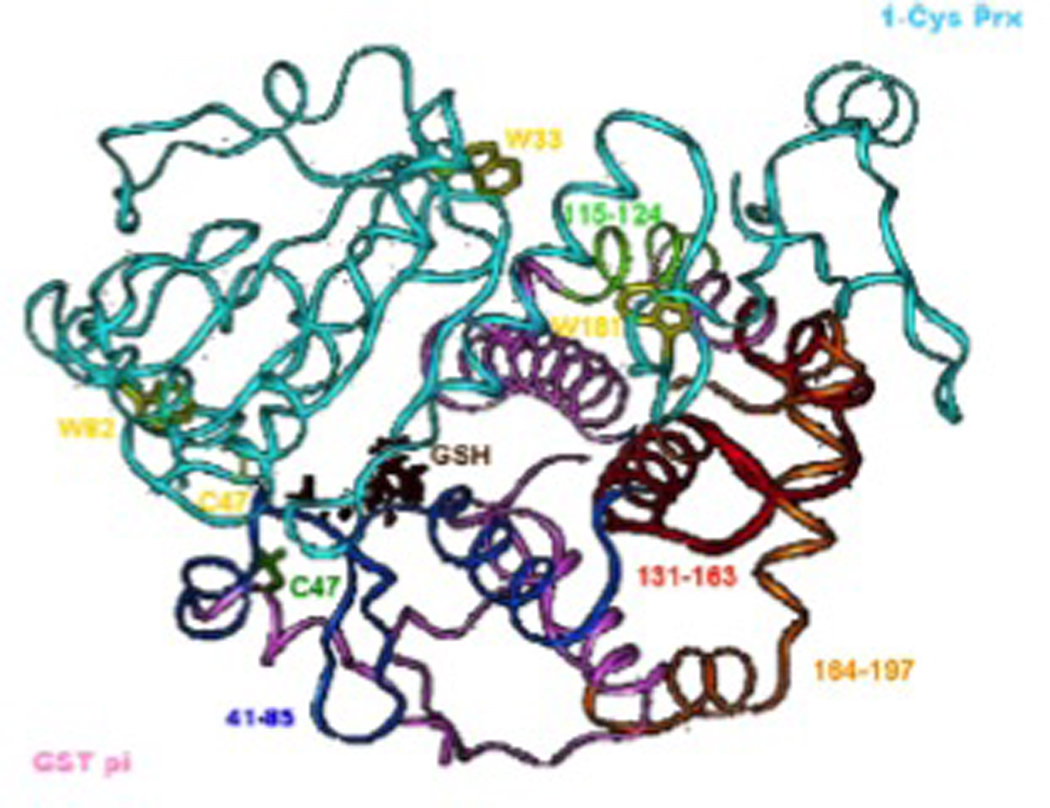
Model of the heterodimer of GSTP and 1-Cys Prdx (PrdxVI) showing relative locations of the four GSTP peptide fragments that bind 1-Cys Prdx (PrdxVI) and that inhibit complex formation. Ribbon representation of 1-Cys Prdx (PrdxVI PDB 1PRDX) complexed with GSTP (PDB 19GS). The backbone of subunit B of 1-Cys Prdx (PrdxVI) is cyan with tryptophan 33, 82 and 181 highlighted in yellow; the backbone of subunit A of GSTP is pink with peptide 131–1163 highlighted in red; peptide 164–197 is highlighted in orange. GSH is shown in brown, Cys47 of GSTP is green and Cys47of 1-Cys Prdx (PrdxVI) is in yellow. Taken from [49].
Nitric Oxide Synthase
Two recent reports implicate S-glutathionylation involvement in the control of nitric oxide mediated signaling events. The active form of eNOS is a homodimer with tetrahedral zinc ions coordinated to two pairs of symmetrical cysteines (Cys94 and Cys99 in each monomer [58]). These cysteine residues are in a basic environment and have a low pK and may be subject to S-glutathionylation and indeed, nitrosylation of some of these cysteines results in dissociation of homodimers into inactive monomers [59]. eNOS is palmitoylated and therefore attached to an inner leaf of the plasma membrane. Thus, its activation results in an NO burst close to the plasma membrane. As well as an NADPH oxidase, there is a chloride ion channel-3 (CIC-3) [60] in the plasma membrane. Ca2+ fluxes could activate NADPH oxidase [61] and superoxideradical generated outside the cells could influx through CIC-3 channels [62]. When spatially close, the eNOS and CIC-3 channels may generate peroxynitrite (ONOO−) that together with high levels of GSH can induce eNOS S-glutathionylation. Treatment with the NO releasing prodrug, PABA/NO causes changes in Ca2+/NO homeostasis that start as an extracellular NO-mediated surface protein-thiol modification [63]. Intracellularly the drug generates NO and a stable nitro-aromatic GSH-conjugate (PABA-SG), the latter of which inhibits SERCA initiating an intracellular Ca2+ increase, activating calmodulin and consequently eNOS with the resultant NO burst [64]. It seems likely that PABA/NO causes intracellular NO levels to rise above a certain threshold through eNOS activation with a subsequent conversion of S-nitrosylated to S-glutathionylated cysteine(s). There is evidence to suggest that two distinct pools of S-nitrosylated proteins can exist, one GSH stable and another GSH labile and subject to rapid conversion to a S-glutathionylated product [65, 66]. Two mechanisms of NO-mediated protein S-glutathionylation have been considered. The first through GSNO (activated glutathione thiol) formation and subsequent reaction with protein-thiol [67]; the second through an intermediate protein-thiol nitrosylation (activated protein-thiol: analogue of sulfenic acid) and its subsequent reaction with the most abundant intracellular redox buffer, GSH [68]. Although the exact mechanism of eNOS modification is unknown, in vivo experiments have shown that eNOS activation in aortas and iNOS transgenic expression in mouse heart both result in NO-induced protein S-glutathionylation [69]. Overall, indications are that PABA/NO-mediated eNOS activation results in its S-glutathionylation in HL60 and HDMVE cells [64]. This dynamic modification may serve to physiologically down regulate eNOS by NO under normal conditions [70]. Conversely, eNOS deglutathionylation may cause its up-regulation, maintaining physiological NO levels. Under normal physiological conditions the NO increase might be controlled by S-nitrosylation/glutathionylation of eNOS as an immediate response or by similar modification/activation of SERCA in steady-state regulation. Alternatively, in tumor cells with high levels of GSTP expression, the rate of PABA/NO-mediated NO increase could be substantially faster [38], outcompeting eNOS down-regulation through S-nitrosylation/glutathionylation and this could contribute to the cytotoxicity of the drug. Recently, Chen et al [71] showed that S-glutathionylation of eNOS reversibly decreases NOS activity with an increase in superoxide generation primarily from its reductase domain, in which two highly conserved cysteine residues (689 and 908) are identified as sites of S-glutathionylation and found to be critical for redox-regulation of eNOS function. eNOS S-glutathionylation in endothelial cells caused loss of NO and gain of superoxide generation and was associated with impaired endothelium-dependent vasodilation. In hypertensive vessels, eNOS S-glutathionylation was enhanced with impaired endothelium-dependent vasodilation rescued by thiol-specific reducing agents, which reversed the S-glutathionylation. They concluded that S-glutathionylation of eNOS provides redox regulation of cellular signaling, endothelial function and vascular tone.
Protein Disulfide Isomerase
Protein disulfide isomerase (PDI) is one of the most abundant ER proteins and maintains a sentinel function in organizing accurate protein folding. Release of NO from PABA/NO caused S-glutathionylation of PDI in tumor cells [72]. Drug treatment leads to translational attenuation as measured by the phosphorylation and activation of the ER trans-membrane kinase, PERK, and its downstream effector eIF2. Cleavage of the transcription factor, XBP-1 and transcriptional activation of the ER resident proteins, BiP, PDI, GRP94 and ERO1 (5–10 fold induction) occur concomitantly with PDI S-glutathionylation following drug treatment. Mass spectrometry identified a single cysteine residue within each of the catalytic sites of PDI with a mass increase [+305.3 Da] consistent with S-glutathionylation. Circular dichroism confirmed that S-glutathionylation of PDI alters the alpha-helix content of PDI, a process concurrent with inhibition of its isomerase activity. These results would seem to be consistent with the conclusion that S-glutathionylation of PDI is an upstream signaling event in the unfolded protein response (UPR), a process linked with the cytotoxicity of PABA/NO and presumably other drugs like it. As a corollary to the PDI data, general accumulation of S-glutathionylated proteins appears to be emerging as a key factor in human disease pathologies. There are now associations between protein S-glutathionylation and ER stress in human diseases such as ischemia/cardiovascular disease and Friedreich’s ataxia, Alzheimers disease, type 2 diabetes, cystic fibrosis, cataracts and sickle cell anemia (for discussion see [35]). Since the ER has a GSSG:GSH ratio that is ten-fold more oxidized than the cytosol (10:1 vs100:1), the more oxidized environment of this localized organelle may be relevant to this observation.
GSTP, Ligand binding and signaling
Stress kinases and GSTP
Jun-terminal kinases (JNK’s) comprise a family of stress kinases transiently activated in response to oxidative or nitrosative stress, heat or osmotic shock or inflammatory cytokines [73]. In concert with DNA damage, JNK activation may be mediated by a number of potential upstream signaling components, including cdc42, p21PAK, ASK1, MLK, MEKK1, SEK1/MKK4, MKK7 [74]. Different forms of stress mediate JNK activation via various cellular pathways with resultant JNK mediated phosphorylation of the transcription factors c-Jun, ATF2, p53 and ELK-1 and stimulation of downstream events that directly contribute to the stress response through changes in the cell cycle, DNA repair or apoptosis [73, 75]. In cells under normal growth conditions, basal activity of JNK is necessarily maintained at a low level. However activity can be enhanced in response to growth factors [76] and hence should also be observed in cells proliferating under normal growth conditions. Mouse embryo fibroblast (MEF) cells null for GSTP contain higher levels of JNK activity than their wild type counterparts [28]. Regulation of JNK activity in response to stress is independent of transcript and protein levels of the kinase, but several studies implicated the existence of endogenous JNK inhibitors in normal growing cells. Earlier studies attempting to understand those factors/mechanisms responsible for the regulation of JNK before and immediately after stress identified GSTP as an endogenous negative regulatory switch [54].
Figure 6 illustrates in cartoon form how in non-stressed cells, low JNK activity is maintained as a consequence of sequestration of the kinase in a multi-protein complex which includes GSTP1-JNK [54]. In cells exposed to oxidative stress (or drug treatment), GSTP1 dissociates from the complex, accumulating as GSTP oligomers [54], with resultant activation of released JNK impacting subsequent downstream events, which contingent upon cell or tissue type can include such divergent events as proliferation or apoptosis [77]. It seems reasonable to assume that JNK-dependent stress-induced apoptosis might be suppressed during tumor development. In this sense, high levels of expression of GSTP1 may serve such a purpose by enhancing sequestration of JNK in an inactive form. Such a mechanism could explain why increased GSTP levels occur in resistant tumor cells even when the selecting drug is not a substrate for GSH conjugation. They could reflect the increased functional role of GSTP in attempting to maintain JNK activity at more basal levels. Within the GST super family, ligand binding can be promiscuous and functional redundancies are not uncommon. The homology between GST A and P family members helps to explain why GSTA1 is also capable of suppressing JNK signaling caused by inflammatory cytokines or oxidative stress [78] - by a mechanism similar to that shown for the GSTP.
Figure 6.
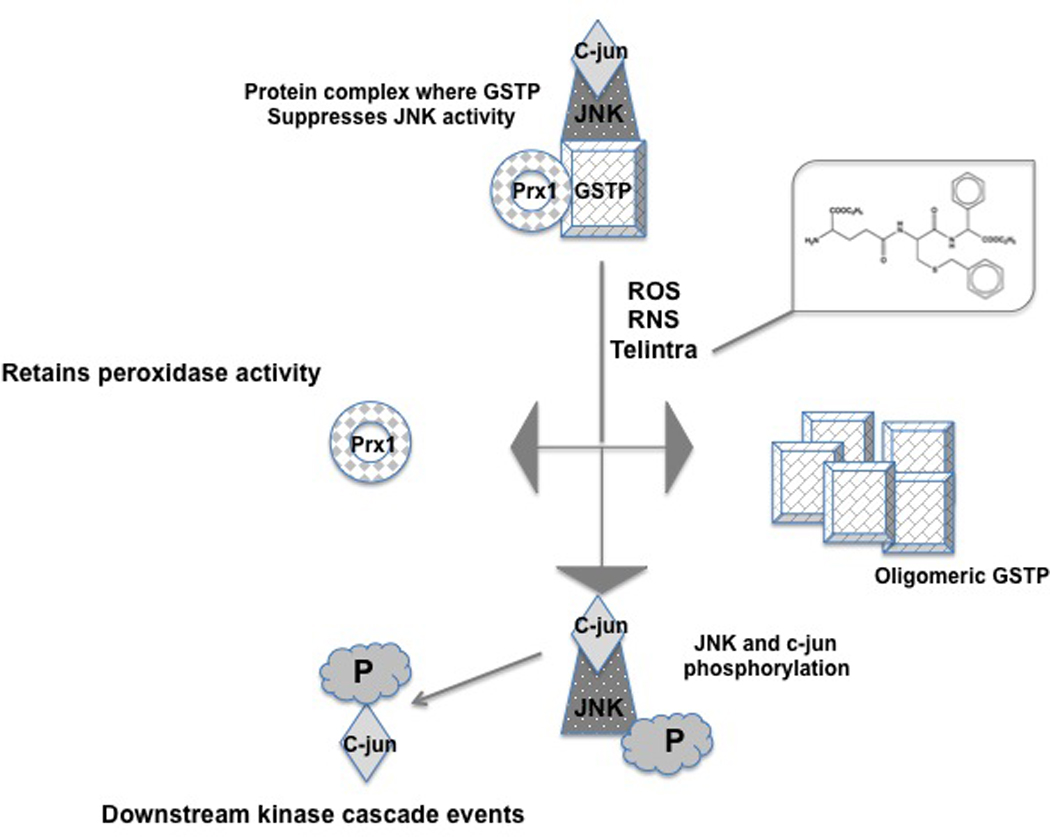
Cartoon model of JNK-GSTP-Prdx1 interactions initially maintaining JNK in an inactive state and activation of downstream signaling cascades through exposure to ROS/RNS or the GSTP inhibitor Telintra. GSTP either as a monomer or homodimer can become loaded with GSH and act as an S-glutathionylation donor. By altering the reduction status of key cysteine residues within the GSTP structure, the complex dissociates with consequent activation of first JNK, and then c-jun by phosphorylation. Prdx1, part of this complex as a mono(multi)mer maintains its peroxidase activity after release.
Peroxiredoxins
Peroxiredoxin1 (Prdx1) is also a binding partner for GSTP and associates with the GSTP/JNK complex (Figure 6). The GSTP JNK heterodimer forms either because of, or as a consequence of the presence of GSH bound to the “G-site” of GSTP [79]. However, subsequent S-glutathionylation of JNK could occur as an outcome of formation of this complex [35]. As part of a larger multi-protein complex, Prdx1 binding to the JNK-GSTP heterodimer occurs through GSTP and is mediated, at least in part, by Prdx1 catalytic Cys52, independently from the redox status of this protein [79]. Thus, in forming this complex the affinity of GSTP for the thioredoxin-fold of Prdx1 is independent of its catalytic reduction and/or reactivation. There will be significant merit in interpreting further studies that are designed to interrogate the stability and functional implications of this protein complex, particularly as on and off rates may be important in determining the overall rates of response to stress conditions or drug treatment. In this regard, increased expression of Prdx1 (and PrdxII or PrdxIV) has been associated with resistance to irradiation and a suppression of ionizing radiation induced JNK activation and subsequent apoptosis [80, 81]. Mutating the Cys52 residue attenuated the peroxidase activity of Prdx1 and reduced the JNK activation; nevertheless, both proteins still co-immunoprecipitated with the GSTP/JNK complex, implying that Prdx1 may have a role in suppressing apoptosis through inhibition of JNK activation.
Other Signaling Events
GSTP has also been implicated in regulating tumor necrosis factor-alpha (TNF-alpha) signaling primarily through a physical association with tumor necrosis factor receptor-associated factor 2 (TRAF2) [82]. High GSTP levels inhibit: (i) TRAF2-induced activation of both JNK and p38, but not NFkB, (ii) attenuated TRAF2-enhanced apoptosis signal regulating kinase 1 (ASK1) auto-phosphorylation and (iii) inhibited TRAF2-ASK1-induced apoptosis by suppressing the interaction of these two proteins. Low levels of GSTP increased TNF-alpha-dependent TRAF2-ASK1 association, activating both ASK1 and JNK. Compared to wild type, GSTP engineered without the TRAF binding motif had a reduced binding to TRAF2 with subsequent lower impact on TRAF2-ASK1 signaling. The basic catalytic activity of GSTP is unaffected by protein binding suggesting that the kinase effects are mediated at sites distant to those regions of GSTP that are involved in GSH or substrate binding.
An example of plausible functional redundancy within the GST family is provided by the observations that GSTM1 binds to, and inhibits the activity of Apoptosis Signaling Kinase1 (ASK1) [52]. Further evidence of redundancy of this activity is the fact that thioredoxin can mediate the same suppression [83]. Mechanistically similar to GSTP:JNK, the interaction of the GSTM1:ASK1 complex is dissociated under oxidative stress or heat shock, oligomerizing GSTM and leading to activation of ASK1 [53]. Since ASK1 is a MAP kinase kinase kinase that activates the JNK and p38 pathways, this disassociation can lead to cytokine- and stress-induced apoptosis [84]. Altered expression of GSTM1 has been linked with impaired clinical response to some tumor types. Although not as prevalent as s with GSTP, there are some reports of increased GSTM1 expression in drug resistance and once again, these may not be linked to detoxification, but may be supplanted and/or augmented by its role in kinase regulation [85].
A recent study has extended those regulatory role(s) ascribable to GSTP through protein:protein interactions. In this instance, physical interactions between the HPV-16 E7 viral factor and GSTP1 can enact survival capabilities of host cells. The authors [86] propose a mechanism by which GSTP1 is activated via physical interaction with HPV-16 E7. HPV-16 E7 decreases the levels of oxidized GSTP1, thereby increasing levels of the reduced form, interfering with JNK signaling. Concomitant increases in GSH in HPV-16 E7-infected cells can then also increase the concentration of reduced GSTP1. The model also predicts that HPV-16 E7 will occupy the space between Cys47 and Cys101 of GSTP1 and prevent intra- and inter-molecular disulfide formation. As a consequence, HPV-16 E7 binding might protect GSTP1 against inactivation via oxidative attacks at Cys47 and Cys101. Furthermore, because these two different complexes use essentially overlapping regions of the enzyme surface the binding of HPV-16 E7 to GSTP1 and GSTP1 homodimerization should be mutually exclusive events. The conformation of this complex has characteristics that resemble the pattern of intermolecular contacts in the GSTP1 homodimer with the prediction that HPV-16 E7 functions to establish a subset of GSTP1 molecules inaccessible to oxidative attack, thereby creating a reservoir of reduced monomeric GSTP1.
GSTP polymorphisms
GSTP and Cancer Drug response
Apart from aberrant protein expression level, mounting evidence has directed attention towards to the association of GSTP1 polymorphisms with a variety of clinical outcomes in cancer. The genetic polymorphisms in the GSTP1 gene arise from nucleotide transitions that change codon 105 from Ile to Val and codon 114 from Ala to Val, thus generate four GSTP1 alleles: wild-type GSTP1*A (Ile105/Ala114), GSTP1*B (Val105/Ala114), GSTP1*C (Val105/Val114) and GSTP1*D (Ile105/Val114) [87, 88]. Structural analyses of variant GSTP1 proteins reveal that Ile 105→Val105 and Ala114→Val114 substitutions, without affecting the glutathione-binding affinity, cause a steric change at the substrate-binding site of the enzyme [87, 89]. As a consequence of this, the enzymatic activities of GSTP1B and GSTP1C are significantly affected, depending on the various substrates used in the assay. For 1-chloro-2,4-dinitrobenzene (CDNB), chlorambucil and thiotepa, GSTP1B and GSTP1C exhibit significantly lower activity than GSTP1A, with GSTP1C being the least effective [87, 88, 90, 91]. However, in glutathione conjugation of cis-platin, carboplatin, 4-hydroxyifosfamide and diol epoxides of polycyclic aromatic hydrocarbons, the opposite is true with GSTP1C being the most protective against these compounds [92–95]. The altered conformation of the substrate-binding site(s) may contribute to final substrate specificities. The hydrophobicity and size of residue 114 could serve as an important determinant of the substrate specificity of each of the GSTP1 isozymes [95]. On the other hand, since GSTP1D, bearing Ile105 and Val114, has enzyme activity toward CDNB comparable to GSTP1A, Val105 may circumvent the influence of Val114 [88]. Because different GSTP1 proteins differ in their ability to catalyze specific detoxification reactions, the polymorphisms in GSTP1 will likely impact response to therapy. Examples of clinical drugs include, cis-platin, carboplatin, chlorambucil, ethacrynic acid, melphalan, nitrogen mustard, phosphoramide mustard. Thioether conjugates of these drugs with GSH can be catalyzed by GST’s, however the catalytic constants for these reactions are generally not impressive. At least one exception exists, GSTP1*A has a role in the acquisition of cis-platin resistance reportedly through enhancing the formation of platinum–glutathione conjugates [92]. Individuals with the GSTP1*B allele (a single nucleotide (A:G) substitution at position 313 produces an isoleucine to valine conversion) substantially reduced catalytic activity [92, 93] correlates with a diminished potential to detoxify the drug. Additionally, homozygocity for GSTP1*B is linked with a diminished capacity to detoxify a number of platinum based anticancer drugs [93]. GSTP1*C is an allelic variant predominant in malignant glioma cells and differs from other GSTP1 variants by two transitions resulting in Ile105Val and Ala114Val [96, 97].
GSTP and Disease etiology
The allele frequencies for GSTP1*A, *B and *C in Caucasian populations are 0.685, 0.262 and 0.0687, respectively [98]. The wild-type genotype GSTP1*A has been correlated with the development and progression of Hodgkin's and non-Hodgkin’s lymphoma [99–101]. Moreover, the GSTP1 Val105 polymorphism is generally associated with higher susceptibility to a variety of malignancies (see Table 2). However, multiple factors such as ethnicity, gender, age and general statistical significance of population size serve to complicate the analyses. For example, a meta-analysis of 30 published case-control studies including 15,901 cases and 18,757 controls showed that GSTP1 Val105 polymorphism was not associated with breast cancer susceptibility. However, in a sub-group analysis by ethnicity, a significant association within Asian populations was found [102]. From the limited reports on GSTP1 Val114 polymorphism in cancer risk, it is Val114, not Val105 that apparently contributes to esophageal cancer susceptibility [103]. This may reflect a geographic susceptibility difference to environmental carcinogen exposure as a consequence of different detoxification profiles for GSTP1 isozymes. Without a clear understanding of why, GSTP1*C has been correlated with lower incidence of breast cancer [102].
Table 2.
GSTP1 polymorphisms in cancer susceptibility and therapy
| Allele | ↑ Cancer risk | Variation & survival following therapy | Refs |
|---|---|---|---|
| I105/A114 (GSTP1*A) |
Hodgkin’s Lymphoma |
Val 105 ------------5-year survival↑ | [100, 183] |
| Val105 (GSTP1*B and GSTP1*C) |
Breast cancer | Val105 ------------OS↑ | [102, 184] |
| CML | Val105 ------------ Poor or minor response | [185] | |
| Endometrial cancer | No report | [186] | |
| HCC | Val105 ------------OS↓ | [187] | |
| Pancreatic cancer | Val105/Val114 ------------OS↑ | [188, 189] |
|
| Val114 (GSTP1*C and GSTP1*D) |
Esophageal cancer | Val105, Val11 4 ------------OS↓, Recurrence rate↑ |
[190– 193] |
| No definitive correlation with GSTP1 polymorphisms |
Colorectal cancer | Val105 ------------ Controversial | [104, 194, 195] |
| Val114 ------------Neurotoxicity↑ | |||
| Gastric cancer | Val105 ------------OS↑, Neurotoxicity↓ | [103, 196] |
|
| Glioma | Ile105/Ala114 -----------OS↑, Toxicity↑ | [96, 97, 107] |
|
| Val105/Val114 ------------OS↑, Toxicity↓ | |||
| Lung cancer | Val114 ------------OS↑ | [197– 199] |
|
| Multiple Myeloma | Val105 ------------OS↑ | [200, 201] |
|
| Ovarian cancer | Val105 ------------OS↑ | [104, 202] |
OS, overall survival; CML, chronic myeloid leukemia; HCC, hepatocellular carcinoma.
In most, but not all, the cases listed in Table 2, the GSTP1 Val105 polymorphism is associated with longer overall survival in patients with different malignancies who are treated with alkylating agents and/or platinum compounds [104]. However, the correlations for colorectal cancer chemotherapy remain imperfect. One group reported that the GSTP1 Val105 polymorphism was associated in a dose-dependent fashion with increased survival of patients with advanced colorectal cancer receiving 5-FU/oxaliplatin chemotherapy [105], whereas another showed the opposite [106]. These conflicting results were considered by the authors to be related to the differences in ethnicity and age of the patients, treatment, and follow-up periods included in the two studies. Another controversy is from primary malignant glioma, with GSTP1*A and GSTP1*C conferring the survival advantage in separate studies [96, 107]. Since GSTP1A and GSTP1C show the most difference in substrate specificity among GSTP1 isozymes, it was speculated that they react with different components of the chemotherapy regimens, which might contribute to the improved survival of patients with brain tumors.
These limited examples under represent an extensive literature of correlative associations between GST expression patterns and cancer epidemiology. In more recent times, there have been more focused efforts to correlate polymorphic expression patterns of GSTP with disease occurrence. While such clinical and epidemiological correlations require considerably more proof, further information on what is presently known can be found in Table 2 and Ref. [104]. The complexity of multivariant analyses of the large number of factors that contribute to accurate prognosis and clinical outcome will probably continue to confound epidemiological analyses and complicate our understanding of the large number of existing literature references in this area.
GSTP activated prodrugs
Over the last two decades, GSTP has been a targeted platform for drug discovery and development efforts, particularly in designing prodrugs. TLK286 or Telcyta [γ-glutamyl-α-amino-β-(2-ethyl-N,N,N0 ,N0 –tetrakis (2- chloroethyl)phosphorodiamidate)-sulfonyl-propionyl-(R)- (−) phenylglycine] is the most advanced lead clinical candidate from a group of rationally designed glutathione analogues designed to exploit high GSTP levels in solid tumors and drug-resistant cells [108]. Selective targeting of susceptible tumor phenotypes is a strategy that results in release of more active drug in malignant cells compared with normal tissue, thereby enhancing therapeutic index. Published preclinical studies have confirmed the mechanism of action of this drug [109]. In a series of Phase II clinical trials, TLK-286 was initially shown to have clinical activity and a favorable toxicity profile as a single agent in the salvage setting in ovarian, non-small cell lung, breast and colorectal cancers. More recently, Phase III trials in NSCLC have not provided definitive increased response rates and as a consequence, further clinical testing is in progress. Although no pharmacogenetic biomarkers were included in the trial design, unveiling any correlations between GSTP1 polymorphisms and response rates could help to establish a GST platform for targeted drug design and individualized therapy. There are a number of candidate GST targeted drugs at various stages of preclinical development (including PABA/NO) and these are reviewed more comprehensively elsewhere [85, 110]. Consideration of genetic polymorphisms of GSTP could provide a more rational basis for their clinical testing and protocol design might benefit from inclusion of such correlative analyses.
Redox, GSTP and bone marrow
While the aberrant redox potential of tumor cells is well established, it is also apparent that many normal tissues are sensitive to changes in physiological redox homeostasis. In particular, the maintenance of normal hematopoiesis is quite dependent upon thiol regulation. Although the importance of thiols and redox in the regulation of bone marrow cell proliferation has been appreciated for some 50 years [111], it is only recently that some mechanistic explanations for the linkage have been forthcoming. Long-term, self-renewing hematopoietic stem cells (HSC) have low levels of intracellular ROS and mice deficient in ROS regulating genes have HSC that retain neither quiescence nor self-renewal capacities [112]. It appears that two distinct populations of HSC can be identified based upon their baseline ROS. While both populations have similar cell surface markers, ROSlow HSC retain self-renewal capabilities in serial transplantations, whereas this is diminished in ROShigh HSC. N-acetyl cysteine (NAC) mediated thiol antioxidant treatment can rescue HSC self-renewal [113] indicating that redox manipulation of sub-populations may influence migration and differentiation potentials. Moreover, AIDS patients have decreased GSH levels in their blood cells [114] and NAC has also been shown to be effective in the management of patients with HIV, influencing Th1 versus Th2 cytokine response patterns [115].
Telintra
The peptidomimetic inhibitor of GSTP, TLK 199 [γ-glutamyl-S-(benzyl)-cysteinyl-R-(−) phenyl glycine diethyl ester], now named Telintra exemplifies the principles of serendipity in drug discovery. While early preclinical testing focused on overcoming GSTP-associated drug resistance, further rodent studies revealed the drug also caused an increase in circulating blood cells of all lineages [28]. Telintra increased peripheral white blood cell number in wild type mice when compared to GSTP-deficient mice. Genetic ablation (i.e. GSTP-null animals) produced a phenotype characterized by an increase in myeloid cell differentiation and proliferation, evidenced by elevated numbers of circulating leukocytes. This could be interpreted as a consequence of increased numbers of bone marrow progenitor cells that (re)populate circulating mature blood cells [116]. The concordance of genetic and pharmacological ablation of GSTP can be understood in a mechanistic manner. For example, these observations are consistent with the capacity of Telintra to dissociate GSTP from JNK, allowing kinase phosphorylation, activation and downstream myeloproliferative effects (Figures 6 and 7). Further downstream, the drug’s myeloproliferative properties have been associated with activation of STAT proteins in GSTP-deficient mice [116]. Isozymes of the GSTA family are generally expressed at low levels in marrow, but their potential to suppress stress-induced activation of JNK signaling serves to emphasize GST promiscuity [78]. Moreover, GSTP may also directly influence the S-glutathionylation of a number of proteins involved in myeloproliferative events (such as JNK and SHP-1 and SHP-2).
Figure 7.
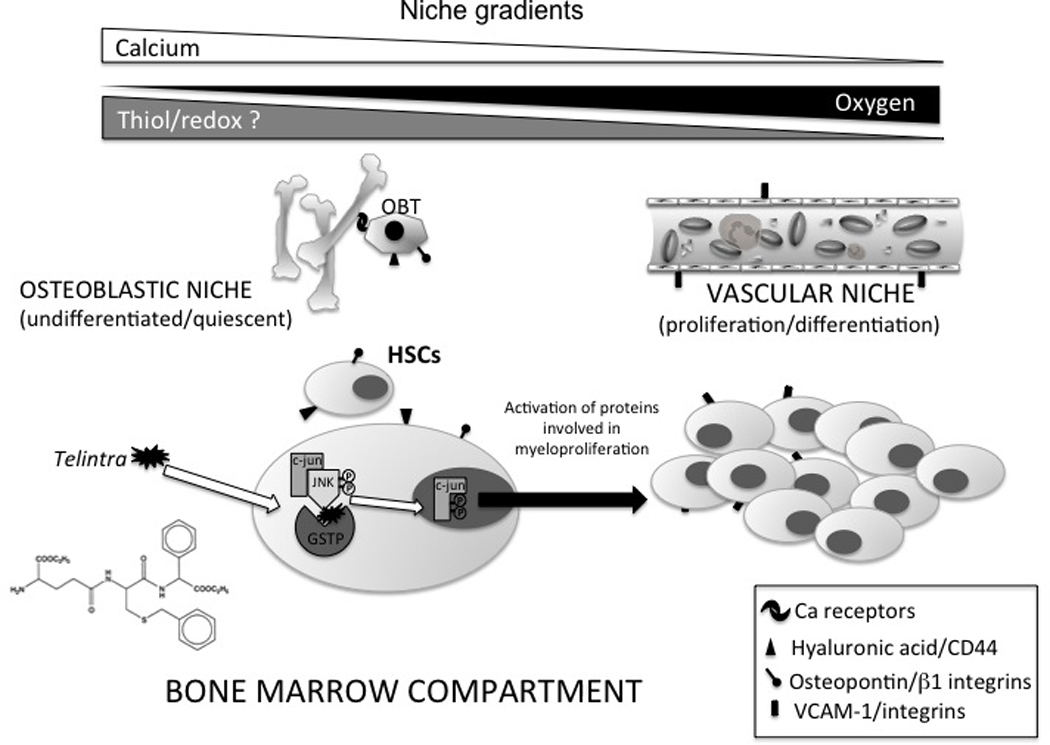
Representative model of the effects of Telintra, a peptidomimetic inhibitor of GSTP on hematopoietic stem cells in the bone marrow microenvironment. Hematopoietic stem cells (HSCs) are located either at the osteoblastic (with the osteoblasts (OBT)) or the vascular niche. HSCs in the osteoblastic niche maintain self-renewal and quiescent capabilities while temporal expression of adhesive molecules cytokines or chemokines can influence migration to the vascular niche. Drug treatment may effect proliferation through kinase pathways and influence such migration. Movement from one niche to another may involve some combination of calcium, oxygen and plausibly thiol/redox gradients. See text for additional details.
Inhibition of GSTP with Telintra causes changes in the bone marrow compartment that are consistent with altered redox status and like NAC, Telintra is in clinical testing. There are presently ongoing Phase 2 clinical trials for myelodysplastic syndrome (MDS) (online company reference: http://www.telik.com/pr/2010/pr_2010_0608.html), a stem cell disorder characterized by ineffective blood cell production and an increased risk for transformation to acute leukemia. Telintra treated patients with low to intermediate-1 risk MDS demonstrated multilineage hematologic improvement including decreased requirements for red blood cell, platelet, and growth factor support. Additional clinical trials are focusing on the use of Telintra in the treatment of chronic idiopathic neutropenia and additional blood disorders. The potential for FDA approval and registration of this drug awaits further clinical examination, but does serve to emphasize the impact of GSTP and redox in regulating myeloproliferative pathways.
Redox as Part of the Marrow Environment
All differentiated hematopoietic cells traverse to the peripheral blood from distinct microenvironments where HSC, hematopoetic progenitor cells (HPC) and mature plasma cells occupy their own niches (Figure 7). A variety of local factors can maintain and influence HSC number and destiny. Committed progenitors tend to localize to the bone marrow center [117]. Approximately 75% of HSC are not actively cycling at any given time [118] and these quiescent cells may reside at the osteoblastic niche. Conversely, HSC in the vascular niche favors proliferation and differentiation [117, 119–122] (Figure 7). Each population can be defined by the expression of adhesive molecules, cytokines and chemokines (such as CXCR4 or CXCL12; [123, 124]). Osteoclast and osteoblast-mediated bone remodeling results in a Ca2+ gradient in the endosteum, enabling calcium-sensitive HSCs to sense and migrate appropriately [125]. Although oxygen levels are higher closer to the vascular niche, the bone marrow is relatively hypoxic (1% to 2% O2) [126]. The hypoxic osteoblastic environment encourages HSC quiescence and movement along the oxygen gradient to the vascular niche promotes HSC differentiation [121]. While both oxygen and calcium gradients are involved in this process, it seems probable that a redox gradient may also influence HSC migration and differentiation. Older mice accumulate HSC more distantly in the endosteum and have increased levels of endogenous DNA damage in their HSC [127, 128]. This correlates with increasing numbers, but decreasing function of aged HSC [129] and perhaps may be explained by accumulation of oxidative stress associated with the aging process.
There are other links between redox homeostasis and bone marrow function. For example, Nrf2 is a redox activated transcription factor that regulates antioxidant Phase I and Phase II enzymes [130, 131] and HSC from Nrf2null mice have increased sensitivity to oxidative stress. In addition, the forkhead O (FoxO) family of transcription factors can protect quiescent HSC from oxidative stress through regulation of ROS detoxifying genes. FoxOs are expressed commensurate with the transition of HSC to myeloid progenitors and conditional silencing of FoxO increases ROS and decreases repopulating capacities of HSC. Therapeutically, treatment with NAC restores the FoxO transcriptional program [132]. These observations are consistent with the fact that altered redox conditions regulate HSC differentiation. A more detailed review of redox in HSC function exists elsewhere [133].
Conclusion
The association of high levels of GSTP with malignant diseases and drug resistant cancers may not be a straightforward reflection of the protein’s ability to participate in detoxification reactions. Recent reports have detailed unexpected protein interactions either through catalysis of S-glutathionylation of cysteines in target proteins or through ligand binding to regulate kinase pathways. Such functions serve to emphasize the functional promiscuity and flexibility of usage that can be ascribed to the GST isozyme family and to GSTP in particular.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jakobsson PJ, et al. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent inducible enzyme, constituting a potential novel drug target. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics. 2003;3(3):157–172. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mannervik B, Danielson UH. Glutathione transferases--structure and catalytic activity. CRC Crit Rev Biochem. 1988;23(3):283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 4.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Critical reviews in biochemistry and molecular biology. 1995;30(6):445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 5.Martin JL. Thioredoxin--a fold for all reasons. Structure. 1995;3(3):245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 6.Martin JL, Bardwell JC, Kuriyan J. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature. 1993;365(6445):464–468. doi: 10.1038/365464a0. [DOI] [PubMed] [Google Scholar]
- 7.Bushweller JH, et al. The nuclear magnetic resonance solution structure of the mixed disulfide between Escherichia coli glutaredoxin(C14S) and glutathione. J Mol Biol. 1994;235(5):1585–1597. doi: 10.1006/jmbi.1994.1108. [DOI] [PubMed] [Google Scholar]
- 8.Epp O, Ladenstein R, Wendel A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur J Biochem. 1983;133(1):51–69. doi: 10.1111/j.1432-1033.1983.tb07429.x. [DOI] [PubMed] [Google Scholar]
- 9.Schroder E, Ponting CP. Evidence that peroxiredoxins are novel members of the thioredoxin fold superfamily. Protein Sci. 1998;7(11):2465–2468. doi: 10.1002/pro.5560071125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mannervik B. The isoenzymes of glutathione transferase. Advances in enzymology and related areas of molecular biology. 1985;57:357–417. doi: 10.1002/9780470123034.ch5. [DOI] [PubMed] [Google Scholar]
- 11.Reinemer P, et al. The three-dimensional structure of class pi glutathione S-transferase in complex with glutathione sulfonate at 2.3 A resolution. EMBO J. 1991;10(8):1997–2005. doi: 10.1002/j.1460-2075.1991.tb07729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji X, et al. The three-dimensional structure of a glutathione S-transferase from the mu gene class. Structural analysis of the binary complex of isoenzyme 3–3 and glutathione at 2.2-A resolution. Biochemistry. 1992;31(42):10169–10184. doi: 10.1021/bi00157a004. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong RN. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chemical research in toxicology. 1997;10(1):2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 14.Strange RC, et al. The development expression of alpha-, mu- and pi-class glutathione S-transferases in human liver. Biochimica et biophysica acta. 1989;993(2–3):186–190. doi: 10.1016/0304-4165(89)90162-1. [DOI] [PubMed] [Google Scholar]
- 15.Hiley C, et al. Differential expression of alpha and pi isoenzymes of glutathione S-transferase in developing human kidney. Biochimica et biophysica acta. 1989;990(3):321–324. doi: 10.1016/s0304-4165(89)80052-2. [DOI] [PubMed] [Google Scholar]
- 16.Sinning I, et al. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J Mol Biol. 1993;232(1):192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 17.Sheehan D, et al. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360(Pt 1):1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Litwack G, Ketterer B, Arias IM. Ligandin: a hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions. Nature. 1971;234(5330):466–467. doi: 10.1038/234466a0. [DOI] [PubMed] [Google Scholar]
- 19.Prade L, et al. Structures of class pi glutathione S-transferase from human placenta in complex with substrate, transition-state analogue and inhibitor. Structure. 1997;5(10):1287–1295. doi: 10.1016/s0969-2126(97)00281-5. [DOI] [PubMed] [Google Scholar]
- 20.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54(16):4313–4320. [PubMed] [Google Scholar]
- 21.Wang AL, Tew KD. Increased glutathione-S-transferase activity in a cell line with acquired resistance to nitrogen mustards. Cancer Treat Rep. 1985;69(6):677–682. [PubMed] [Google Scholar]
- 22.Ciaccio PJ, Tew KD, LaCreta FP. Enzymatic conjugation of chlorambucil with glutathione by human glutathione S-transferases and inhibition by ethacrynic acid. Biochem Pharmacol. 1991;42(7):1504–1507. doi: 10.1016/0006-2952(91)90468-k. [DOI] [PubMed] [Google Scholar]
- 23.Batist G, et al. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J Biol Chem. 1986;261(33):15544–15549. [PubMed] [Google Scholar]
- 24.Barrett WC, et al. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38(20):6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 25.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42(34):10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 26.Henderson CJ, Wolf CR. Disruption of the glutathione transferase pi class genes. Methods Enzymol. 2005;401:116–135. doi: 10.1016/S0076-6879(05)01007-4. [DOI] [PubMed] [Google Scholar]
- 27.Henderson CJ, et al. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci U S A. 1998;95(9):5275–5280. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruscoe JE, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther. 2001;298(1):339–345. [PubMed] [Google Scholar]
- 29.Elsby R, et al. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J Biol Chem. 2003;278(25):22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- 30.Bakker J, Lin X, Nelson WG. Methyl-CpG binding domain protein 2 represses transcription from hypermethylated pi-class glutathione S-transferase gene promoters in hepatocellular carcinoma cells. J Biol Chem. 2002;277(25):22573–22580. doi: 10.1074/jbc.M203009200. [DOI] [PubMed] [Google Scholar]
- 31.Jones DP. Redox sensing: orthogonal control in cell cycle and apoptosis signalling. J Intern Med. 268(5):432–448. doi: 10.1111/j.1365-2796.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000;17(8):1232–1239. doi: 10.1093/oxfordjournals.molbev.a026406. [DOI] [PubMed] [Google Scholar]
- 33.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295(4):C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcaraz LD, et al. The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment. Proc Natl Acad Sci U S A. 2008;105(15):5803–5808. doi: 10.1073/pnas.0800981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7(6):313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Townsend DM, Pazoles CJ, Tew KD. NOV-002, a mimetic of glutathione disulfide. Expert Opin Investig Drugs. 2008;17(7):1075–1083. doi: 10.1517/13543784.17.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saavedra JE, et al. PABA/NO as an anticancer lead: analogue synthesis, structure revision, solution chemistry, reactivity toward glutathione, and in vitro activity. J Med Chem. 2006;49(3):1157–1164. doi: 10.1021/jm050700k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsend DM, et al. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69(2):501–508. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graminski GF, Kubo Y, Armstrong RN. Spectroscopic and kinetic evidence for the thiolate anion of glutathione at the active site of glutathione S-transferase. Biochemistry. 1989;28(8):3562–3568. doi: 10.1021/bi00434a062. [DOI] [PubMed] [Google Scholar]
- 40.Atkins WM, et al. The catalytic mechanism of glutathione S-transferase (GST). Spectroscopic determination of the pKa of Tyr-9 in rat alpha 1-1 GST. J Biol Chem. 1993;268(26):19188–19191. [PubMed] [Google Scholar]
- 41.Ghezzi P. Regulation of protein function by glutathionylation. Free Radic Res. 2005;39(6):573–580. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 42.Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal. 2004;6(1):63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 44.Findlay VJ, et al. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66(13):6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiaschi T, et al. Redox regulation of beta-actin during integrin-mediated cell adhesion. J Biol Chem. 2006;281(32):22983–22991. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- 46.Chen FC, Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol Cell Physiol. 2006;290(3):C719–C727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 47.Townsend DM, et al. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284(1):436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peltoniemi MJ, et al. Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the gamma-linkage present in glutathione. J Biol Chem. 2006;281(44):33107–33114. doi: 10.1074/jbc.M605602200. [DOI] [PubMed] [Google Scholar]
- 49.Ralat LA, et al. Characterization of the complex of glutathione S-transferase pi and 1-cysteine peroxiredoxin. Arch Biochem Biophys. 2008;474(1):109–118. doi: 10.1016/j.abb.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ralat LA, et al. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45(2):360–372. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 51.Pettigrew NE, Colman RF. Heterodimers of glutathione S-transferase can form between isoenzyme classes pi and mu. Arch Biochem Biophys. 2001;396(2):225–230. doi: 10.1006/abbi.2001.2629. [DOI] [PubMed] [Google Scholar]
- 52.Cho SG, et al. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276(16):12749–12755. doi: 10.1074/jbc.M005561200. [DOI] [PubMed] [Google Scholar]
- 53.Dorion S, Lambert H, Landry J. Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione S-transferase Mu from Ask1. J Biol Chem. 2002;277(34):30792–30797. doi: 10.1074/jbc.M203642200. [DOI] [PubMed] [Google Scholar]
- 54.Adler V, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18(5):1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101(11):3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cromer BA, et al. From glutathione transferase to pore in a CLIC. Eur Biophys J. 2002;31(5):356–364. doi: 10.1007/s00249-002-0219-1. [DOI] [PubMed] [Google Scholar]
- 57.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38(11):1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 58.Raman CS, et al. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95(7):939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 59.Ravi K. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. PNAS. 2004;101(8):2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang BHN, Lui B, Nakayama Y, Kitagawa K, Sumita K, Inagaki C. Expression and roles of Cl- channel CIC-5 in cell cycles of myeloid cells. Biochemical and Biophysical Research Communications. 2004;317:192–197. doi: 10.1016/j.bbrc.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 61.Banfi BMG, Maturana A, Steger K, Hegodus B, Demaurex N, Krause K-H. A Ca2+-activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. Journal of Biological chemistry. 2001;40:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 62.Hawkins BJMM, Kirkpatrick CJ, Fisher AB. Superoxide Flux in Endothelial Cells via the Chloride Channel-3 Mediates Intracellular Signalling. MBC. 2007;18(6):2002–2012. doi: 10.1091/mbc.E06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Townsend DM, et al. NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Res. 2008;68(8):2870–2877. doi: 10.1158/0008-5472.CAN-07-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manevich Y, et al. Diazeniumdiolate mediated nitrosative stress alters nitric oxide homeostasis through intracellular calcium and S-glutathionylation of nitric oxide synthetase. PLoS One. 5(11):e14151. doi: 10.1371/journal.pone.0014151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paige JS, et al. Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem Biol. 2008;15(12):1307–1316. doi: 10.1016/j.chembiol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forrester MT, et al. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27(6):557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohr S, et al. Nitric Oxide-induced S-Glutathionylation and Inactivation of Glyceraldehyde-3-phosphate Dehydrogenase. The Journal of Biological Chemistry. 1999;274(14):9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 68.Martínez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: Convergences and divergences. Cardiovascular Research. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 69.West M, et al. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. FASEB Journal. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- 70.Patel J, Zhang J, Block E. Nitric oxide-induced inhibition of lung endothelial cell nitric oxide synthase via interaction with allosteric thiols: role of thioredoxin in regulation of catalytic activity. Amrican Journal Respiratory Cell Molecular Biology. 1996;15(3):410–419. doi: 10.1165/ajrcmb.15.3.8810647. [DOI] [PubMed] [Google Scholar]
- 71.Chen CA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 468(7327):1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Townsend DM, et al. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69(19):7626–7634. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kyriakis JM, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369(6476):156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 74.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Current opinion in cell biology. 1998;10(2):205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 75.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 76.Minden A, et al. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266(5191):1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 77.Yin Z, et al. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000;60(15):4053–4057. [PubMed] [Google Scholar]
- 78.Romero L, et al. Human GSTA1-1 reduces c-Jun N-terminal kinase signalling and apoptosis in Caco-2 cells. Biochem J. 2006;400(1):135–141. doi: 10.1042/BJ20060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim YJ, et al. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66(14):7136–7142. doi: 10.1158/0008-5472.CAN-05-4446. [DOI] [PubMed] [Google Scholar]
- 80.Chen WC, et al. Induction of radioprotective peroxiredoxin-I by ionizing irradiation. J Neurosci Res. 2002;70(6):794–798. doi: 10.1002/jnr.10435. [DOI] [PubMed] [Google Scholar]
- 81.Park JJ, et al. Peroxiredoxin IV protects cells from radiation-induced apoptosis in head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2009;73(4):1196–1202. doi: 10.1016/j.ijrobp.2008.10.070. [DOI] [PubMed] [Google Scholar]
- 82.Wu Y, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25(42):5787–5800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- 83.Saitoh M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ichijo H, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 85.Tew KD. Redox in redux: Emergent roles for glutathione S-transferase P (GSTP) in regulation of cell signaling and S-glutathionylation. Biochem Pharmacol. 2007;73(9):1257–1269. doi: 10.1016/j.bcp.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 86.Mileo AM, et al. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One. 2009;4(10):e7254. doi: 10.1371/journal.pone.0007254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ali-Osman F, et al. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272(15):10004–10012. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]
- 88.Watson MA, et al. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19(2):275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 89.Zimniak P, et al. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. Eur J Biochem. 1994;224(3):893–899. doi: 10.1111/j.1432-1033.1994.00893.x. [DOI] [PubMed] [Google Scholar]
- 90.Pandya U, et al. Activity of allelic variants of Pi class human glutathione S-transferase toward chlorambucil. Biochem Biophys Res Commun. 2000;278(1):258–262. doi: 10.1006/bbrc.2000.3787. [DOI] [PubMed] [Google Scholar]
- 91.Srivastava SK, et al. Differential catalytic efficiency of allelic variants of human glutathione S-transferase Pi in catalyzing the glutathione conjugation of thiotepa. Arch Biochem Biophys. 1999;366(1):89–94. doi: 10.1006/abbi.1999.1217. [DOI] [PubMed] [Google Scholar]
- 92.Goto S, et al. Overexpression of glutathione S-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radic Res. 1999;31(6):549–558. doi: 10.1080/10715769900301121. [DOI] [PubMed] [Google Scholar]
- 93.Ishimoto TM, Ali-Osman F. Allelic variants of the human glutathione S-transferase P1 gene confer differential cytoprotection against anticancer agents in Escherichia coli. Pharmacogenetics. 2002;12(7):543–553. doi: 10.1097/00008571-200210000-00006. [DOI] [PubMed] [Google Scholar]
- 94.Hu X, et al. Differential protection against benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced DNA damage in HepG2 cells stably transfected with allelic variants of pi class human glutathione S-transferase. Cancer Res. 1999;59(10):2358–2362. [PubMed] [Google Scholar]
- 95.Hu X, et al. Activity of four allelic forms of glutathione S-transferase hGSTP1-1 for diol epoxides of polycyclic aromatic hydrocarbons. Biochem Biophys Res Commun. 1997;238(2):397–402. doi: 10.1006/bbrc.1997.7311. [DOI] [PubMed] [Google Scholar]
- 96.Okcu MF, et al. Glutathione S-transferase polymorphisms and survival in primary malignant glioma. Clin Cancer Res. 2004;10(8):2618–2625. doi: 10.1158/1078-0432.ccr-03-0053. [DOI] [PubMed] [Google Scholar]
- 97.Lai R, Crevier L, Thabane L. Genetic polymorphisms of glutathione S-transferases and the risk of adult brain tumors: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1784–1790. doi: 10.1158/1055-9965.EPI-05-0105. [DOI] [PubMed] [Google Scholar]
- 98.Garte S, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10(12):1239–1248. [PubMed] [Google Scholar]
- 99.Chiu BC, et al. Association of NAT and GST polymorphisms with non-Hodgkin's lymphoma: a population-based case-control study. Br J Haematol. 2005;128(5):610–615. doi: 10.1111/j.1365-2141.2004.05358.x. [DOI] [PubMed] [Google Scholar]
- 100.Lourenco GJ, et al. Polymorphisms of glutathione S-transferase Mu 1, glutathione S-transferase theta 1 and glutathione S-transferase Pi 1 genes in Hodgkin's lymphoma susceptibility and progression. Leuk Lymphoma. 2009;50(6):1005–1009. doi: 10.1080/10428190902878455. [DOI] [PubMed] [Google Scholar]
- 101.Kim HN, et al. Polymorphisms of drug-metabolizing genes and risk of non-Hodgkin lymphoma. Am J Hematol. 2009;84(12):821–825. doi: 10.1002/ajh.21556. [DOI] [PubMed] [Google Scholar]
- 102.Lu S, et al. Glutathione S-transferase P1 Ile105Val polymorphism and breast cancer risk: a meta-analysis involving 34,658 subjects. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-0969-x. [DOI] [PubMed] [Google Scholar]
- 103.Li QF, et al. Genetic polymorphism of GSTP1: prediction of clinical outcome to oxaliplatin/5-FU-based chemotherapy in advanced gastric cancer. J Korean Med Sci. 2010;25(6):846–852. doi: 10.3346/jkms.2010.25.6.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ekhart C, et al. An overview of the relations between polymorphisms in drug metabolising enzymes and drug transporters and survival after cancer drug treatment. Cancer Treat Rev. 2009;35(1):18–31. doi: 10.1016/j.ctrv.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 105.Stoehlmacher J, et al. Association between glutathione S-transferase P1, T1, and M1 genetic polymorphism and survival of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2002;94(12):936–942. doi: 10.1093/jnci/94.12.936. [DOI] [PubMed] [Google Scholar]
- 106.Sun XF, et al. Polymorphisms in sulfotransferase 1A1 and glutathione S-transferase P1 genes in relation to colorectal cancer risk and patients' survival. World J Gastroenterol. 2005;11(43):6875–6879. doi: 10.3748/wjg.v11.i43.6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kilburn L, et al. Glutathione S-transferase polymorphisms are associated with survival in anaplastic glioma patients. Cancer. 2010;116(9):2242–2249. doi: 10.1002/cncr.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tew KD. TLK-286: a novel glutathione S-transferase-activated prodrug. Expert opinion on investigational drugs. 2005;14(8):1047–1054. doi: 10.1517/13543784.14.8.1047. [DOI] [PubMed] [Google Scholar]
- 109.Rosario LA, et al. Cellular response to a glutathione S-transferase P1-1 activated prodrug. Molecular pharmacology. 2000;58(1):167–174. doi: 10.1124/mol.58.1.167. [DOI] [PubMed] [Google Scholar]
- 110.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. 2009;11(12):3013–3069. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baldini M, Sacchetti C. [Effect of cystine and cysteine on human bone marrow cultured in medium deficient in amino acids.] Rev Hematol. 1953;8(1):3–19. [PubMed] [Google Scholar]
- 112.Naka K, Ohmura M, Hirao A. Regulation of the self-renewal ability of tissue stem cells by tumor-related genes. Cancer Biomark. 2007;3(4–5):193–201. doi: 10.3233/cbm-2007-34-504. [DOI] [PubMed] [Google Scholar]
- 113.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roberts RL, Aroda VR, Ank BJ. N-acetylcysteine enhances antibody-dependent cellular cytotoxicity in neutrophils and mononuclear cells from healthy adults and human immunodeficiency virus-infected patients. J Infect Dis. 1995;172(6):1492–1502. doi: 10.1093/infdis/172.6.1492. [DOI] [PubMed] [Google Scholar]
- 115.Peterson JD, et al. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A. 1998;95(6):3071–3076. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gate L, et al. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279(10):8608–8616. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 117.Lo Celso C, Wu JW, Lin CP. In vivo imaging of hematopoietic stem cells and their microenvironment. J Biophotonics. 2009;2(11):619–631. doi: 10.1002/jbio.200910072. [DOI] [PubMed] [Google Scholar]
- 118.Cheshier SH, et al. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A. 1999;96(6):3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nilsson SK, Johnston HM, Coverdale JA. Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood. 2001;97(8):2293–2299. doi: 10.1182/blood.v97.8.2293. [DOI] [PubMed] [Google Scholar]
- 120.Calvi LM, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 121.Iwasaki H, Suda T. Cancer stem cells and their niche. Cancer Sci. 2009;100(7):1166–1172. doi: 10.1111/j.1349-7006.2009.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 123.Sugiyama T, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 124.Arai F, et al. Niche regulation of hematopoietic stem cells in the endosteum. Ann N Y Acad Sci. 2009;1176:36–46. doi: 10.1111/j.1749-6632.2009.04561.x. [DOI] [PubMed] [Google Scholar]
- 125.Adams GB, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439(7076):599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- 126.Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82(7):2031–2037. [PubMed] [Google Scholar]
- 127.Kohler A, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. 2009;114(2):290–298. doi: 10.1182/blood-2008-12-195644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rossi DJ, et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 129.Chambers SM, et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fahey JW, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002;99(11):7610–7615. doi: 10.1073/pnas.112203099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramos-Gomez M, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98(6):3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 133.Naka K, et al. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal. 2008;10(11):1883–1894. doi: 10.1089/ars.2008.2114. [DOI] [PubMed] [Google Scholar]
- 134.Katti SK, LeMaster DM, Eklund H. Crystal structure of thioredoxin from Escherichia coli at 1.68 A resolution. J Mol Biol. 1990;212(1):167–184. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- 135.Nulton-Persson AC, et al. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry. 2003;42(14):4235–4242. doi: 10.1021/bi027370f. [DOI] [PubMed] [Google Scholar]
- 136.Cabiscol E, Levine RL. The phosphatase activity of carbonic anhydrase III is reversibly regulated by glutathiolation. Proc Natl Acad Sci U S A. 1996;93(9):4170–4174. doi: 10.1073/pnas.93.9.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Borges CR, et al. Dopamine biosynthesis is regulated by S-glutathionylation. Potential mechanism of tyrosine hydroxylast inhibition during oxidative stress. J Biol Chem. 2002;277(50):48295–48302. doi: 10.1074/jbc.M209042200. [DOI] [PubMed] [Google Scholar]
- 138.Cappiello M, et al. Modulation of aldose reductase activity through S-thiolation by physiological thiols. Chem Biol Interact. 2001;130–132(1–3):597–608. doi: 10.1016/s0009-2797(00)00286-6. [DOI] [PubMed] [Google Scholar]
- 139.Konorev EA, Kalyanaraman B, Hogg N. Modification of creatine kinase by S-nitrosothiols: S-nitrosation vs. S-thiolation. Free Radic Biol Med. 2000;28(11):1671–1678. doi: 10.1016/s0891-5849(00)00281-1. [DOI] [PubMed] [Google Scholar]
- 140.Reddy S, et al. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem J. 2000;347(Pt 3):821–827. [PMC free article] [PubMed] [Google Scholar]
- 141.Eaton P, et al. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J Mol Cell Cardiol. 2002;34(11):1549–1560. doi: 10.1006/jmcc.2002.2108. [DOI] [PubMed] [Google Scholar]
- 142.Mohr S, et al. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274(14):9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 143.Ghezzi P, et al. Protein glutathionylation: coupling and uncoupling of glutathione to protein thiol groups in lymphocytes under oxidative stress and HIV infection. Mol Immunol. 2002;38(10):773–780. doi: 10.1016/s0161-5890(01)00114-6. [DOI] [PubMed] [Google Scholar]
- 144.Davis DA, et al. Reversible oxidative modification as a mechanism for regulating retroviral protease dimerization and activation. J Virol. 2003;77(5):3319–3325. doi: 10.1128/JVI.77.5.3319-3325.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fratelli M, et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99(6):3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lind C, et al. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406(2):229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 147.Haendeler J. Thioredoxin-1 and posttranslational modifications. Antioxid Redox Signal. 2006;8(9–10):1723–1728. doi: 10.1089/ars.2006.8.1723. [DOI] [PubMed] [Google Scholar]
- 148.Dalle-Donne I, et al. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34(1):23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 149.Dalle-Donne I, et al. Actin S-glutathionylation: evidence against a thiol-disulphide exchange mechanism. Free Radic Biol Med. 2003;35(10):1185–1193. doi: 10.1016/s0891-5849(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 150.Starke DW, Chock PB, Mieyal JJ. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem. 2003;278(17):14607–14613. doi: 10.1074/jbc.M210434200. [DOI] [PubMed] [Google Scholar]
- 151.Caplan JF, et al. Regulation of annexin A2 by reversible glutathionylation. J Biol Chem. 2004;279(9):7740–7750. doi: 10.1074/jbc.M313049200. [DOI] [PubMed] [Google Scholar]
- 152.Rossi R, et al. Membrane skeletal protein S-glutathionylation and hemolysis in human red blood cells. Blood Cells Mol Dis. 2006;37(3):180–187. doi: 10.1016/j.bcmd.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 153.Ward NE, et al. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39(33):10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 154.Humphries KM, Juliano C, Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277(45):43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 155.Hehner SP, et al. Enhancement of T cell receptor signaling by a mild oxidative shift in the intracellular thiol pool. J Immunol. 2000;165(8):4319–4328. doi: 10.4049/jimmunol.165.8.4319. [DOI] [PubMed] [Google Scholar]
- 156.Lee SR, et al. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273(25):15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 157.Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381(Pt 3):675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol. 2005;68(3):847–854. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 159.Burgoyne JR, et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317(5843):1393–1397. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 160.Leonberg AK, Chai YC. The functional role of cysteine residues for c-Abl kinase activity. Mol Cell Biochem. 2007;304(1–2):207–212. doi: 10.1007/s11010-007-9501-y. [DOI] [PubMed] [Google Scholar]
- 161.Velu CS, et al. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46(26):7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Klatt P, et al. Novel application of S-nitrosoglutathione-Sepharose to identify proteins that are potential targets for S-nitrosoglutathione-induced mixed-disulphide formation. Biochem J. 2000;349(Pt 2):567–578. doi: 10.1042/0264-6021:3490567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Klatt P, et al. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999;13(12):1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 164.Pineda-Molina E, et al. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40(47):14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 165.Qanungo S, et al. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282(25):18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 166.Reynaert NL, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103(35):13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cao X, et al. Glutathionylation of two cysteine residues in paired domain regulates DNA binding activity of Pax-8. J Biol Chem. 2005;280(27):25901–25906. doi: 10.1074/jbc.M411443200. [DOI] [PubMed] [Google Scholar]
- 168.Kim SO, et al. OxyR: a molecular code for redox-related signaling. Cell. 2002;109(3):383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- 169.Adachi T, et al. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279(28):29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 170.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476(1–2):52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 171.Aracena P, et al. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278(44):42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- 172.Wang W, et al. Reversible silencing of CFTR chloride channels by glutathionylation. J Gen Physiol. 2005;125(2):127–141. doi: 10.1085/jgp.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Adachi T, et al. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10(11):1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 174.Cohen RA, Adachi T. Nitric-oxide-induced vasodilatation: regulation by physiologic s-glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med. 2006;16(4):109–114. doi: 10.1016/j.tcm.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 175.Goch G, et al. Affinity of S100A1 protein for calcium increases dramatically upon glutathionylation. FEBS J. 2005;272(10):2557–2565. doi: 10.1111/j.1742-4658.2005.04680.x. [DOI] [PubMed] [Google Scholar]
- 176.Zhukova L, et al. Redox modifications of the C-terminal cysteine residue cause structural changes in S100A1 and S100B proteins. Biochim Biophys Acta. 2004;1742(1–3):191–201. doi: 10.1016/j.bbamcr.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 177.Mieyal JJ, et al. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10(11):1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Taylor ER, et al. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem. 2003;278(22):19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 179.Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem. 2005;280(11):10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 180.Chen CL, et al. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry. 2007;46(19):5754–5765. doi: 10.1021/bi602580c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cotton NJ, Stoddard B, Parson WW. Oxidative inhibition of human soluble catechol-O-methyltransferase. J Biol Chem. 2004;279(22):23710–23718. doi: 10.1074/jbc.M401086200. [DOI] [PubMed] [Google Scholar]
- 182.Odin JA, et al. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. 2001;108(2):223–232. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hohaus S, et al. Glutathione S-transferase P1 genotype and prognosis in Hodgkin’s lymphoma. Clin Cancer Res. 2005;11(6):2175–2179. doi: 10.1158/1078-0432.CCR-04-1250. [DOI] [PubMed] [Google Scholar]
- 184.Yang G, et al. Genetic polymorphisms in glutathione-S-transferase genes (GSTM1, GSTT1, GSTP1) and survival after chemotherapy for invasive breast carcinoma. Cancer. 2005;103(1):52–58. doi: 10.1002/cncr.20729. [DOI] [PubMed] [Google Scholar]
- 185.Sailaja K, et al. Association of the GSTP1 gene (Ile105Val) Polymorphism with Chronic Myeloid Leukemia. Asian Pac J Cancer Prev. 2010;11(2):461–464. [PubMed] [Google Scholar]
- 186.Chan QK, et al. Single nucleotide polymorphism of pi-class glutathione s-transferase and susceptibility to endometrial carcinoma. Clin Cancer Res. 2005;11(8):2981–2985. doi: 10.1158/1078-0432.CCR-04-2038. [DOI] [PubMed] [Google Scholar]
- 187.Chen YL, et al. Glutathione S-Transferase P1 (GSTP1) gene polymorphism increases age-related susceptibility to hepatocellular carcinoma. BMC Med Genet. 2010;11:46. doi: 10.1186/1471-2350-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vrana D, et al. The association between glutathione S-transferase gene polymorphisms and pancreatic cancer in a central European Slavonic population. Mutat Res. 2009;680(1–2):78–81. doi: 10.1016/j.mrgentox.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 189.Jiao L, et al. Glutathione S-transferase gene polymorphisms and risk and survival of pancreatic cancer. Cancer. 2007;109(5):840–848. doi: 10.1002/cncr.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee JM, et al. Association of GSTP1 polymorphism and survival for esophageal cancer. Clin Cancer Res. 2005;11(13):4749–4753. doi: 10.1158/1078-0432.CCR-04-2333. [DOI] [PubMed] [Google Scholar]
- 191.Li D, Dandara C, Parker MI. The 341C/T polymorphism in the GSTP1 gene is associated with increased risk of oesophageal cancer. BMC Genet. 2010;11:47. doi: 10.1186/1471-2156-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Malik MA, et al. Association of xenobiotic metabolizing enzymes genetic polymorphisms with esophageal cancer in Kashmir Valley and influence of environmental factors. Nutr Cancer. 2010;62(6):734–742. doi: 10.1080/01635581003605904. [DOI] [PubMed] [Google Scholar]
- 193.Wu X, et al. Genetic variations in radiation and chemotherapy drug action pathways predict clinical outcomes in esophageal cancer. J Clin Oncol. 2006;24(23):3789–3798. doi: 10.1200/JCO.2005.03.6640. [DOI] [PubMed] [Google Scholar]
- 194.McLeod HL, et al. Pharmacogenetic predictors of adverse events and response to chemotherapy in metastatic colorectal cancer: results from North American Gastrointestinal Intergroup Trial N9741. J Clin Oncol. 2010;28(20):3227–3233. doi: 10.1200/JCO.2009.21.7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gao Y, et al. Glutathione S-transferase P1 Ile105Val polymorphism and colorectal cancer risk: a meta-analysis and HuGE review. Eur J Cancer. 2009;45(18):3303–3314. doi: 10.1016/j.ejca.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 196.Nguyen TV, et al. Genetic polymorphisms in GSTA1, GSTP1, GSTT1, and GSTM1 and gastric cancer risk in a Vietnamese population. Oncol Res. 2010;18(7):349–355. doi: 10.3727/096504010x12626118080064. [DOI] [PubMed] [Google Scholar]
- 197.Cote ML, et al. Meta- and pooled analysis of GSTP1 polymorphism and lung cancer: a HuGE-GSEC review. Am J Epidemiol. 2009;169(7):802–814. doi: 10.1093/aje/kwn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lu C, et al. Association between glutathione S-transferase pi polymorphisms and survival in patients with advanced nonsmall cell lung carcinoma. Cancer. 2006;106(2):441–447. doi: 10.1002/cncr.21619. [DOI] [PubMed] [Google Scholar]
- 199.Ada AO, et al. CYP and GST polymorphisms and survival in advanced non-small cell lung cancer patients. Neoplasma. 2010;57(6):512–521. doi: 10.4149/neo_2010_06_512. [DOI] [PubMed] [Google Scholar]
- 200.Maggini V, et al. Lack of association of NQO1 and GSTP1 polymorphisms with multiple myeloma risk. Leuk Res. 2008;32(6):988–990. doi: 10.1016/j.leukres.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 201.Dasgupta RK, et al. Polymorphic variation in GSTP1 modulates outcome following therapy for multiple myeloma. Blood. 2003;102(7):2345–2350. doi: 10.1182/blood-2003-02-0444. [DOI] [PubMed] [Google Scholar]
- 202.Coughlin SS, Hall IJ. Glutathione S-transferase polymorphisms and risk of ovarian cancer: a HuGE review. Genet Med. 2002;4(4):250–257. doi: 10.1097/00125817-200207000-00003. [DOI] [PubMed] [Google Scholar]