

Abstract
Aims
Ventricular myocytes isolated from hearts of streptozotocin (STZ)-diabetic rats exhibit increased spontaneous Ca2+ release. Studies attribute this defect to an enhancement in activity of type 2 ryanodine receptor (RyR2). To date, underlying reasons for RyR2 dysregulation remain undefined. This study assesses whether the responsiveness of RyR2 following stimulation by intrinsic ligands is being altered during experimental type 1 diabetes (T1D).
Methods and results
M-mode echocardiography established a cardiomyopathy in 8 weeks STZ-diabetic rats. Confocal microscopy confirmed an increase in the spontaneous Ca2+ release in isolated ventricular myocytes. Western blots revealed no significant change in steady-state levels of the RyR2 protein. When purified to homogeneity and incorporated into planar lipid bilayers, RyR2 from STZ-diabetic rats (dRyR2) exhibited reduced current amplitude at ±35 mV. dRyR2 was also more responsive to intrinsic cytoplasmic activators Ca2+, adenosine triphosphate, and cyclic adenosine diphosphate ribose and less responsive to the cytoplasmic deactivator Mg2+. Threshold for the activation of RyR2 by trans (luminal) Ca2+ was also reduced. These changes were independent of phosphorylation at Ser2808 and Ser2814. Two weeks of insulin treatment starting after 6 weeks of diabetes blunted the phenotype change, indicating that the gain of function is specific to the diabetes and not the result of STZ interacting directly with RyR2.
Conclusion
These data show, for the first time, that RyR2 is acquiring a gain-of-function phenotype independent of its phosphorylation status during T1D and provides new insights for the enhanced spontaneous Ca2+ release in myocytes from T1D rats.
Keywords: Cardiac ryanodine receptor, Type 1 diabetes, Gain of function, Diabetic cardiomyopathy
1. Introduction
More than 12 million children, adolescents, and young adults worldwide have type 1 diabetes (T1D) mellitus,1 a syndrome that results from loss of insulin-producing pancreatic beta cells. In the USA, about 1.2 million individuals have T1D.2 These individuals rely on a combination of exogenous insulin, food management, and moderate exercise to regulate their blood glucose levels on a daily basis.3 However, for a significant number of them, tittering their blood glucose (even with an insulin pump) before and after meals to physiologic levels remains challenging and the consequences of this glucose dysregulation are devastating. They develop cardiovascular diseases including heart failure at rates three to five times higher than that of the general population.4,5 A significant percentage of adolescents and young adults (∼6%) with T1D also succumb to sudden cardiac death as a result of ventricular arrhythmias triggered by iatrogenic insulin-induced hypoglycaemia.6 Hypoglycaemia-induced arrhythmias can also be precipitated if the insulin dose is not lowered and/or carbohydrate intake increased prior to intense exercise training.7 To date, mechanisms responsible for heart failure in individuals with TID remain incompletely understood.
Excitation–contraction coupling is the fundamental cellular process that keeps the heart beating in a rhythmic manner. Following depolarization, L-type Ca2+ channels on invaginated T-tubules activate, allowing the influx of a small amount of Ca2+ into the myocyte. This influxed Ca2+ binds to and simultaneously activates multiple, juxtaposed type 2 ryanodine receptor (RyR2) on the sarcoplasmic reticulum (SR), elevating cytosolic-free Ca2+ ≥10-fold and triggering contraction. Contraction is terminated when released Ca2+ is returned to the SR via sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) and the influxed Ca2+ extruded via the sarcolemmal Na+–Ca2+ exchanger.8,9
Using the streptozotocin (STZ)-induced type 1 diabetic rat model, several groups including ours have found perturbation in myocyte intracellular Ca2+ handling including reductions in the rate of Ca2+ release from the SR and the amplitude of evoked Ca2+ release during T1D.10–14 Using confocal microscopy in the line-scan mode, we and others also discovered enhanced spontaneous Ca2+ release in ventricular myocytes isolated from hearts of T1D rats13–15 and attributed it in part to an increase in the activity of RyR2. Unregulated or aberrant release of Ca2+ from the SR is especially deleterious as it not only reduces the SR Ca2+ content and the beat-to-beat force of cardiac contraction but can also induce delayed after depolarization (DAD) and ventricular arrhythmias.16,17 Initially, the ‘leakiness’ of RyR2 during T1D was attributed to an increase in phosphorylation at Ser2808 and dissociation of FKBP12.6 (calstabin-2) from the RyR2 complex.13,14 We later concluded that increased phosphorylation of RyR2 at Ser2808 and Ser2814 and/or dissociation of FKBP12.6 per se are unlikely to be principal causes for the enhanced activity of RyR2 during experimental T1D.15 This study was designed to assess whether the responsiveness of RyR2 to intrinsic ligands is being altered during T1D, i.e. is diabetes altering the phenotype of RyR2?
2. Methods
2.1. Chemicals and drugs
[3H]Ryanodine (specific activity 87 Ci/mmol) was purchased from GE Life Sciences (Boston, MA, USA). Phosphatidylserine, phosphatidylcholine, and phosphatidylethanolamine were obtained from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Dialysis membranes were obtained from Spectrum Laboratories Inc. (Rancho Dominguez, CA, USA). STZ, Na2-adenosine triphosphate (ATP) and cyclic ADP-ribose were obtained from Sigma-Aldrich (St Louis, MO, USA). Insulin pellets were obtained from LinShin Canada Inc. (Scarborough, Canada). Other reagents and buffers used were of the highest grade commercially available. Phospho-RyR2(Ser2808) and phospho-RyR2(Ser2814) antibodies were obtained from Dr Xander Wehrens, Baylor College of Medicine, Houston, TX, USA.
2.2. Induction and verification of type 1 diabetes
Animals used in this investigation adhered to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23)18 and were approved for use by the Institutional Animal Care and Use Committee, University of Nebraska Medical Center. After induction of anaesthesia with inhaled isoflurane, 90 rats were injected with freshly prepared STZ in cold 0.1 m citrate buffer, pH = 4.5 (45 mg/kg, i.v., 150 µL volume) and another 45 rats were injected with citrate buffer only to serve as controls. Animals with blood glucose of >18 mmol/L 3 days following STZ injection were considered diabetic.13,15,19 Animals with blood glucose levels <15 mmol/L were injected with a second dose of STZ (usually 22 mg/kg, i.v.). Six weeks after STZ injections, diabetic rats were randomly divided into two groups. One group was treated with insulin pellets (0.5–0.75 mm × 5 mm) inserted subcutaneously to attain the euglycemic state. The other diabetic group remained untreated.
2.3. In vivo assessment of ventricular function
M-mode echocardiography was performed at the end of the 8-week protocol in anaesthetized animals (100 mg/kg ketamine/10 mg/kg acepromazine, i.p.) as described earlier to assess left ventricular function.13,15,19 Left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), left ventricular end-diastolic volume (LVEDV), and end-systolic volume (LVESV) were measured. Percent fractional shortening (FS) and percent ejection fraction (EF) were derived (EF = [(LVEDV – LVESV)/LVEDV] × 100 and FS = [(LVEDD − LVESD)/LVEDD] × 100).
2.4. Isolation of ventricular myocytes
Ten minutes prior to euthanasia, animals were injected with heparin (1000 U/kg, i.p.). After euthanasia, chest cavities were opened, and hearts were rapidly removed and placed in Krebs-Henseleit buffer. Hearts were cannulated and perfused retrograde in a Langendorff's apparatus with collagenase. Cells were used within 5–6 h following isolation.13,15,19
2.5. Measurement of spontaneous and evoked Ca2+ releases
Spontaneous and evoked Ca2+ releases were assessed as described previously.13,15 Briefly, ventricular myocytes in DMEM-F12 were incubated for 1 h at 37°C in petri dishes containing glass cover slips that had been previously coated with laminin. After incubation, unbound cells were gently removed by suction, and DMEM-F12 was replaced with Tyrode solution. Cells were then loaded with fluo-3 (5 μM) for 30 min at 37°C. Spontaneous Ca2+ sparks were recorded in the line-scan mode with the use of a Zeiss LSM 510 confocal microscope equipped with an argon-krypton laser (25 mW, 5% intensity) with a ×60 objective. Fluo-3 was excited by light at 488 nm, and fluorescence was measured at wavelengths of >515 nm. Evoked Ca2+ release was assessed by field stimulating cells at 1 Hz (10 V for 10 ms) and rates of Ca2+ rise, decay constants and changes in fluorescence intensities (ΔF) determined. Sparks and Ca2+ transient kinetics were analysed using LSM 5 Meta (Zeiss), GraphPad Prism 4.0, and Microsoft Excel.
2.6. Preparation of junctional SR membrane vesicles (JSRV)
SR membranes were prepared from left ventricular tissues isolated from control (C), STZ-diabetic (D), and insulin-treated animals (Ins-D) as described earlier,13,15,19 except that phosphatase inhibitors were excluded from isolation buffers to promote dephosphorylation of RyR2, and 2 mM dithiothreitol (DTT) and 5 mM reduced glutathione were included to reverse-oxidized hyperreactive cysteine residues on RyR2. Fifteen hearts from each group were pooled for preparation of SR membrane vesicles. Junctional SR vesicles (JSRV) were prepared by fractionating SR membranes using discontinuous sucrose (0.6 M, 0.8 M, 1.0 M, 1.5 M) gradient centrifugation (103 745 x gav for 2 h) and vesicles at the 1.0 to 1.5 M sucrose interface were collected.20,21
2.7. RyR2 content and phosphorylation status at Ser2808 and Ser2814
JSRV (30 µg) from C, D, and Ins-D rat hearts were solubilized in the gel dissociation medium, electrophoresed on a 4–15% gradient Tris–glycine polyacrylamide gel (150 V for 210 min), and transferred overnight onto polyvinylidene fluoride membrane as described earlier.13,15 Western blots were then conducted to assess relative levels of the RyR2 protein and phosphorylation status at Ser2808 and Ser2814. β-Actin levels served as the internal control to correct for any variations in sample loading.
2.8. RyR2 purification and reconstitution into proteoliposomes
Proteoliposomes containing RyR2 were prepared as described previously with some modifications.20–22 Briefly, JSRV (1.5 mg/mL) from C, D, and Ins-D animals were solubilized with CHAPS and fractionated by using linear sucrose gradient centrifugation. After centrifugation, aliquots were collected and evaluated for RyR2 content using polyacrylamide gel electrophoresis. Fractions containing RyR2 proteoliposomes were pooled, dialysed, quick-frozen in liquid nitrogen, and stored frozen in the vapour phase of liquid nitrogen until use. Additional details for this procedure are listed in the Supplemental method.
2.9. Single channel studies
Single channel studies were performed as described previously.20,22 Phosphatidylethanolamine, phosphatidylserine, and phosphatidylcholine in a ratio of 5:3:2 (35 mg/mL of lipid) in n-decane were painted across a 200 µm diameter hole of the bilayer cup. Proteoliposomes containing RyR2 from C, D, or Ins-D animals (i.e. cRyR2, dRyR2, or InsRyR2) were then fused to the bilayer. The side of the bilayer to which the proteoliposomes were added was designated the cis side (cytoplasmic). The trans side (lumen of SR) served as ground. Channel activities were recorded in symmetric KCl buffer solution (0.25 mM KCl, 20 mM K-Hepes, pH 7.4) and the response of RyR2 to the intrinsic ligands, Ca2+ (0.45 µM–16 mM), ATP (1–5 mM), cyclic adenosine diphosphate ribose (cADPR, 1–10 µM), and Mg2+ (1–6 mM) were determined. Ligands were added as a bolus to the desired chamber (cis or trans) at 200× higher than the final concentration and chambers were vigorously stirred for 30 s prior to 8 min recording. Experiments were carried out at room temperature (23–25°C). Electrical signals were filtered at 2 kHz, digitized at 10 kHz, and analysed as described previously.20,22 Data acquisition was performed using commercially available instruments and software (Axopatch 1D, Digidata 1322A and pClamp 10.0, Axon Instruments, Burlingame, CA, USA). Data analyses were performed using pClamp 10.0 (Axon Instruments, Burlingame, CA , USA) and Sigma Plot 10.0 (Stystat Software Inc, Chicago, IL, USA).
2.10. Measurement of cADPR levels
Macgregor et al.23 found that cADPR increases Ca2+ spark frequency in guinea pig ventricular myocytes. Sitsapesan et al.24 also showed that cADPR is an activator of RyR2. This prompted us to assay cADPR levels in myocytes from control and STZ-diabetic rats. For this, ventricular myocytes were pulverized in liquid nitrogen and extracted with 0.6 M perchloric acid at 4°C. Perchloric acid was neutralized with 1,1,2 trichlorotrifluoroethane and tri-n-octylamine and vortexing. The aqueous phase was then separated and pH adjusted to 8.0 using 20 mM sodium phosphate. Contaminating nucleotides were removed by adding nucleotide pyrophosphatase, alkaline phosphatase, NADase, and MgCl2 and incubating overnight at 37°C. Radioimmunoassays were then used to measure cADPR levels as described earlier.25
2.11. Ability of cADP-ribose to alter binding of [3H]ryanodine to RyR2
[3H]Ryanodine assays were also used as an indirect measurement of the ability of cADPR to modulate RyR2. For this, membrane vesicles (0.1 mg/ml) prepared from C, D, and Ins-D rats were incubated in binding buffer containing 30 µM Ca2+ or 200 µM Ca2+ for 2 h at 37°C and various amounts of cADPR (up to 50 µM). After incubation, vesicles were filtered and washed, and the amount of [3H]ryanodine bound to RyR2 was determined using liquid scintillation counting. Non-specific binding was determined simultaneously by incubating vesicles with 1 µM unlabeled ryanodine. Additional details for this procedure are listed in the Supplemental method.
2.12. Statistical analyses
Differences among values from the three JSRV groups (cRyR2, dRyR2, InsRyR2) were evaluated using ANOVA employing GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA). Data shown are means ± SEM. Results were considered significantly different if P< 0.05 (95% confidence interval).
3. Results
3.1. Establishing diabetic cardiomyopathy in vivo
General characteristics of animals used in this study are listed in Supplementary material online, Table S1. Heart rate, FS, EF, LVEDD, LVESD, and cardiac output of STZ-diabetic animals were less than that of control animals, consistent with our prior studies13,15,19 (Supplementary material online, Figure S1 and table therein). Two-weeks of insulin treatment after 6 weeks of diabetes blunted cardiac function loss, indicating that the cardiomyopathy resulted from the diabetes and not from STZ itself.
3.2. Ventricular myocytes from STZ-diabetic rats exhibit increased spontaneous Ca2+ release
Myocytes from diabetic hearts exhibited 2.5× more spontaneous Ca2+ sparks than control myocytes (8.6 ± 0.9 sparks/50 µm/s vs. 2.3 ± 0.3 sparks/50 µm/s, P< 0.05, Supplementary material online, Figure S2 and table therein). Intensity, duration, rate of Ca2+ rise, and decay of sparks in diabetic myocytes were also less than that in control myocytes, but full width at half maximum of Ca2+ sparks remained unchanged. Mean data are listed in the table of Supplementary material online, Figure S2. Insulin treatment blunted the increase in spontaneous Ca2+ sparks, indicating that these defects were due to diabetes and not from the direct action of the diabetogenic agent, STZ acting on intracellular Ca2+ cycling proteins.
3.3. Spontaneous Ca2+ release in between evoked stimulated Ca2+ release
Line-scan confocal microscopy also showed that spontaneous Ca2+ release in diabetic myocytes persisted in-between evoked Ca2+ releases (Supplementary material online, Figure S4, white arrows). In addition to the increase in spontaneous Ca2+ release, myocytes from 8-week STZ-diabetic animals also exhibited characteristic prolongation in time to Ca2+-transient decay, indicative of defects in SERCA2a and/or sodium–Ca2+ exchanger. Insulin treatment blunted abnormal myocyte intracellular Ca2+ handling.
3.4. Relative levels and phosphorylation status of RyR2
No significant change in steady-state levels of the RyR2 protein was seen after 8 weeks of diabetes (Figure 1A). Excluding phosphatase inhibitors from isolation buffers resulted in similar levels of phosphorylation at Ser2808 and Ser 2814 on cRyR2, dRyR2, and InsRyR2 (Figure 1A). We also anticipate similar levels of phosphorylation at Ser2030, although this was not specifically assayed. Had phosphatase inhibitors been included in the isolation buffer, RyR2 from diabetic animals (dRyR2) would have shown an increase in phosphorylation at Ser2808 and Ser2814.13,15 Figure 1B shows a typical silver-stained electrophoretogram of JSRV fractions collected following linear gradient sucrose centrifugation and electrophoresed on 4–15% polyacrylamide gels for 3 h at 150 V. Fractions 3–6 also bound significant amounts of [3H]ryanodine, indicating that the purified RyR2 was functional (i.e. in tetrameric form). Fractions 3–5 were collected, dialysed, and used for all subsequent single-channel studies.
Figure 1.

Purification of RyR2 and assessment of gating and conductance. (A) Representative autoradiograms of total RyR2 and their phosphorylation status at Ser2808 and Ser2814 after isolation from control (C), diabetic (D), and insulin-treated diabetic (Ins-D) animals. (B) A typical silver-stained electrophoretogram of fractions collected after linear sucrose gradient purification. (C) Left show representative 1 s single-channel recordings of RyR2 purified from control animals (cRyR2), diabetic animals (dRyR2), and insulin-treated animals (InsRyR2). Mean current amplitude for ≥12 channels from three separate preparations are shown in (C), right. (D) Mean current–voltage relationship for cRyR2, dRyR2, and InsRyR2. *Denotes significantly different from control (P< 0.05).
3.5. dRyR2 exhibits enhanced activity but reduced conductance
Having concluded from prior studies13,15 that an increase in phosphorylation of RyR2 and/or dissociation of FKBP12.6 per se are unlikely to be underlying causes for the enhanced activity of RyR2 during experimental T1D, RyR2 from control and STZ-diabetic hearts were purified to homogeneity, reconstituted into planar lipid bilayers, and their molecular characteristics were compared. When reconstituted into planar lipid bilayers and 1.0 µM Ca2+ added to cis chamber, mean open probability (Po) of cRyR2 was 0.21 ± 0.01 at +35 mV holding potential (HP, Figure 1C, upper left panel). Dwell times in the open and closed states were 0.91 ± 0.2 ms and 4.05 ± 1.7 ms, respectively. Mean current amplitude of 17 channels obtained from three separate preparations was 26.03 ± 0.61pA at +35 mV (conductance = 744 ± 17 pS, Figure 1C, right panel, upper). dRyR2 was activated to a greater extent by 1 µM cis Ca2+ (Po = 0.85 ± 0.02, n= 12, Figure 1C, left middle panel). Dwell times in the open and closed states for dRyR2 were also significantly altered: 7.57 ± 1.31 ms and 0.23 ± 0.12 ms, respectively. Mean current amplitude of dRyR2 at +35 mV was 20% less than that of cRyR2 (Figure 1C, middle right panel, 20.72 ± 0.72pA, conductance = 592 ± 21pS). Treating STZ-diabetic rats with insulin blunted these changes. Current–voltage relationships for cRyR2, dRyR2, and InsRyR2 are shown in Figure 1D. Ryanodine (10–25 µM) added to the cis chamber induced the characteristic subconductance state, confirming that the channels under study were indeed RyR2 (data not shown). At a HP of ±35 mV, channels were stable for up to 1 h and responses to ligands were typically seen within 2 min after addition of the agent.
3.6. dRyR2 showed enhanced responsiveness to high nanomolar/low micromolar Ca2+
The enhanced responsiveness of dRyR2 to 1.0 µM cis Ca2+ prompted us to compare the responsiveness of cRyR2 and dRyR2 over a wide Ca2+ concentration. Figure 2A and B shows representative 1s single-channel recordings of cRyR2 and dRyR2 exposed to various cis [Ca2+] at +35 mV. Mean data are shown in Figure 2D. dRyR2 was activated to a greater extent by low micromolar cis Ca2+ (0.45 –3.3µM), but its response was similar to that of cRyR2 at higher cis Ca2+ (10 µM to 16 mM). Insulin treatment attenuated the enhanced low micromolar Ca2+ response (Figure 2C).
Figure 2.

Comparison of the responsiveness of cRyR2, dRyR2, and InsRyR2 to various cis [Ca2+]. (A) The response of cRyR2 to increasing cis Ca2+, (B) the response of dRyR2 to increasing cis Ca2+, and (C) the response of InsRyR2 to increasing cis Ca2+. (D) Mean ± SEM for ≥8 channels per group. *Denotes significantly different from control (P< 0.05).
3.7. dRyR2 exhibits enhanced responsiveness to ATP
In addition to Ca2+, RyR2 is also activated by ATP.26 McNeill and colleagues27 also found that myocytes from STZ-diabetic rats release more intracellular Ca2+ when challenged with ATP compared with myocytes from control animals. This prompted us to assess whether the responsiveness of RyR2 to ATP is being enhanced during T1D. When the Po of dRyR2 and cRyR2 was adjusted to similar levels by adjusting cis Ca2+ at +35 mV HP and 1 mM ATP added to the cis chamber, the Po of dRyR2 increased from 0.26 ± 0.06 to 0.48 ± 0.05 while that of cRyR2 increased from 0.20 ± 0.02 to 0.32 ± 0.04, P< 0.05, Figure 3D. Increasing ATP concentrations further to 2 and 5 mM increased the Po of dRyR2 to 0.53 ± 0.03 and 0.57 ± 0.03 and the Po of cRyR2 to 0.42 ± 0.03 and 0.44 ± 0.02, respectively (P< 0.05). The responsiveness of cRyR2 to increasing ATP was also investigated with 0.45 µM cis Ca2+ and representative 1 s recordings are shown in Figure 3A, further emphasizing that cRyR2 was less responsive than dRyR2 to ATP stimulation. Treating T1D animals with insulin blunted the enhanced responsiveness of RyR2 to ATP (Figure 3C).
Figure 3.

Comparison of the responsiveness of cRyR2, dRyR2, and InsRyR2 to cis [ATP]. (A) The response of cRyR2 to increasing cis ATP, (B) the response of dRyR2 to increasing cis ATP, and (C) the response of InsRyR2 to increasing cis ATP. (D) Mean ± SEM for ≥8 channels per group. *Denotes significantly different from control (P< 0.05).
3.8. Myocytes cADPR content is elevated during T1D
Another intrinsic ligand that activates RyR2 is cADPR. Recently, Kim et al.28 found elevated levels of cADPR and the enzyme that synthesizes it, ADP-ribosyl cyclase in glomeruli of STZ-diabetic mice. This prompted us to assess whether cADPR is also being elevated in rat ventricular myocytes during T1D. Ventricular myocytes from STZ-diabetic rats contained 3× more cADPR than ventricular myocytes from control animals (2.1 ± 0.78 fmol/mg protein vs. 0.57 ± 0.14 fmol/mg protein P< 0.05). At this time, it remains to be determined whether the increase in myocyte cADPR reflects an increase in expression and/or activity of ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase (CD38). Although not assayed, we anticipate the cADPR content of myocytes isolated from insulin-treated diabetic rats to be lower than that of diabetic animals.
3.9. cADPR potentiates the binding of [3H]ryanodine to RyR2
A target for cADPR is the RyR2. As such, [3H]ryanodine binding assays were performed to assess if the increase in cADPR is of pathophysiological importance. There was no significant difference in the RyR2 content of hearts from C, D, and Ins-D animals (Figure 1A). Consistent with prior studies,29 dRyR2 also bound 35.2 ± 6.2% less [3H]ryanodine (in binding buffer contained reduced glutathione and DTT) compared with cRyR2 (data not shown). Interestingly, while cADPR (5 –50 µM) potentiated the binding of [3H]ryanodine to dRyR2 in a low Ca2+ (30 µM) and in an optimal Ca2+ (200 µM) binding buffer (Supplementary material online, Figure S4), it did not increase [3H]ryanodine to cRyR2. InsRyR2 was less responsive to cADPR than dRyR2.
3.10. dRyR2 exhibits enhanced responsiveness to cADP ribose
Planar lipid bilayer assays were then used to assess in more detail, mechanisms underlying cADPR actions on dRyR2. At similar Po levels, dRyR2 was activated to a greater extent by 1, 2, and 10 µM cis cADPR compared with cRyR2 (Figure 4D). The responsiveness of cRyR2 to increasing cADPR was also investigated in the presence of 0.45 µM cis Ca2+ and representative 1 s recordings are shown in Figure 4A emphasizing only modest enhancement in activation (Po increased from 0.14 ± 0.01 to 0.22 ± 0.01, 0.19 ± 0.03 and 0.16 ± 0.04, respectively, Figure 4A and D).24 Insulin treatment attenuated the enhanced responsiveness of RyR2 to cADPR (Figure 4C).
Figure 4.

Comparison of the responsiveness of cRyR2, dRyR2, and InsRyR2 to cis [cADPR]. (A) The response of cRyR2 to increasing cis cADPR, (B) the response of dRyR2 to increasing cis cADPR, and (C) the response of InsRyR2 to increasing cis cADPR. (D) Mean ± SEM for ≥8 channels per group. *Denotes significantly different from control (P< 0.05).
3.11. dRyR2 exhibits enhanced luminal Ca2+ sensitivity
The activity of RyR2 is also regulated in part by luminal [Ca2+].30,31 Figure 5A, B and D shows that unlike cRyR2 which was only modestly activated by up to 16 mM trans (luminal) Ca2+, significant activation of dRyR2 began with as low as 500 µM luminal Ca2+ and persisted with higher trans Ca2+. Thus, the threshold for activation of dRyR2 by luminal Ca2+ was significantly lower than that of cRyR2. Insulin treatment attenuated the reduction in luminal Ca2+ threshold for RyR2 (Figure 5C).
Figure 5.

Comparison of the responsiveness of cRyR2, dRyR2, and InsRyR2 to increasing trans Ca2+ (cis Ca2+ = 1.0 µM). (A) The response of cRyR2 to increasing trans Ca2+, (B) the response of dRyR2 to increasing trans Ca2+, and (C) the response of InsRyR2 to increasing trans Ca2+. (D) Mean ± SEM for ≥8 channels per group. *Denotes significantly different from control (P< 0.05). **Denotes significantly different from STZ-diabetic (P< 0.05).
3.12. dRyR2 exhibits reduced responsiveness to Mg2+ inactivation
An increase in activity of RyR2 could also result from the inability of intrinsic deactivators to close the channel. One such inactivator ligand is Mg2+.26,32 To compare their responses to Mg2+ inactivation, the Po of cRyR2 and dRyR2 was adjusted to ∼0.9 using cis Ca2+: 3.3 µM for dRyR2 and 7.7 µM for cRyR2, and increasing [Mg2+] were added to the cis chamber at +35 mV HP. Under these conditions, addition of 1 mM Mg2+ to the cis chamber reduced the Po of dRyR2 from 0.90 ± 0.04 to 0.78 ± 0.05 and the Po of cRyR2 from 0.85 ± 0.03 to 0.70 ± 0.02, P> 0.05, Figure 6D. Increasing the concentrations of cis Mg2+ further to 3 and 6 mM attenuated the Po of dRyR2 to 0.72 ± 0.04 and 0.66 ± 0.03, respectively (Figure 6B and D), while that of cRyR2 was reduced to 0.42 ± 0.03 and 0.23 ± 0.03, respectively (P< 0.05, Figure 6D). The ability of Mg2+ to inactivate cRyR2 was also assessed in 3.3 µM cis Ca2+ and representative 1 s recordings are shown in Figure 6A and D, further emphasizing that cRyR2 was more sensitive to Mg2+ blockade than dRyR2. Treating STZ-diabetic animals with insulin blunted the loss of responsiveness of RyR2 to Mg2+ inactivation (Figure 6B and D).
Figure 6.

Comparison of the responsiveness of cRyR2, dRyR2, and InsRyR2 to cis [Mg2+]. (A) The response of cRyR2 to increasing cis Mg2+, (B) the response of dRyR2 to increasing cis Mg2+, and (C) the response of InsRyR2 to increasing cis Mg2+. (D) Mean ± SEM for ≥8 channels per group. *Denotes significantly different from control (P< 0.05).
4. Discussion
Efficient and rhythmic ventricular contractions require the orchestrated actions of a number of proteins residing on the plasma membrane and inside the myocyte.7,8 Untimely, activation/inactivation of any one of these proteins will reduce the efficiency of this process leading to heart failure. Of particular interest in this study is RyR2, whose activity was found to be enhanced in ventricular myocytes isolated from hearts of STZ-diabetic rats. Several signalling cues, including kinases, phosphatases, oxidant (e.g. NADPH oxidases), and anti-oxidant proteins (superoxide dismutases, catalase) to name a few, contribute to produce the net activity of RyR2 in myocytes.33 As far as we know, no prior studies have attempted to ascertain whether the enhanced activity of RyR2 during T1D is independent of these cues. To address this, RyR2 from control and STZ-diabetic rat hearts was purified to homogeneity in buffers lacking phosphatase inhibitors (so as to achieve similar degrees of phosphorylation) but with reduced glutathione and DTT (to reduce oxidized hyperreactive cysteine residues), and their responsiveness to intrinsic ligands were compared.
4.1. The model
The extensively characterized STZ-induced T1D rat model was used in this study.34 This model recapitulates many of the cardiac defects seen in patients with T1D including bradycardia, changes in early and late transmitral flow velocities, reduction in contraction, and long QT syndrome.35,36 Insulin pellets inserted subcutaneously were used to maintain blood glucose levels between 19–21 mmol during the study.19 Heart failure is induced in >95% of animals and this provides a reproducible model to investigate mechanisms underlying abnormal myocyte Ca2+ handling during T1D.
4.2. Altered responsiveness of RyR2 following stimulation by cytoplasmic ligands
The principal finding of the present study is that the responsiveness of dRyR2 following stimulation by activator ligands (Ca2+, ATP, and cADPR) is potentiated during T1D while its responsiveness to Mg2+ inactivation is blunted. This gain of function of dRyR2 is independent of phosphorylation at Ser2808 and Ser2814 and oxidation of hyperreactive cysteine residues. Earlier, we showed that disulfide bonds formed on hyperreactive cysteines on dRyR2 could be reduced by treatment with DTT,29 which along with reduced glutathione was present in isolation buffers. Since basal cytoplasmic Ca2+ is elevated in rat ventricular myocytes during T1D,15 and the threshold for the activation of RyR2 by cis (cytoplasmic) Ca2+ is reduced, these new data provide additional mechanistic insights for the increased spontaneous Ca2+ release seen in ventricular myocytes during T1D. These new data also suggest that by increasing the activity of RyR2, cADPR is playing a role in the pathogenesis of diabetic cardiomyopathy. Since, in our proteoliposome preparations, RyR2 used are devoid of FKBP12.6 (calstabin2), these data further emphasize that the increase in spontaneous Ca2+ sparks seen in diabetic myocytes are likely to be via mechanisms that are independent of reduced FKBP12.6 (calstabin2).13,14,37–39
In this study, we found that in nanomolar Ca2+, dRyR2 exhibited enhanced responsiveness to cis ATP stimulation. Whether this is a compensatory mechanism in response to the low ATP production in the heart during T1D40,41 remains to be determined. We also found elevated levels of cADPR in ventricular myocytes during T1D and this is likely to be due in part to an increase in activity of ADP-ribosyl cyclase triggered by diabetes-induced increase in angiotensin II production.42,43 In addition to being present in larger quantities, cis cADPR also potentiated the activity of dRyR2 in low micromolar Ca2+. This enhancement was shown using both binding assays and single assays. In addition to direct cis activation, since cADPR increases accumulation of Ca2+ inside the SR44 and since the threshold for luminal Ca2+ activation of RyR2 is lowered during T1D, this nucleotide may also result in activation of dRyR2 from the trans side.
4.3. Reduced conductance of RyR2 during T1D
In addition to the enhanced sensitivity to low micromolar Ca2+, the current amplitude (conductance) of dRyR2 was also 20% less than that of cRyR2 at ±35 mV HP and persisted at ±20, ±40, and ±60 mV HP. This decrease in dRyR2 conductance could be an underlying cause for reduction in Ca2+ spark amplitude seen in diabetic myocytes. Treating diabetic animals with insulin for 2 weeks blunted the reduction in RyR2 current amplitude (conductance), indicating that metabolic factors may be responsible. As far as we are aware, this is the first study to show reduced conductance of RyR2 during T1D.
4.4. Reduced threshold for activation by luminal Ca2+
In addition to cytoplasmic ligands, the activity of RyR2 is also regulated by luminal Ca2+.30,31 A drop in intra-SR Ca2+ has been proposed to ‘turn off’ RyR2 to facilitate reloading of the SR. In the present study, we found that the threshold for the activation of RyR2 by trans (luminal) Ca2+ was reduced during T1D. At this time, it is not clear whether diabetes is altering Ca2+ binding sites on the luminal surface itself or altering the interactions of RyR2-associated intra-SR proteins such as calsequestrin and/or junction with RyR2.45 It should be mentioned that we and others found reduced SR Ca2+ in myocytes isolated from hearts of STZ-diabetic rats.11,13,14
4.5. Reduced responsiveness to inactivation by Mg2+
Cytoplasmic Mg2+ serves to inactivate RyR2, restricting its opening (Po) under resting and activated conditions.46,47 In the present study, we found that dRyR2 was less sensitive to blockade by Mg2+ than cRyR2. Unlike cRyR2 whose activity was restricted in 6 mM Mg2+ (Po = 0.2 in 3.3 µM Ca2+), the activity of dRyR2 was essentially unhindered in 6 mM Mg2+ (Po = 0.7 in 3.3 µM Ca2+). Since the phosphorylation status of cRyR2 and dRyR2 was similar and phosphorylation of RyR2 has been shown to reduce the responsiveness of RyR2 to both ATP and Mg2+,46 these data further emphasizes that differences between dRyR2 and cRyR2 cannot be attributed to increased phosphorylation.
4.6. Summary
In the present study, we show, for the first time, a gain of function of RyR2 is a contributing cause for the increase in spontaneous Ca2+ sparks seen in rat ventricular myocytes during T1D. During T1D, the response of RyR2 to intrinsic cytoplasmic activators is potentiated, while the response of RyR2 to physiological inhibitors is blunted. We also found that the threshold for luminal Ca2+ activation of RyR2 is lowered during T1D. This phenotype change is independent of phosphorylation at Ser2808, Ser2814, and oxidation of hyperreactive cysteines residues. What causes the gain of function of RyR2 during T1D is not clear at this time. The enhanced activity of RyR2 is especially deleterious as Ca2+ release from one RyR2 could diffuse to adjacent RyR2s initiating more Ca2+ release from the SR, and generation/propagation of Ca2+ waves.47 Spontaneous Ca2+ waves can activate Ca2+-sensitive inward currents (INa/Ca, 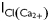 ,
, 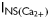 ) which can prematurely depolarize ventricular myocytes leading to DAD.47,48 If of sufficient magnitude, DAD will depolarize the cardiomyocyte above threshold, resulting in a single or repetitive premature heartbeat and the generation of arrhythmia. In a very recent study, Kauferstein et al.49 found a novel V4299M mutation on cardiac RyR2 in a patient with long QT syndrome. This patient did not have any mutations in the major long QT syndrome-related genes SCN5A, KCNH2, KCNQ1, KCNE1, KCNE2, KCNI2. Could this gain of function of RyR2 be an underlying cause for the increased incidence of long QT syndrome and the stress-induced ventricular arrhythmias in patients with T1D?5
) which can prematurely depolarize ventricular myocytes leading to DAD.47,48 If of sufficient magnitude, DAD will depolarize the cardiomyocyte above threshold, resulting in a single or repetitive premature heartbeat and the generation of arrhythmia. In a very recent study, Kauferstein et al.49 found a novel V4299M mutation on cardiac RyR2 in a patient with long QT syndrome. This patient did not have any mutations in the major long QT syndrome-related genes SCN5A, KCNH2, KCNQ1, KCNE1, KCNE2, KCNI2. Could this gain of function of RyR2 be an underlying cause for the increased incidence of long QT syndrome and the stress-induced ventricular arrhythmias in patients with T1D?5
4.7. Limitations of the study
Caution should be used when correlating our data to in vivo settings. Our experiments using isolated hearts, ventricular myocytes, and purified RyR2 were performed at room temperature and ambient O2 tension (20%) as opposed to physiological 37°C temperature and 5% O2 levels. The former conditions are known to affect the redox status of Ca2+ cycling proteins and intracellular Ca2+ handling. Earlier, we proposed two population of RyR2 in diabetic myocytes.13,15 In this study, only Ca2+-responsive dRyR2 was assessed.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported in part by grants from National Institutes of Health (HL-085061) and Nebraska Redox Biology Center (P20-RR 17675) to K.R.B.
Supplementary Material
Acknowledgements
We thank Dr Gerhard Meissner, Dr Le Xu and Daniel Pasek, University of North Carolina, Chapel Hill for help with bilayers set up and proteoliposome preparation.
Conflict of interest: none declared.
References
- 1.International Diabetes Federation: IDF Diabetes Atlas. http://www.diabetesatlas.org/ (accessed 20 January 2011) [Google Scholar]
- 2.American Diabetes Association: All about Diabetes. http://www.diabetes.org/about-diabetes.jsp. (accessed 20 January 2011. [Google Scholar]
- 3.Skyler JS. Prediction and prevention of type 1 diabetes: progress, problems, and prospects. Clin Pharmacol Ther. 2007;8:768–771. doi: 10.1038/sj.clpt.6100179. [DOI] [PubMed] [Google Scholar]
- 4.Blendea MC, McFarlane SI, Isenovic ER, Gick G, Sowers JR. Heart disease in diabetic patients. Curr Diab Rep. 2003;3:223–229. doi: 10.1007/s11892-003-0068-z. [DOI] [PubMed] [Google Scholar]
- 5.Gallego M, Alday A, Urrutia J, Casis O. Transient outward potassium channel regulation in healthy and diabetic hearts. Can J Physiol Pharmacol. 2009;87:77–83. doi: 10.1139/Y08-106. [DOI] [PubMed] [Google Scholar]
- 6.Koltin D, Daneman D. Dead-in-bed syndrome—a diabetes nightmare. Pediatr Diabetes. 2008;9:504–507. doi: 10.1111/j.1399-5448.2008.00404.x. [DOI] [PubMed] [Google Scholar]
- 7.Norman E. Exercise and blood sugar management in type 1 diabetes. BC Endocrine. Research Foundation, Summer Solstice. 2002;4:1–3. [Google Scholar]
- 8.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. 2010;106:659–673. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen PS, Joung B, Shinohara T, Das M, Chen Z, Lin SF. The initiation of the heart beat. Circ J. 2010;74:221–225. doi: 10.1253/circj.cj-09-0712. [DOI] [PubMed] [Google Scholar]
- 10.Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol. 1997;272:H148–H158. doi: 10.1152/ajpheart.1997.272.1.H148. [DOI] [PubMed] [Google Scholar]
- 11.Choi KM, Zhong Y, Hoit BD, Grupp IL, Hahn H, Dilly KW, et al. Defective intracellular Ca(2+) signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am J Physiol Heart Circ Physiol. 2002;283:H1398–H1408. doi: 10.1152/ajpheart.00313.2002. [DOI] [PubMed] [Google Scholar]
- 12.Lacombe VA, Viatchenko-Karpinski S, Terentyev D, Sridhar A, Emani S, Bonagura JD, et al. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1787–R1797. doi: 10.1152/ajpregu.00059.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shao CH, Rozanski GJ, Patel KP, Bidasee KR. Dyssynchronous (non-uniform) Ca2+ release in myocytes from streptozotocin-induced diabetic rats. J Mol Cell Cardiol. 2007;42:234–246. doi: 10.1016/j.yjmcc.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Yaras N, Ugur M, Ozdemir S, Gurdal H, Purali N, Lacampagne A, et al. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–3088. doi: 10.2337/diabetes.54.11.3082. [DOI] [PubMed] [Google Scholar]
- 15.Shao CH, Wehrens XH, Wyatt TA, Parbhu S, Rozanski GJ, Patel KP, et al. Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J Appl Physiol. 2009;106:1280–1292. doi: 10.1152/japplphysiol.91280.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190:1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- 17.Nam GB, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 19.Shao CH, Rozanski GJ, Nagai R, Stockdale FE, Patel KP, Wang M, et al. Carbonylation of myosin heavy chains in rat heart during diabetes. Biochem Pharmacol. 2010;80:205–217. doi: 10.1016/j.bcp.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bidasee KR, Xu L, Meissner G, Besch HR., Jr. Diketopyridylryanodine has three concentration-dependent effects on the cardiac calcium-release channel/ryanodine receptor. J Biol Chem. 2003;278:14237–14248. doi: 10.1074/jbc.M208372200. [DOI] [PubMed] [Google Scholar]
- 21.Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1998;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 22.Tian C, Shao CH, Fenster DS, Mixan M, Romberger DJ, Toews ML, et al. Chloroform extract of hog barn dust modulates skeletal muscle ryanodine receptor calcium-release channel (RyR1) J Appl Physiol. 2010;109:830–839. doi: 10.1152/japplphysiol.00123.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macgregor AT, Rakovic S, Galione A, Terrar DA. Dual effects of cyclic ADP-ribose on sarcoplasmic reticulum Ca2+ release and storage in cardiac myocytes isolated from guinea-pig and rat ventricle. Cell Calcium. 2007;41:537–546. doi: 10.1016/j.ceca.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Sitsapesan R, McGarry SJ, Williams AJ. Cyclic ADP-ribose, the ryanodine receptor and Ca2+ release. Trends Pharmacol Sci. 1995;16:386–391. doi: 10.1016/s0165-6147(00)89080-x. [DOI] [PubMed] [Google Scholar]
- 25.Graeff R,, Lee HC. A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem J. 2002;361:379–384. doi: 10.1042/bj3610379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hymel L, Schindler H, Inui M, Fleischer S. Reconstitution of purified cardiac muscle calcium release channel (ryanodine receptor) in planar bilayers. Biochem Biophys Res Commun. 1988;152:308–314. doi: 10.1016/s0006-291x(88)80715-0. [DOI] [PubMed] [Google Scholar]
- 27.Yu JZ, Quamme GA, McNeill JH. Altered [Ca2+]i mobilization in diabetic cardiomyocytes: responses to caffeine, KCl, ouabain, and ATP. Diabetes Res Clin Pract. 1995;30:9–20. doi: 10.1016/0168-8227(95)01144-7. [DOI] [PubMed] [Google Scholar]
- 28.Kim SY, Park KH, Gul R, Jang KY, Kim UH. Role of kidney ADP-ribosyl cyclase in diabetic nephropathy. Am J Physiol Renal Physiol. 2009;296:F291–F297. doi: 10.1152/ajprenal.90381.2008. [DOI] [PubMed] [Google Scholar]
- 29.Bidasee KR, Nallani K, Besch HR, Jr, Dincer UD. Streptozotocin-induced diabetes increases disulfide bond formation on cardiac ryanodine receptor (RyR2) J Pharmacol Exp Ther. 2003;305:989–998. doi: 10.1124/jpet.102.046201. [DOI] [PubMed] [Google Scholar]
- 30.Lukyanenko V, Györke I, Györke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflugers Arch. 1996;432:1047–1054. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- 31.Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawano S. Dual mechanisms of Mg2+ block of ryanodine receptor Ca2+ release channel from cardiac sarcoplasmic reticulum. Receptors Channels. 1998;5:405–416. [PubMed] [Google Scholar]
- 33.Turan B, Vassort G. Ryanodine receptor: a new therapeutic target to control diabetic cardiomyopathy. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3725. doi:10.1089/ars.2010.3725. Published online ahead of print 21 November 2011. [DOI] [PubMed] [Google Scholar]
- 34.Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech. 2009;2:454–466. doi: 10.1242/dmm.001941. [DOI] [PubMed] [Google Scholar]
- 35.Retnakaran R, Zinman B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371:1790–1799. doi: 10.1016/S0140-6736(08)60767-9. [DOI] [PubMed] [Google Scholar]
- 36.Heller SR. Abnormalities of the electrocardiogram during hypoglycaemia: the cause of the dead in bed syndrome? Int J Clin Pract Suppl. 2002;129:27–32. [PubMed] [Google Scholar]
- 37.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 38.Lehnart SE, Wehrens XH, Marks AR. Calstabin deficiency, ryanodine receptors, and sudden cardiac death. Biochem Biophys Res Commun. 2004;322:1267–1279. doi: 10.1016/j.bbrc.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 39.Yaras N, Bilginoglu A, Vassort G, Turan B. Restoration of diabetes-induced abnormal local Ca2+ release in cardiomyocytes by angiotensin II receptor blockade. Am J Physiol Heart Circ Physiol. 2007;292:H912–H920. doi: 10.1152/ajpheart.00824.2006. [DOI] [PubMed] [Google Scholar]
- 40.Mokhtar N, Lavoie JP, Rousseau-Migneron S, Nadeau A. Physical training reverses defect in mitochondrial energy production in heart of chronically diabetic rats. Diabetes. 1993;42:682–687. doi: 10.2337/diab.42.5.682. [DOI] [PubMed] [Google Scholar]
- 41.Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol. 1996;271:H192–H202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 42.Gul R, Kim SY, Park KH, Kim BJ, Kim SJ, Kim MJ, et al. A novel signaling pathway of ADP-ribosyl cyclase activation by angiotensin II in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H77–H88. doi: 10.1152/ajpheart.01355.2007. [DOI] [PubMed] [Google Scholar]
- 43.Higashida H, Zhang J, Hashii M, Shintaku M, Higashida C, Takeda Y. Angiotensin II stimulates cyclic ADP-ribose formation in neonatal rat cardiac myocytes. Biochem J. 2000;352:197–202. [PMC free article] [PubMed] [Google Scholar]
- 44.Lukyanenko V, Györke I, Wiesner TF, Györke S. Potentiation of Ca(2+) release by cADP-ribose in the heart is mediated by enhanced SR Ca(2+) uptake into the sarcoplasmic reticulum. Circ Res. 2001;89:614–622. doi: 10.1161/hh1901.098066. [DOI] [PubMed] [Google Scholar]
- 45.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, et al. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;31:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uehara A, Yasukochi M, Mejía-Alvarez R, Fill M, Imanaga I. Gating kinetics and ligand sensitivity modified by phosphorylation of cardiac ryanodine receptors. Pflugers Arch. 2002;444:202–212. doi: 10.1007/s00424-002-0791-3. [DOI] [PubMed] [Google Scholar]
- 47.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian S, Viatchenko-Karpinski S, Lukyanenko V, Gyorke S, Wiesner TF. Underlying mechanisms of symmetric calcium wave propagation in rat ventricular myocytes. Biophys J. 2001;80:1–11. doi: 10.1016/S0006-3495(01)75991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kauferstein S, Kiehne N, Erkapic D, Schmidt J, Hamm CW, Bratzke H, et al. A novel mutation in the cardiac ryanodine receptor gene (RyR2) in a patient with an unequivocal LQTS. Int J Cardiol. 2011;146:249–250. doi: 10.1016/j.ijcard.2010.10.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.