

Abstract
Background
Lung inflammation precedes the development of hypoxia-induced pulmonary hypertension (HPH); however its role in the pathogenesis of HPH is poorly understood. We sought to characterize the hypoxic inflammatory response and elucidate its role in the development of HPH. We also aimed to investigate the mechanisms by which heme oxygenase-1 (HO-1), an anti-inflammatory enzyme, is protective in HPH.
Methods and Results
We generated bitransgenic mice that overexpress human HO-1 under doxycycline (dox) control in an inducible, lung-specific manner. Hypoxic exposure of mice in the absence of dox resulted in early transient accumulation of monocytes/macrophages in the bronchoalveolar lavage. Alveolar macrophages acquired an alternatively activated phenotype (M2) in response to hypoxia, characterized by the expression of Found in Inflammatory Zone-1, Arginase-1 and Chitinase-3-like-3. A brief, two-day pulse of dox delayed but did not prevent the peak of hypoxic inflammation, and could not protect from HPH. In contrast, a seven-day dox treatment sustained high HO-1 levels during the entire period of hypoxic inflammation, inhibited macrophage accumulation and activation, induced macrophage IL-10 expression, and prevented the development of HPH. Supernatants from hypoxic M2 macrophages promoted proliferation of pulmonary artery smooth muscle cells while treatment with carbon monoxide, a HO-1 enzymatic product, abrogated this effect.
Conclusions
Early recruitment and alternative activation of macrophages in hypoxic lungs is critical for the later development of HPH. HO-1 may confer protection from HPH by effectively modifing macrophage activation state in hypoxia.
Keywords: pulmonary hypertension, hypoxia, heme oxygenase-1, alternative macrophage activation, regulatory macrophages
Pulmonary arterial hypertension (PAH) is a devastating disease, characterized by vasoconstriction and vascular wall remodeling, with resultant right ventricular hypertrophy and eventual failure. Despite significant progress in this field, the mechanisms underlying the development of PAH are still obscure. Recently, in an increasing number of studies, lung inflammation has been implicated as a potential maladaption underlying the development of PAH. Infiltrates of leukocytes as well as inflammatory mediators have been detected in patients with PAH 1-3 and have been reported to contribute to pulmonary vascular remodeling in animal models of disease.4-6 Tissue hypoxia, a well-known stimulus for pulmonary hypertension, has been also demonstrated by our group and others to induce an inflammatory response that precedes the development of hypoxia-induced pulmonary hypertension (HPH).7-9
Among the inflammatory cells implicated in PAH, those of the monocyte/macrophage lineage have been more often correlated with disease.1, 2, 8-10 However, macrophages efficiently respond to environmental signals with remarkable plasticity and undergo different forms of polarized activation that can be roughly categorized as classically-activated (M1), alternatively-activated (M2), and anti-inflammatory (regulatory) macrophages.11, 12 M1 are “effector” phagocytes, activated by interferon-γ (INF-γ) and tumor necrosis factor (TNF-α). They produce inducible nitric oxide synthase (iNOS) and IL-12 and exhibit enhanced microbicidal or tumoricidal capacity.11 On the other hand, M2-polarized macrophages are activated mostly by IL-4 or IL-13 and, as recently discovered, by CCL2 and IL-6,13 and express Found in Inflammatory Zone-1 (Fizz1), arginase-1 (Arg1), and chitinase-3-like-3 (Ym1) and mannose receptor, C type lectin-1 (MR).11, 12, 14 M2 macrophages have been implicated in the pathogenesis of lung and other disorders via their ability to promote trophic, profibrotic, and angiogenic functions.15, 16 The major characteristic of the third population, regulatory macrophages, is the production of high IL-10 and low IL-12 levels and the promotion of immunosuppression.11, 14 In the case of PAH, the activation state of the recruited macrophages and their contribution to disease has remained up to now unclear.
Heme oxygenase-1 (HO-1) is a major antioxidant and cytoprotective enzyme that catalyzes the degradation of heme to three enzymatic end-products: carbon monoxide (CO), free Fe2+ and biliverdin.17 HO-1 and its enzymatic product, CO, have been reported by our group and others to be protective in HPH.7, 18-20 This protection, up to now, has been mainly attributed to the relaxation of vascular tone and inhibition of vascular smooth muscle cell (VSMC) proliferation by CO.21 However, it has been demonstrated that HO-17, 17, 22, 23 and CO 24, 25 have potent anti-inflammatory properties, some of which may be exerted via the upregulation of the anti-inflammatory cytokine, IL-10 25, 26. Moreover, HO-1-deficient mice develop a chronic oxidative inflammatory state that progresses with age 27 and have a maladaptive response to hypoxia with right ventricular dilation, fibrosis, and inflammation.18, 28 Therefore, we hypothesized that immunomodulation is a key mechanism of HO-1 protection in PAH.
In order to characterize in detail the lung inflammatory response caused by hypoxia, assess its role in pulmonary hypertension, and investigate the protective properties of HO-1 in this context, we generated a bitransgenic mouse model with doxycycline (dox) - inducible, lung-specific expression of HO-1. We report here that hypoxic exposure in the absence of dox provoked a significant monocyte/macrophage accumulation in the bronchoalveolar lavage fluid (BALF) which manifested a phenotype consistent with alternative activation, with upregulated expression of Fizz1, Arg1, Ym1 and MR. HO-1 overexpression by dox treatment inhibited hypoxic macrophage recruitment and activation and resulted in upregulation of IL-10 in macrophages. Supernatants from hypoxic cultures of M2 macrophages promoted proliferation of pulmonary artery smooth muscle cells while CO treatment abrogated this effect. By modulating the timing and duration of HO-1 expression with dox, we were able to either delay or suppress lung inflammation and macrophage activation and, in the latter case, abolish HPH.
Methods
Bitransgenic mice were generated by crossing Balb/c transgenic mice that harbor the tetracycline transcriptional activator (rtTA, tetON system) under the control of the Clara Cell Secretory Protein (CCSP) promoter with FVB transgenic mice that carry the human HO-1 (hHO-1) transgene under the control of the tetracycline response element (TRE) (Figure 1A). Expression of hHO-1 in the lung was achieved by the addition of 1 mg/ml dox in the drinking water. The CCTA mouse line that lacks the hHO-1 transgene was treated with dox and served as control to eliminate any potential effects imparted by dox itself, independent of hHO-1. All animal procedures were approved by the Children’s Hospital Boston Animal Care and Use Committee. An expanded Methods section is available in the Online Data Supplement at http://circ.ahajournals.org.
Figure 1. Lung-specific, doxycycline-regulated expression of human HO-1.

[A] Bitransgenic mice were generated by crossing lines CCTA and TH77. CCTA harbors the reverse tetracycline transactivator (rtTA) under the control of the 2.3kb rat CCSP (Clara Cells Secretory Protein) promoter. TH77 harbors a human HO-1 (hHO-1) transgene under the control of seven copies of the tet operator linked to a minimal CMV promoter. [B] Bitransgenic mice (CC77) were treated with 0.2 mg/ml or 1 mg/ml dox in the drinking water for 2 to 12 days and semi-quantitative PCR analysis on total lung RNA for hHO1 and the housekeeping gene, GADPH is depicted. The following mouse strains, SHO1: transgenics constitutively expressing hHO-1 in lung epithelium, and FVB: wild-type, served as positive and negative controls, respectively. The primers used target a divergent region on HO-1 mRNA and do not amplify endogenous murine HO-1 transcripts. [C] Western Blot analysis of HO-1 protein in total lung extracts of CC77 and CCTA mice. Note that the antibody used detects both the dox-regulated human HO-1 and the endogenous murine HO-1. HO-2 was used as an internal control.
Statistical analysis
All values were expressed as mean ± SD. Comparison of results between different groups was performed by one-way analysis of variance or Mann Whitney U test, where appropriate, using GraphPad InStat (GraphPad Software, San Diego, CA). P value <0.05 was considered significant.
Results
Lung specific, inducible expression of hHO-1
Based on the design of the bitransgenic model (designated as CC77) (Figure 1A), the hHO-1 transgene is under the control of both dox and the CCSP promoter and therefore, it is inducibly expressed in the lung epithelium. Semi-quantitative PCR analysis on total lung RNA using hHO1-specific primers indicates that hHO-1 levels were upregulated with dox treatment in a dose-dependent manner while they remained undetectable in the absence of dox (Figure 1B). Using an antibody that detects both human and murine HO-1, we detected profoundly elevated protein levels of HO-1 in the lungs of dox-treated CC77 mice but not in CCTA mice (Figure 1C).
Sustained induction of HO-1 prevents HPH
The development of PAH in our model was assessed by the measurement of right ventricular systolic pressure (RVSP), Fulton’s Index (FI) and the medial wall thickness index (MWTI). FI, the ratio of right ventricle weight to left ventricle plus septum weight [RV/(LV+S)], represents a hallmark of right ventricular hypertrophy resulting from increased right ventricle pressure afterload. The MWTI was estimated based on the histological sections of pulmonary arterioles stained with alpha-smooth muscle actin (α-SMA). RVSP, FI and MWTI were significantly elevated as early as seven days of hypoxia both in bitransgenics (CC77) and controls (CCTA) (Figure 2A-C). Dox administration for the entire course of hypoxia prevented the increase of RVSP, FI, and MWTI in the bitransgenic mice but not in the controls (Figure 2).
Figure 2. Sustained Expression of HO-1 Prevents HPH.

[A] Right Ventricular Systolic Pressure (RVSP), [B] Fulton’s Index, and [C] Medial Wall Thickness Index were determined at the indicated times of hypoxic exposure, in the presence or absence of 1 mg/ml dox in the drinking water of CC77 and CCTA mice. [D] Representative images of vascular remodeling in lung sections of mice exposed to hypoxia for 21 days and stained for alpha-smooth muscle actin (α-SMA). CC77: bitransgenic mice (CCSP-rtTA × TH77). CCTA: transgenic mice lacking HO-1 (CCSP-rtTA). Numbers represent mean +/−SD, n≥6 per group. Scale bar is representative of 25 μm. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #:relative to hypoxia –dox; #p<0.05, ##p<0.01, ###p<0.001.
Immunostaining of pulmonary arterioles for α-SMA revealed thickened and remodeled medial vascular walls in lung sections of hypoxic mice, while this pathology was absent in hypoxic mice treated with dox (Figure 2D).
Hypoxia induces monocyte/macrophage infiltration and lung cytokine production that is ameliorated by HO-1 and its enzymatic product, CO
To track the inflammatory response at the initial stages of hypoxic exposure and prior to the development of hypertension, animals were exposed to hypoxia and a temporal profile of the cell content of BAL was performed. More than 95% of the isolated BAL cells were CD45 positive leukocytes (Supplemental Figure 1A). The cells expressing the macrophage-specific cell surface antigens F4/80 and CD11c remained the predominant population (>98%) irrespective of hypoxic exposure or dox-treatment (Supplemental Figure 1B and Figure 3A). Under these conditions, only a subtle increase in BALF neutrophils and T lymphocytes was observed (data not shown). Upon commencing the hypoxic exposure, the numbers of monocytes/macrophages were significantly increased in the BALF of control hypoxic mice, reaching a peak at two days of hypoxia and dropping significantly by seven days, although remaining slightly elevated compared with normoxic animals (Figure 3B). Dox administration had a suppressive effect on the accumulation of cells at all time intervals investigated (Figure 3B). Dox treatment of the control mice (CCTA) had no impact on monocyte/macrophage accumulation establishing that inhibition of cell infiltrate is specifically due to HO-1 overexpression and not an artifact of dox treatment (Figure 3C).
Figure 3. Hypoxia induces early monocyte/macrophage infiltration in the lungs that is ameliorated by HO-1.

[A] Flow cytometric analysis of CD45 (+) leukocytes isolated from BALF demonstrates expression of CD11c and F4/80 macrophage markers (≈98%). [B] Accumulation of monocytes/macrophages (CD11c+, F4/80+) is depicted over time in the BALF of hypoxic mice in the absence (−) or presence (+) of 1 mg/ml dox in the drinking water. [C] Dox treatment suppressed the hypoxia-induced 2-day peak of macrophage accumulation in the BALF of CC77 mice but not in the CCTA controls. The effect of the HO-1 products (Biliverdin i.p (Bil), inhaled carbon monoxide (CO), and Bil + CO, combined) on macrophage accumulation is also shown. [D] Morphologic alteration of a population of BALF-isolated macrophages is observed within four days of hypoxia in untreated mice, a phenotype absent in dox-treated mice. Numbers represent mean +/−SD, n≥6 mice per group. Scale bar is representative of 25um. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #:relative to hypoxia –dox; #p<0.05, ##p<0.01, ###p<0.001.
To identify the specific enzymatic product of HO-1, CO and/or biliverdin, that is responsible for suppressing the peak of inflammation at two days of hypoxia, we exposed animals to either intermittent inhalation of CO (250 ppm for one hour twice/day) and/or biliverdin injections (50 μmol/kg, i.p, twice/day). PBS i.p injections served as control. Only inhaled CO or CO plus biliverdin, but not biliverdin alone, were effective in inhibiting the inflammatory cell infiltrate in the BALF at levels comparable to dox treatment (Figure 3C).
In addition to the accumulation of macrophages, several cytokines/chemokines were also upregulated in the BALF of hypoxic mice (Supplemental Figure 2). In as early as two and four days of hypoxia, upregulation of FGFβ, IL-1β, MIP-1α, IL-17 and IL-2 as well as of Th2-related cytokines, IL-13 and IL-4, was observed. The Th1-related cytokines, IL-12, TNF-α remained unaffected (Supplemental Figure 2), and INF-γ levels were undetectable (not shown). Dox administration effectively suppressed FGFβ, IL-1β, MIP-1α and IL-2 at both, two and four days of hypoxia, and suppressed IL-17, IL-13, and IL-4 only after four days of continuous administration (Supplemental Figure 2).
Interestingly, a striking alteration of macrophage morphology in cytospin preparations of BALF-isolated macrophages was observed at four days of hypoxia, characterized by cytoplasmic enlargement in a population of cells. This phenotype is consistent with activation and it was not detected in any of the macrophages isolated from dox-treated mice (Figure 3D).
Hypoxia induces alternative activation of macrophages: suppressive effect of HO-1
The observation that macrophage morphology was altered in response to hypoxia led us to further investigate the potential activation state of hypoxic macrophages. Q-PCR analysis of BALF-isolated alveolar macrophages from CC77 bitransgenic mice revealed an induction of well-defined markers of M2 macrophages in hypoxic mice, including Arg1, Fizz1, Ym1 (Figure 4A) and MR (data not shown). The peak expression occurred at four days of hypoxia and remained upregulated for at least fourteen days. Fizz1 was also secreted in the BALF of hypoxic mice (Figure 4B). In contrast, there was no change in the mRNA levels of markers of M1 macrophage phenotype, such as iNOS, TNF-α and IL-12β (IL- 12p40) or the costimulatory molecules, CD80/86, essential in the process of antigen presentation (Supplemental Figure 3). Urea production, indicative of arginase activity, was also upregulated in in vivo hypoxic alveolar macrophages (Figure 4C) and this increase in enzymatic activity was due to Arg1, since Arg1 mRNA levels were induced 9.1 ± 3.4-fold after four days of hypoxic exposure, whereas Arg2 mRNA levels were 0.6 ± 0.1 of their normoxic value at this time point. iNOS activity, as assessed by nitrite and nitrate production in the BALF, remained unchanged (Figure 4D). Dox administration effectively suppressed all markers of alternative activation (Figures 4A - C) whereas these markers were not suppressed in the CCTA line treated with dox (Figure 4B, C and Supplemental Figure 4A). Immunostaining revealed that 10.8 ± 2.7 % (35.5±8.9×103) (mean±SD) of the macrophages were Fizz1-positive, while in the presence of dox, this number was reduced to 2.17±0.6% (5.1±1.4×103) (mean±SD, p<0.01) (Figure 5A). Immunofluorescent staining confirmed the localization of Fizz1 and the absence of iNOS in the cytoplasm of hypoxic macrophages (Figure 5B,C).
Figure 4. Hypoxia induces alternatively-activated macrophages: the suppressive effect of HO-1.

[A] qPCR analysis of hypoxic alveolar macrophage mRNA isolated from bitransgenic mice (CC77) revealed increased Arg1, Fizz1, and Ym1 levels that were suppressed with dox. [B] Western blot analysis for Fizz1 on BALF from normoxic mice (Nrm) and mice exposed to hypoxia for 4 days -dox (Hyp−dox) or with dox treatment (Hyp+dox). IgA served as internal control. [C] Arginase activity (U/L) was assessed by urea formation in alveolar macrophages from normoxic and hypoxic animals. [D] iNOS activity was estimated by the levels of nitrite and nitrate in the BALF of hypoxic mice. Supernatants from RAW 264.7 macrophages stimulated with 100 μg/ml LPS E.coli and 100 U/ml INF-γ for 48 hours served as positive controls. Mean ± SD is depicted for n≥6 mice per group. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #:relative to hypoxia –dox; #p<0.05, ##p<0.01, ###p<0.001.
Figure 5. M2 and M1 expression profile of hypoxic alveolar macrophages.

Fizz1 expression in alveolar macrophages from normoxic mice or mice exposed to hypoxia in the absence or presense of dox were assessed by [A] Flow cytometry and [B] Immunofluorescence (FITC). [C] iNOS (FITC) staining in alveolar macrophages. Primary alveolar macrophages stimulated with 100 μg/ml LPS E.coli and 100 U/ml INF-γ for 48 hours served as positive controls. Nuclei were counterstained with DAPI. Scale bar is representative of 25 μm.
Apart from the slight elevation of the Th2 cytokines, IL-13 and IL-4, in the BALF of hypoxic mice, we investigated the potential presence of other non-canonical inducers of M2 polarization. Thus, we assessed the mRNA levels of CCL2 and IL-6 in total lung extracts by qPCR. CCL2 and IL-6 mRNA was robustly upregulated soon after hypoxic exposure but was significantly suppressed in the presence of dox (Supplemental Figure 5A). In the CCTA control line lacking the HO-1 transgene, dox treatment did not suppress CCL2 and IL-6 levels (Supplemental Figure 5B). Interestingly, primary alveolar macrophages cultured in vitro under hypoxic conditions (0.5% O2) also manifested the M2 phenotype with increased levels of Fizz1 and Ym1 but not IL-12 and TNF-α (Figure 8C and Supplemental Figure 6), suggesting that M2 polarization induced by hypoxia is a cell autonomous phenomenon.
Figure 8. The role of macrophage activation profile in the development of HPH.

[A] After a two-day pulse of dox and four days after HO-1 dropped to baseline, a pattern of alternative macrophage activation ensues (increased expression of Fizz-1, Arg1, Ym1), whereas IL-10 levels remained low. [B] After a seven-day pulse with dox, even when HO-1 levels return to baseline, high IL-10 expression persists. Shaded areas represent levels of hHO-1 transgene expression. [C] Fizz1 and IL-10 mRNA levels are shown in primary alveolar macrophages exposed to hypoxia or IL-4 (20 ng/ml) with or without CO. [D] Effect of macrophage supernatants from different treatments on PASMC proliferation. PDGF-BB (25 ng/ml) was used as positive control and cell culture medium (DMEM) or medium exposed to 0.5% Oxygen for 48 hours (hypoxic DMEM) as negative controls. Numbers represent mean +/−SD, n≥6 animals or wells per group. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #: relative to hypoxia-dox; #p<0.05, ##p<0.01.
HO-1 promotes the expression of IL-10 in alveolar macrophages
In an effort to further evaluate the effect of HO-1 on macrophage phenotype, mRNA and protein levels of IL-10, a well-documented anti-inflammatory mediator, were directly assessed in freshly isolated alveolar macrophages. IL-10 was significantly elevated in alveolar macrophages derived from hypoxic mice treated with dox (Figure 6). Dox treatment in the CCTA line failed to upregulate IL-10 (Supplemental Figure 4B), establishing that the observed effect is HO-1-dependent. Furthermore, the number of regulatory macrophages expressing IL-10 (CD11c+, IL-10+) under hypoxia were assessed by flow cytometry to be increased four to nine-fold with dox treatment, comprising slightly less than 10% of the total macrophage population in BALF (data not shown).
Figure 6. HO-1 induction promotes the expression of IL-10 in alveolar macrophages.
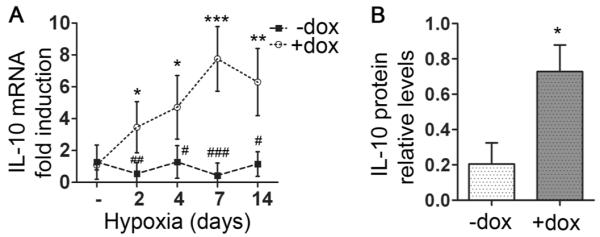
Alveolar macrophages were isolated from animals exposed to hypoxia for the indicated times, IL-10 mRNA expression was assessed by qPCR and IL-10 protein levels by Western blot followed by densitometric analysis. Relative IL-10 mRNA levels at the indicated time points [A] and IL-10 protein levels at day 7 of hypoxia [B] are shown normalized to β-actin. Numbers represent mean +/−SD, n≥5 mice per group. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #:relative to hypoxia +dox; #p<0.05, ##p<0.01, ###p<0.001.
Early monocyte/macrophage accumulation is critical for the later development of pulmonary hypertension
To determine whether this early inflammatory response is essential for the later development of HPH and whether inducible expression of HO-1 at defined internals during this process modulates the disease, we exposed the bitransgenic mice to dox for varying time periods. In addition to control animals exposed to hypoxia for three weeks in the absence of dox, one group of littermates received dox continuously for three weeks and in two other groups, dox was removed from the drinking water at either two days or seven days of hypoxia, respectively. In all groups, animals remained in hypoxia for the entire three-week period. Upon removal of dox, hHO- 1 mRNA levels returned to baseline within three days (Figure 7A, B).
Figure 7. Early monocyte/macrophage accumulation is critical for the later development of HPH.

HO-1 was transiently over-expressed in hypoxic mouse lung. [A] Dox administered at days −2 to +2 of hypoxic exposure (2d) delays but does not prevent the peak of macrophage accumulation when HO-1 levels return to baseline. [B] Dox administered at days −2 to +7 of hypoxic exposure (7d) prevents the peak of macrophage accumulation for more than twelve days after HO-1 levels have returned to baseline. Mφ: macrophage numbers. [C] RVSP, [D] FI, and [E] MWTI were elevated in mice that received the two-day pulse but not in mice that received the seven-day pulse of dox. [F] Representative immunostaining for α-SMA of lung sections from mice exposed to hypoxia for 21 days who had previously received dox either for two days or seven days, in comparison with normoxia. Numbers represent mean +/−SD, n≥6 animals per group. *: relative to normoxia; *p<0.05, **p<0.01, ***p<0.001. #:relative to hypoxia –dox; ##p<0.01, ###p<0.001.
A short two-day pulse of dox at the onset of hypoxia caused a delay in the peak of macrophage recruitment in the BALF from two days to seven days, a time when HO-1 levels were reduced to baseline (Figure 7A). In the same group of animals, development of pulmonary hypertension, as assessed by RVSP, FI, MWTI and histology of vascular remodeling, was not prevented (Figure 7C-F). In the case of a more prolonged, yet still transient administration of dox for seven days, there was no macrophage influx even when HO-1 had reached baseline low levels, and pulmonary hypertension was completely prevented at three weeks (Figure 7B-F).
Alternative macrophage activation is associated with the development of HPH in vivo and enhances PASMC proliferation in vitro
In agreement with macrophage numbers, M2 markers followed a similar pattern (Figure 8A). In the group where dox was removed after two days of hypoxic exposure and where HPH was not prevented, mRNA levels of Fizz1, Arg1, and Ym1 increased four days after HO-1 levels had fallen to baseline (Figure 8A). However, in the group that received dox for the first seven days of hypoxia and where the later development of hypertension was prevented, levels of Fizz1, Arg1, and Ym1 remained suppressed for all time periods examined (Figure 8B). Interestingly, only in the seven-day dox-treatment group, the anti-inflammatory marker, IL-10, remained sustainably elevated above baseline (Figure 8A-B). IL-10 mRNA levels were also upregulated in primary alveolar macrophages cultured in vitro under hypoxic conditions (0.5% O2) and treated with CO (500 ppm) (Figure 8C), suggesting that CO release upon HO-1 induction in this system is the trigger for IL-10 induction in macrophages.
Supernatants of primary alveolar macrophages that were cultured in vitro under hypoxic conditions (0.5% Oxygen) or normoxic macrophages treated with IL-4 (20 ng/ml), had high levels of Fizz1 and low levels of IL-10 and were able to stimulate PASMC proliferation (Figure 8D). However, supernatants from CO-treated hypoxic macrophages had reduced levels of Fizz1 and elevated IL-10 and had no proliferative effect on PASMCs (Figure 8D). Exogenous administration of IL-10 on PASMCs treated with hypoxic macrophage supernatants had no direct suppressive effect on their proliferation (Supplemental Figure 7). Arg1 and Ym1 mRNA was upregulated in both IL-4 or hypoxia-stimulated macrophages whereas neither PDGF-BB, nor the M1 specific markers, IL-12 and TNF-α were affected (Supplemental Figure 6, and data not shown). CO treatment suppressed the upregulation of the above M2 markers in both the IL-4 and hypoxia-stimulated macrophages (Supplemental Figure 6, and data not shown).
Discussion
In our bitransgenic mouse model, we demonstrate that hypoxia provokes an accumulation of alternatively activated alveolar macrophages that precedes the development of pulmonary hypertension and appears to play a critical role in the pathogenesis of disease. HO-1 overexpression induced a switch of macrophage polarity towards an anti-inflammatory phenotype and this effect was associated with protection from HPH.
Hypoxia resulted in alveolar inflammation that consisted predominantly of macrophages. These findings correlate with the fact that macrophages tend to accumulate in poorly vascularized areas with low oxygen tension29 and with previous studies in HPH which highlighted the predominant role of the monocyte/macrophage lineage in modulating vascular remodeling.8 Additionally, we found that hypoxia in vivo and in vitro polarized the population of alveolar macrophages towards the M2 phenotype. Hypoxic microenvironment is also a hallmark feature of tumors and, similar to the hypoxic macrophages in our model, the tumor-associated macrophages exhibit a M2-like phenotype.11, 29 The cell autonomous M2 polarization in in vitro hypoxic conditions, the upregulation in vivo of mRNA levels of two recently recognized non-canonical inducers of M2 polarization, CCL2 and IL-6,13 as well as the increased IL-13 and IL-4 cytokine levels in the BALF of hypoxic mice, support the M2-like activation in our hypoxic model. Furthermore, Fizz1, a M2 specific marker, is a hypoxia-inducible molecule, also designated as Hypoxia-Induced Mitogenic Factor (HIMF).30 However, contrary to our findings, two previous studies reported upregulation of TNF- α,31 IL-12 and INF-γ 32 in hypoxic macrophages. We believe that our in vivo studies most closely approximate the disease physiology, since the first of the two previous studies was performed only in vitro, using isolated rat alveolar macrophages in a sepsis-induced hypoxia model, and the second study was conducted on peritoneal macrophages.
Our finding that the presence of M2 macrophages is associated with the development of HPH in vivo and PASMC proliferation in vitro, suggests that these polarized trophic macrophages may play a significant role in the later development of pulmonary hypertension. Indeed, enhanced polyamine and L-proline synthesis due to Arg1 has been shown to contribute to vascular damage and remodeling and elevated Arg1 in lungs of hypoxic mice has been associated with increased severity of PAH. 33, 34 Fizz1 has been recently reported to have mitogenic, angiogenic, and vasoconstrictive properties that are associated with pulmonary vascular remodeling.5, 30, 35 Its human homologue, resistin-like molecule beta (RETNLB), has also been detected in patients with scleroderma-associated pulmonary hypertension.36 However, to the authors’ knowledge, this is the first study that proposes a link between alternatively activated macrophages and the development of HPH. One potential mechanism of action of alternatively activated macrophages may be via the secretion of Fizz1 whose overexpression has been reported to lead to PAH.5 Further studies are required to determine the specific mediators from the secretome of M2 macrophages that may be contributing factors in the signaling cascade leading to HPH. In agreement with our findings, previous studies have shown that Th2 cytokines, as well as IL-6 and CCL2, induce M2, and have been implicated in human pulmonary disease and animal models of PAH.3, 6, 37
HO-1 overexpression in our bitransgenic model provoked a robust anti-inflammatory effect. It suppressed macrophage accumulation, M2 activation, and cytokine production in the lungs and prevented the subsequent development of pulmonary hypertension. CO appeared to be the key HO-1 effector inhibiting macrophage accumulation in the BALF and suppressing the expression of M2 markers in vitro. This observation is in agreement with previous studies, where HO-1 and CO were reported to have potent anti-inflammatory effects.7, 25
Endogenous HO-1 is upregulated in hypoxia as a compensatory mechanism19 but its brief upregulation is not adequate to prevent the hypoxia-induced inflammation and HPH, and only a more sustained enhancement of HO-1 expression can be protective in HPH. 7 For this reason, it is not surprising that a two-day upregulation of HO-1 with dox only postponed inflammation and did not protect from the development of HPH. Interestingly, HO-1 induction for seven days, covering the entire period of hypoxia-induced inflammation, was sufficient to prevent the later development of HPH. Indeed, seven-day upregulation of HO-1 suppressed the inflammatory response even two weeks after the return of HO-1 to baseline levels despite continuous hypoxia.
Since the switch in macrophage phenotype occured within the first four days of hypoxia, we hypothesize that the enhancement of HO-1 during this critical period may act as a pivot to shift the balance of immune response from pro-inflammatory towards immunosuppressive. In support of this, lung HO-1 overexpression upregulated IL-10 in hypoxic macrophages and increased the number of IL-10 expressing, regulatory macrophages in the BALF. Since CO could upregulate IL-10 in the in vitro cultured hypoxic macrophages it seems to be a major effector molecule of HO-1 immunomodulation. In agreement with our findings, HO-1 and exogenous CO have been previously reported to increase IL-10 expression in macrophages in vivo and in vitro.25, 38-40
Interestingly, IL-10 remained elevated even two weeks after HO-1 expression returned to baseline levels. These findings point to a switch in immunoregulation triggered by HO-1 during an early critical period and whose presence was no longer essential, at least for the subsequent two weeks of hypoxia. Elevated IL-10 levels were also associated with protection from HPH, and IL-10 expression has been reported to protect from monocrotaline-induced PAH in rats.4 We show that macrophages were the source of IL-10 but it remains unclear if they are also a target of this cytokine. Since IL-10 is a pleiotrophic cytokine it may act in an autocrine and paracrine manner to affect many different cell types, besides macrophages. However, IL-10 did not have direct anti-proliferative effects on PASMCs in our in vitro model, indicating that HO-1 and CO may also have anti-inflammatory functions independently of IL-10. Further studies are required to decipher the role of IL-10 pathway in the protection from HPH development, and the mechanism of sustained protection from HPH conferred by a transient immunomodulatory event.
In summary, the present study demonstrates a link between macrophage accumulation and M2 activation and the promotion of HPH. Based on our findings, targeting the elimination or inactivation of this subset of macrophages may ameliorate the outcome of disease and improve the long term prognosis of PAH. Importantly, M2 activation may serve as a biomarker to identify a “therapeutic window” for anti-inflammatory treatments and obviate the need for chronic therapies.
Supplementary Material
CLINICAL PERSPECTIVE.
Pulmonary arterial hypertension (PAH) is a devastating disease whose molecular and cellular underpinnings remain poorly understood, despite substantial progress in the field. Recent studies from several groups, including ours, document an important role for inflammation in the development of pulmonary hypertension but the specific inflammatory cell types and mediators leading to lung vascular remodeling have yet to be characterized. In this report, using a murine model of hypoxia-induced pulmonary hypertension, we show that hypoxia leads to early accumulation of macrophages in the lung which acquire an activated M2 phenotype characterized by overexpression of Fizz1 and Arginase-1. These M2 markers have been recognized as mitogenic, angiogenic, and profibrotic factors and their induction by hypoxia in our model is associated with the later development of PAH. Using transgenic mice with lung specific, inducible expression of heme oxygenase-1 we demonstrated suppression of macrophage accumulation and M2 activation by hypoxia and a shift towards an anti-inflammatory macrophage phenotype that was associated with prevention of PAH. Activated lung macrophages may thus be a significant source of mitogenic and trophic factors that contribute to the remodeling observed in PAH. Importantly, M2 activation may serve as a biomarker of disease progression to identify a critical window for treatment aimed at promoting a switch towards the anti-inflammatory macrophage phenotype that, according to our findings, has antiproliferative and antihypertensive effects on the lung vasculature.
Acknowledgements
We thank Sarah Gately for her expert assistance in the preparation of the manuscript.
Sources of Funding This work was supported by the NIH grants RO1 HL 055454 & RO1 HL085446 (S.K and S.A.M). E.V was supported in part by Propontis Foundation (Athens, Greece) and the State Scholarships Foundation of Greece (E.V) as a graduate student of the Graduate Program in Molecular Basis of Human Disease (University of Crete School of Medicine).
Footnotes
Disclosures None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto RF, Higuchi de LM, Aiello VD. Decreased numbers of T-lymphocytes and predominance of recently recruited macrophages in the walls of peripheral pulmonary arteries from 26 patients with pulmonary hypertension secondary to congenital cardiac shunts. Cardiovasc Pathol. 2004;13:268–275. doi: 10.1016/j.carpath.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2007;176:1041–1047. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 4.Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, Maeda Y, Urabe M, Mizukami H, Kume A, Takahashi M, Ikeda U, Shimada K, Ozawa K. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101:734–741. doi: 10.1161/CIRCRESAHA.107.153023. [DOI] [PubMed] [Google Scholar]
- 5.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Champion HC, Crow MT, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296:L582–593. doi: 10.1152/ajplung.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci U S A. 2001;98:8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashida K, Fujita J, Miyake Y, Kawada H, Ando K, Ogawa S, Fukuda K. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest. 2005;127:1793–1798. doi: 10.1378/chest.127.5.1793. [DOI] [PubMed] [Google Scholar]
- 10.Sahara M, Sata M, Morita T, Nakamura K, Hirata Y, Nagai R. Diverse contribution of bone marrow-derived cells to vascular remodeling associated with pulmonary arterial hypertension and arterial neointimal formation. Circulation. 2007;115:509–517. doi: 10.1161/CIRCULATIONAHA.106.655837. [DOI] [PubMed] [Google Scholar]
- 11.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and IL-6 promote survival of human CD11b+- peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009;284:34342–34354. doi: 10.1074/jbc.M109.042671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Mora AL, Torres-Gonzalez E, Rojas M, Corredor C, Ritzenthaler J, Xu J, Roman J, Brigham K, Stecenko A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:466–473. doi: 10.1165/rcmb.2006-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 18.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–29. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res. 2000;86:1224–1229. doi: 10.1161/01.res.86.12.1224. [DOI] [PubMed] [Google Scholar]
- 20.Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S, Otterbein LE. Carbon monoxide reverses established pulmonary hypertension. J Exp Med. 2006;203:2109–2119. doi: 10.1084/jem.20052267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem. 1997;272:32804–32809. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 22.Wagener FA, Eggert A, Boerman OC, Oyen WJ, Verhofstad A, Abraham NG, Adema G, van Kooyk Y, de Witte T, Figdor CG. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98:1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 23.Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J Exp Med. 2009;206:1167–1179. doi: 10.1084/jem.20081582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 25.Otterbein LE, Bach FH, Alam J, Soares M, Lu H Tao, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 26.Drechsler Y, Dolganiuc A, Norkina O, Romics L, Li W, Kodys K, Bach FH, Mandrekar P, Szabo G. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J Immunol. 2006;177:2592–2600. doi: 10.4049/jimmunol.177.4.2592. [DOI] [PubMed] [Google Scholar]
- 27.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitali SH, Mitsialis SA, Liang OD, Liu X, Fernandez-Gonzalez A, Christou H, Wu X, McGowan FX, Kourembanas S. Divergent cardiopulmonary actions of heme oxygenase enzymatic products in chronic hypoxia. PLoS One. 2009;4:e5978. doi: 10.1371/journal.pone.0005978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis C, Murdoch C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am J Pathol. 2005;167:627–635. doi: 10.1016/S0002-9440(10)62038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res. 2003;92:1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 31.Leeper-Woodford SK, Detmer K. Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-kappaB expression. Am J Physiol. 1999;276:L909–916. doi: 10.1152/ajplung.1999.276.6.L909. [DOI] [PubMed] [Google Scholar]
- 32.Acosta-Iborra B, Elorza A, Olazabal IM, Martin-Cofreces NB, Martin-Puig S, Miro M, Calzada MJ, Aragones J, Sanchez-Madrid F, Landazuri MO. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. J Immunol. 2009;182:3155–3164. doi: 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- 33.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Transforming growth factor-beta(1) stimulates L-arginine transport and metabolism in vascular smooth muscle cells: role in polyamine and collagen synthesis. Circulation. 2001;103:1121–1127. doi: 10.1161/01.cir.103.8.1121. [DOI] [PubMed] [Google Scholar]
- 34.Jin Y, Calvert TJ, Chen B, Chicoine LG, Joshi M, Bauer JA, Liu Y, Nelin LD. Mice deficient in Mkp-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol. 2010;298:H1518–1528. doi: 10.1152/ajpheart.00813.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaji-Kegan K, Su Q, Angelini DJ, Champion HC, Johns RA. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1159–1168. doi: 10.1152/ajplung.00168.2006. [DOI] [PubMed] [Google Scholar]
- 36.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Teng X, Hassoun PM, Yang SC, Champion HC, Tuder RM, Johns RA. Resistin-like molecule-beta in scleroderma-associated pulmonary hypertension. Am J Respir Cell Mol Biol. 2009;41:553–561. doi: 10.1165/rcmb.2008-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, Grunig E, Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue S, Suzuki M, Nagashima Y, Suzuki S, Hashiba T, Tsuburai T, Ikehara K, Matsuse T, Ishigatsubo Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum Gene Ther. 2001;12:967–979. doi: 10.1089/104303401750195926. [DOI] [PubMed] [Google Scholar]
- 39.Xia ZW, Xu LQ, Zhong WW, Wei JJ, Li NL, Shao J, Li YZ, Yu SC, Zhang ZL. Heme oxygenase-1 attenuates ovalbumin-induced airway inflammation by up-regulation of foxp3 T-regulatory cells, interleukin-10, and membrane-bound transforming growth factor-1. Am J Pathol. 2007;171:1904–1914. doi: 10.2353/ajpath.2007.070096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolinay T, Szilasi M, Liu M, Choi AM. Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med. 2004;170:613–620. doi: 10.1164/rccm.200401-023OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.