

Abstract
Small heterodimer partner (SHP) plays important roles in diverse biological processes by directly interacting with transcription factors and inhibiting their activities. SHP has been designated an orphan nuclear receptor, but whether its activity can be modulated by ligands has been a long-standing question. Recently, retinoid-related molecules, including 4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3Cl-AHPC), were shown to bind to SHP and enhance apoptosis. We have examined whether 3Cl-AHPC acts as an agonist and increases SHP activity in the repression of bile acid biosynthetic CYP7A1 and CYP8B1 genes and delineated the underlying mechanisms. Contrary to this expectation, micromolar concentrations of 3Cl-AHPC increased CYP7A1 expression but indirectly via p38 kinase signaling. Nanomolar concentrations, however, repressed CYP7A1 expression and decreased bile acid levels in HepG2 cells, and little repression was observed when SHP was down-regulated by small hairpin RNA. Mechanistic studies revealed that 3Cl-AHPC bound to SHP, increased the interaction of SHP with liver receptor homologue (LRH)-1, a hepatic activator for CYP7A1 and CYP8B1 genes, and with repressive cofactors, Brahma, mammalian Sin3a, and histone deacetylase-1, and, subsequently, increased the occupancy of SHP and these cofactors at the promoters. Mutation of Leu-100, predicted to contact 3Cl-AHPC within the SHP ligand binding pocket by molecular modeling, severely impaired the increased interaction with LRH-1, and repression of LRH-1 activity mediated by 3Cl-AHPC. 3Cl-AHPC repressed SHP metabolic target genes in a gene-specific manner in human primary hepatocytes and HepG2 cells. These data suggest that SHP may act as a ligand-regulated receptor in metabolic pathways. Modulation of SHP activity by synthetic ligands may be a useful therapeutic strategy.
Small heterodimer partner (SHP) (NR0B2) is an unusual orphan nuclear receptor that lacks a DNA-binding domain but contains a putative ligand-binding domain (LBD) (1). SHP acts as transcriptional corepressor by forming nonfunctional heterodimers with a number of DNA binding activators, including liver receptor homologue (LRH)-1, hepatic nuclear factor (HNF)-4, estrogen related receptor, estrogen receptor, forkhead box (Fox)a2, and p53, and inhibiting their transcriptional activities (2–6). Thus, SHP functions as a pleiotropic transcriptional regulator affecting diverse mammalian biological pathways, including lipid and glucose metabolism, energy homeostasis, cell proliferation, apoptosis, and sexual maturation (7–11). Of these reported biological functions, SHP plays a crucial role in maintaining cholesterol and bile acid levels by inhibiting gene expression of cholesterol 7α hydroxylase (CYP7A1) and sterol 12α hydroxylase (CYP8B1), two key enzymes for hepatic conversion of cholesterol to bile acids (9, 12–15). In response to elevated hepatic bile acid levels, SHP directly interacts with LRH-1, a DNA binding hepatic activator for CYP7A1 and CYP8B1, and inhibits expression of these genes (11, 13, 15, 16). We have identified molecular mechanisms by which SHP represses the expression of CYP7A1 by functioning as an epigenetic regulator. SHP coordinately recruits chromatin modifying repressive cofactors, including mammalian Sin3A (mSin3A)/histone deacetylase (HDAC) and nuclear receptor corepressor (N-CoR) corepressors, G9a methyltransferase, and the switch/sucrose nonfermentable (Swi/Snf)-Brahma (Brm) chromatin remodeling complex, which results in sequential histone modification and chromatin remodeling at the CYP7A1 promoter (17–19). We observed that SHP and these repressive cofactors are also recruited to the CYP8B1 gene, resulting in gene repression after bile acid treatment (20). G-protein pathway suppressor-2 (GPS2), a subunit of the N-CoR corepressor complex, was also recently shown to act as a SHP cofactor and to participate in differential regulation of the bile acid biosynthetic genes, CYP7A1 and CYP8B1 (21). We also found that the activity and stability of SHP are increased by posttranslational modifications of SHP in response to elevated bile acid levels in hepatocytes (20, 22).
Because SHP contains a putative LBD, it has been designated as an orphan nuclear receptor (1). However, whether SHP repression activity can be modulated by binding of lipid-soluble ligands has been a long-standing question. Recently, adamantly substituted retinoid-related molecules, 4-(3-(1-adamantyl)-4-hydroxyphenyl)-3-chlorocinnamic acid (3Cl-AHPC) and its derivatives, were reported to be potential SHP ligands in the regulation of cell growth and apoptosis (23, 24). Fontana's and Dawson's groups demonstrated that these atypical retinoid molecules bind to the LBD of SHP and modulate SHP activity in the regulation of cell growth and apoptosis in malignant cells (23–26). In line with these findings, structural and computational molecular modeling, combined with mutation analysis, was used to predict the interaction of these compounds at the putative ligand binding site of SHP (27). However, the molecular mechanisms by which these atypical retinoid compounds regulate hepatic SHP activity and the functional relevance of binding of these molecules to SHP in the regulation of metabolic pathways have not been established.
In this study, we examined whether 3Cl-AHPC directly binds to SHP and increases SHP activity in the repression of hepatic bile acid biosynthetic CYP7A1 and CYP8B1 genes and elucidated the underlying mechanisms. In molecular, biochemical, and functional studies using wild-type and LBD mutants of SHP combined with molecular modeling, we have obtained evidence suggesting that repression of metabolic genes, including CYP7A1 and CYP8B1, by the orphan receptor SHP, can be regulated by ligands.
Results
Effects of 3Cl-AHPC on CYP7A1 expression is dose dependent
A retinoid-related compound, 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN), was reported to bind to the LBD of SHP with high affinity (dissociation constant of 2.8 nm) and its analogs, including 3Cl-AHPC, were shown to effectively compete for the binding of AHPN to SHP (23, 24). Despite the high affinity of these retinoid molecules for SHP, substantially higher concentrations in the range of 1–50 μm were used in the apoptotic studies in tumor cells (23, 24, 26, 28). Therefore, we first examined the dose dependence of 3Cl-AHPC on the expression of a well-known SHP target gene, CYP7A1 (9, 12–15). In quantitative RT-PCR (q-RT-PCR) gene expression studies, nanomolar concentrations of 3Cl-AHPC resulted in decreased mRNA levels of CYP7A1 in a dose-dependent manner. However, opposite to our expectation, treatment with 10 μm 3Cl-AHPC increased mRNA levels of CYP7A1 (Fig. 1A). Retinoids are known to modulate cellular kinase signaling pathways (29, 30); and therefore, we examined effects of high concentrations of 3Cl-AHPC on the activation of p38 and c-Jun N-terminal kinase (JNK) kinases, which have been shown to activate and repress, respectively, CYP7A1 expression (31–35). Treatment with 10 μm 3Cl-AHPC dramatically increased levels of active phospho-p38 kinase, whereas the active form of JNK kinase was only modestly increased (Fig. 1B). Pretreatment with a p38 kinase inhibitor SB203589 markedly decreased the protein levels of active phospho-p38 kinase and largely abolished the 3Cl-AHPC-mediated increase in CYP7A1 mRNA (Fig. 1, C and D). These results demonstrate unexpected biphasic effects of dose on the regulation of CYP7A1 expression by 3Cl-AHPC. Although expression of CYP7A1 is suppressed at nanomolar concentrations of 3Cl-AHPC, activation of p38 kinase at micromolar concentrations resulted in increased CYP7A1 expression. Therefore, nanomolar concentrations of 3Cl-AHPC were used in this study, to determine whether SHP activity is modulated by binding of 3Cl-AHPC to the LBD of SHP.
Fig. 1.

Effect of 3Cl-AHPC on CYP7A1 expression is dose dependent. HepG2 cells were treated with increasing concentrations of 3Cl-AHPC (3-Cl) as indicated for 8 h and collected for q-RT-PCR or Western blot analysis. A, The mRNA levels of CYP7A1 were measured by q-RT-PCR and normalized to those of 36B4, sem (n = 6). B, Phosphorylated or total protein levels of p38 and JNK or tubulin as a loading control were detected by Western blot analysis. Statistical significance was measured using the Student's t test. *, P < 0.05; **, P < 0.01. C, HepG2 cells were pretreated with SB203589, a specific p38 inhibitor, for 3 h and then treated with 10 μm 3Cl-AHPC for 6 h, and cells were collected for q-RT-PCR or Western blot analysis. C, The mRNA levels of CYP7A1 were measured by q-RT-PCR and normalized to those of 36B4, sem (n = 3). D, Phosphorylated or total protein levels of p38 kinase were detected by Western blot analysis.
3Cl-AHPC inhibits CYP7A1 expression in a SHP-dependent manner resulting in decreased bile acid levels
SHP inhibits expression of CYP7A1 by directly interacting with a CYP7A1 activator, LRH-1, and inhibiting its transactivation ability (9, 12–15). As expected, activation of a Gal4 reporter by Gal4 DNA-binding domain-LRH-1, and the coactivator peroxisome proliferator-activated receptor γ coactivator (PGC)-1α was inhibited by SHP in HepG2 cells (Fig. 2A). Nanomolar concentrations of 3Cl-AHPC in the presence of SHP significantly increased repression of transactivation (Fig. 2A, lanes 2–4 and 5–7). Similarly, using the CYP7A1 promoter-luc reporter, SHP repression of CYP7A1 promoter activity was markedly enhanced when cells were treated with 3Cl-AHPC (Fig. 2B, lanes 3–6). Treatment with 3Cl-AHPC also significantly decreased mRNA levels of the endogenous CYP7A1 gene in HepG2 cells in a dose-dependent manner (Fig. 2C). Importantly, the decrease in CYP7A1 mRNA was largely blocked (Fig. 2D, right panel, lanes 2 and 4) when SHP was down-regulated by small hairpin RNA (Fig. 2D, left panel). Moreover, the decreased mRNA levels of CYP7A1 as a result of treatment by 3Cl-AHPC were blocked by 3A-AHPC, an analog of 3Cl-AHPC, which acts as an antagonist for apoptosis induced by 3Cl-AHPC in cancer cells (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) (23, 24). Because 3Cl-AHPC inhibited CYP7A1 gene expression, we determined whether bile acid levels were also decreased. In these experiments, a synthetic agonist for the bile acid receptor farnesoid X receptor, GW4064, was used as a control (Fig. 2E). Treatment with 3Cl-AHPC or GW4064 decreased CYP7A1 expression (Fig. 2F, left panel) and increased SHP expression (Supplemental Fig. 2). Consistent with decreased CYP7A1 expression, bile acid levels were significantly decreased in cells treated with 3Cl-AHPC or GW4064 (Fig. 2F, right panel). These results suggest that 3Cl-AHPC enhances SHP repression of CYP7A1 in a SHP-dependent manner, which results in decreased bile acid levels in HepG2 cells.
Fig. 2.

3Cl-AHPC inhibits CYP7A1 expression in a SHP-dependent manner resulting in decreased bile acid levels. A and B, HepG2 cells were transfected with plasmids as indicated (for plasmid amounts, see Materials and Methods) and treated with 3Cl-AHPC (3-Cl) or vehicle for 10 or 20 h, and reporter assays were performed. The values for firefly luciferase activities were normalized by dividing by values for β-galactosidase activities. The mean and sem are plotted (n = 3). C, HepG2 cells were treated with 3Cl-AHPC as indicated, and relative (rel) mRNA levels of CYP7A1 were measured by q-RT-PCR and normalized to those of 36B4, sem (n = 3). D, HepG2 cells were transfected with small hairpin (sh) pSUPER vector as indicated and 60 h later, cells were treated with 3Cl-AHPC overnight, and mRNA levels of SHP and CYP7A1 were measured by q-RT-PCR and normalized to those of 36B4, sem (n = 3). Consistent results were observed in two independent (each in triplicate) assays. E, Experimental outlines. HepG2 cells were treated with vehicle, 3Cl-AHPC, or GW4064, as a control, for 24 h, and cells were collected for measuring CYP7A1 mRNA levels and bile acid levels (F). A–F, Statistical significance was measured using the Student's t test. *, P < 0.05; **, P < 0.01. NS, Nonsignificant; DMSO, dimethylsulfoxide.
3Cl-AHPC increases SHP interaction with LRH-1 and repressive cofactors, Brm and HDAC1
We have shown that SHP plays an important role in the epigenetic regulation of CYP7A1 by recruiting histone modification and chromatin remodeling enzymes (17–19). In response to bile acid signaling, SHP coordinately recruits chromatin modifying repressive cofactors, HDAC-containing mSin3A and N-CoR corepressors, G9a methyltransferase, and the Brm-containing Swi/Snf chromatin remodeling complex, to the CYP7A1 promoter (17–19). To test whether 3Cl-AHPC alters interaction of SHP with these cofactors, HepG2 cells were treated with 3Cl-AHPC, and the interaction of SHP with Brm, HDAC1, and mSin3A was detected by coimmunoprecipitation (CoIP) assays (Fig. 3A). For Brm, HDAC1, or mSin3A, 3Cl-AHPC substantially increased the amount of flag-SHP present in the immunoprecipitates for each factor, whereas little flag-SHP was detected in the IgG control samples (Fig. 3B). In contrast, SHP interaction with G9a was not enhanced by 3Cl-AHPC treatment, whereas as shown before (19, 20, 36), interaction of SHP with G9a was increased by treatment with a primary bile acid, chenodeoxycholic acid (Fig. 3C). These results suggest that 3Cl-AHPC selectively increased interaction of SHP with its known chromatin modifying cofactors and DNA binding regulatory factors, increasing interaction with Brm, HDAC1, and mSin3A but not with G9a.
Fig. 3.

3Cl-AHPC increases SHP interaction with LRH-1 and repressive cofactors of SHP, Brm, HDAC1, and mSin3A. A, Experimental outlines. B and C, HepG2 cells were treated with 200 nm 3Cl-AHPC or 50 μm chenodeoxycholic acid for 2 h, and Brm, HDAC1, mSin3A, and G9a were immunoprecipitated from HepG2 cell extracts. Flag-SHP in the immunoprecipitates was detected by Western blot analysis using M2 antibody. D, Experimental outlines. E, HepG2 cell extracts were prepared and then treated with 1 μm 3Cl-AHPC in vitro for 30 min on ice to avoid the activation of cellular kinase signaling. Brm was immunoprecipitated from cell extracts, and the presence of flag-SHP was detected by M2 antibody (top). Flag-SHP was immunoprecipitated from cell extracts and the presence of LRH-1 was detected by Western blot analysis (bottom). In all CoIP assays, IgG was used as a negative control. F, GST-SHP or control GST, which had been expressed in bacteria BL21(DE3) and purified by Glutathione Sepharose, was incubated with 1 μm 3Cl-AHPC or vehicle for 30 min on ice and then further incubated with 35S-Brm or LRH-1 synthesized by in vitro transcriptional and translation in reticulocyte lysates. Interaction of GST-SHP with Brm or LRH-1 was detected by autoradiography. Similar amounts of GST-SHP were used in each reaction (data not shown). Band intensities were determined using ImageJ, and the values for control samples treated with vehicle were set to 1.
To directly test whether 3Cl-AHPC binds to SHP and to avoid the potential inadvertent activation of p38 kinase signaling in cells by 3Cl-AHPC, HepG2 cell extracts were prepared first and then treated in vitro with 3Cl-AHPC (Fig. 3D). Impressively, interaction between flag-SHP and Brm or LRH-1 was dramatically increased after treatment with 3Cl-AHPC in vitro (Fig. 3E). Consistent with these results, in glutathione S-transferase (GST) pull down assays, treatment with 3Cl-AHPC in vitro also markedly increased interaction of GST-SHP with 35S-labeled Brm or LRH-1 (Fig. 3F). Further, treatment with 3Cl-AHPC differentially affected the interaction of SHP with other SHP-interacting transcription factors, such as Foxa2 and HNF-4. Although the interaction with HNF-4 or Foxo-1 was not markedly changed, interaction of SHP with Foxa2 was substantially increased (Supplemental Fig. 3). These in vitro and in cell CoIP studies strongly suggest that 3Cl-AHPC binds directly to SHP and increases SHP interaction selectively with LRH-1 and chromatin modifying repressive cofactors, HDAC1, mSin3A, and Brm.
Treatment with 3Cl-AHPC increases occupancy of SHP at the CYP7A1 and CYP8B1 genes
Because 3Cl-AHPC increases the interaction of SHP with LRH-1, a docking protein for association of SHP with the CYP7A1 and CYP8B1 genes (9, 12–15), we next examined whether 3Cl-AHPC treatment increases the occupancy of SHP and its cofactors at the promoter by chromatin immunoprecipitation (ChIP) assays. Treatment of HepG2 cells with 3Cl-AHPC did not change occupancy of LRH-1 but markedly and significantly increased occupancy of flag-SHP and Brm at the CYP7A1 promoter (Fig. 4, A and B). In contrast, increased occupancy of G9a was not observed (Fig. 4A), which is in agreement with the lack of effect of 3Cl-AHPC on the interaction of G9a with SHP (Fig. 3C). In control experiments, occupancy of G9a was markedly increased in HepG2 cells or mouse liver after bile acid treatment (data not shown) (19, 20). Consistent with increased occupancy of flag-SHP and its repressive cofactors, RNA polymerase II occupancy was decreased (Fig. 4, A and B), and CYP7A1 mRNA levels were significantly decreased (Supplemental Fig. 4, lanes 1 and 2). Increased occupancy of adenoviral (Ad)-expressed flag-SHP (Supplemental Fig. 5A) or endogenous SHP at the CYP8B1 gene was also observed, whereas the occupancy at the control glyceraldehyde-3-phosphate dehydrogenase coding region was not detected (Fig. 4, C and D, and Supplemental Fig. 5B). Increased occupancy of HDAC1 and decreased occupancy of RNA polymerase II were also detected at the CYP7A1 and CYP8B1 promoter (data not shown). These results demonstrate that 3Cl-AHPC treatment increased occupancy of SHP and decreased occupancy of RNA polymerase II at the CYP7A1 and CYP8B1 genes, which correlates with repression of these two genes.
Fig. 4.

3Cl-AHPC treatment increases occupancy of SHP at the CYP7A1 and CYP8B1 genes. A and B, HepG2 cells were infected with Ad-flag-SHP; 24 h later, cells were treated with 100 nm 3Cl-AHPC or vehicle for 2 h, and occupancy of SHP and other proteins were examined by ChIP assays. Semi-q-PCR was performed to detect occupancy at the CYP7A1 promoter and the coding region of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a negative control. C and D, HepG2 cells were treated with 200 nm 3Cl-AHPC for 2 h. Occupancy at the CYP7A1 and CYP8B1 genes by endogenous SHP, LRH-1, and RNA polymerase II (Pol II) was examined by ChIP assays followed by semi-q-PCR. Reproducible results were observed from multiple independent ChIP assays as indicated. B and D, Band intensities were determined and the values for control samples treated with vehicle were set to 1.
Leu-100 is predicted to interact with 3Cl-AHPC within the ligand binding pocket of SHP and is important for the enhancement of SHP repression of LRH-1 by 3Cl-AHPC
Molecular modeling studies were performed to predict putative amino acid residues that interact with 3Cl-AHPC within the ligand binding pocket of SHP (Fig. 5A), as previously described, using the Prime program (37). To attempt to reduce the binding of 3Cl-AHPC, we mutated two of the amino acids, Leu-100 and Ala-148, to acidic amino acids. The two residues were also predicted to be in the ligand binding pocket of SHP in earlier modeling studies (27). The effects of these mutations on SHP repression of LRH-1 activity were examined using cell-based reporter assays. Mutation of Ala-148 had little effect on SHP activity in either Gal4-reporter assays (Fig. 5B) or reporter assays with the CYP7A1 promoter (Fig. 5C). In contrast, mutation of Leu-100 blocked the 3Cl-AHPC enhanced repression by SHP of both the Gal4-LRH-1 transactivation (Fig. 5B) and the CYP7A1 promoter (Fig. 5C). Similar expression levels of mutant and wild-type flag-SHP were detected (Fig. 5D). These data suggest that Leu-100 within the putative ligand binding pocket is important for enhancement by 3Cl-AHPC treatment of SHP repression of LRH-1.
Fig. 5.
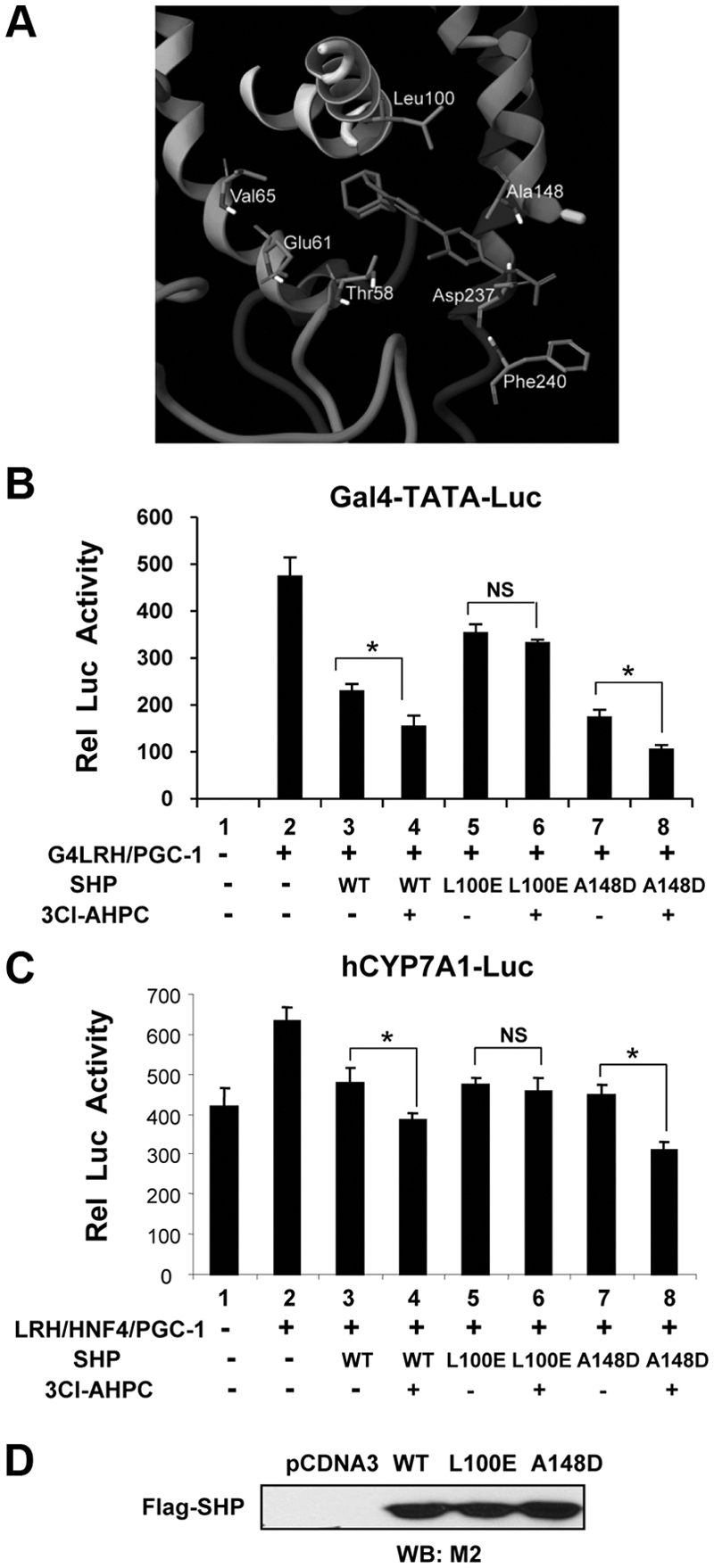
Leu-100 is predicted to contact 3Cl-AHPC within the SHP ligand binding pocket and is important for the enhancement of SHP repression of LRH-1 transactivation by 3Cl-AHPC. A, Predicted structure of the ligand binding pocket of mouse SHP occupied by 3Cl-AHPC with amino acid residues that contact 3Cl-AHPC indicated. The molecular model for the interaction of 3Cl-AHPC with the SHP was built by using the Prime program as described in Materials and Methods. Residues interacting with the ligand and 3Cl-AHPC are shown as stick models. B and C, Two amino acids, Leu-100 and Ala-148, identified from the molecular modeling as ligand interacting residues (A), were selected for functional cell-based reporter assays. HepG2 cells were cotransfected with Gal4-TATA-luc (B) or CYP7A1 promoter-luc (C) reporters along with expression plasmids, including the SHP mutants L100E or A148D as indicated (for plasmid amounts, see Materials and Methods). Twenty-four hours later, cells were treated with 200 nm 3Cl-AHPC or vehicle overnight, and reporter assays were performed. The values for firefly luciferase activities were normalized by dividing by the values for β-galactosidase activities. The mean and sem are plotted (n = 3). Statistical significance was measured using the Student's t test. *, P < 0.05. NS, Nonsignificant. D, Expression levels of flag-SHP wild type (WT) and mutants were detected by Western blot analysis (WB).
The 3Cl-AHPC-mediated enhanced interaction of SHP with LRH-1 is dramatically decreased by mutation of Leu-100
To determine whether impaired functional responses to 3Cl-AHPC after mutation of Leu-100 in SHP might be due to reduced interaction with LRH-1 or the cofactors, Brm and HDAC, the effects of the mutation on these interactions were examined in in vitro CoIP assays (Fig. 6A). The dramatically increased interaction between SHP wild type with LRH-1 after treatment with 3Cl-AHPC was largely abolished with the L100E mutant (Fig. 6B). Protein levels of the SHP L100E mutant were comparable to those of wild-type SHP (Fig. 6B, input). The enhanced interaction between SHP and Brm (Fig. 6C) or HDAC1 (Fig. 6D) after 3Cl-AHPC treatment was also markedly reduced with the L100E mutant. These results suggest that SHP-L100E, but not SHP wild type, is constitutively bound to HDAC1 and Brm and that 3Cl-AHPC does not further increase the interaction of SHP-L100E with the cofactors but does substantially increase the interaction of wild-type SHP with the cofactors. Taken together with those of the cell-based reporter assays (Fig. 5), these results are consistent with decreased binding of the L100E SHP mutant to 3Cl-AHPC. Similar expression levels of the mutant and SHP wild type and the lack of effect of the A148D mutant, which is near Leu-100 in the putative binding pocket and serves as a control, suggest that other effects, such as gross conformational changes, are not likely a major factor in the decreased response of the L100E mutant to 3Cl-AHPC.
Fig. 6.

Increased interaction of SHP with LRH-1 mediated by 3Cl-AHPC is severely impaired by mutation of Leu-100. A, Experimental outlines. B–D, HepG2 cell extracts were prepared and then treated in vitro with 3Cl-AHPC to avoid the activation of cellular kinase signaling, and then CoIP assays were performed. LRH-1, Brm, or HDAC1 was immunoprecipitated from cell extracts, and presence of flag-SHP was detected by M2 antibody. In all in vitro CoIP protein interaction assays, IgG was used as a negative control. Similar levels of LRH-1, Brm, and HDAC1 in the immunoprecipitated samples were detected by Western blot analysis (WB). Similar levels of the L100E mutant and SHP wild type (WT) in input samples were also confirmed by Western blot analysis. Band intensities were determined using ImageJ, and the values for control samples treated with vehicle were set to 1. Kd, Dissociation constant.
3Cl-AHPC inhibits expression of metabolic SHP target genes in a gene-specific manner
To examine the effects of 3Cl-AHPC on the expression of metabolic SHP target genes, primary human hepatocytes (PHH) as well as HepG2 cells were treated with 3Cl-AHPC, and mRNA levels of these genes were examined by q-RT-PCR analysis. Treatment of both PHH and HepG2 cells with 3Cl-APHC decreased mRNA levels of CYP7A1, CYP8B1, and apolipoprotein A1 (ApoA1), although the inhibition of ApoA1 in PHH was not statistically significant (Fig. 7, A and B). The levels of sterol regulatory element-binding protein (SREBP)-1c mRNA were also decreased by 3Cl-APHC treatment in HepG2 cells (Fig. 7B). These results are consistent with previous reports that ApoA1 and SREBP-1c, as well as CYP7A1 and CYP8B1, are repressed by the SHP/LRH-1 pathway (38, 39). In contrast, mRNA levels of the bile salt excretion pump and the gluconeogenic gene, glucose-6-phosphatase, were not significantly decreased in either PHH or HepG2 cells by 3Cl-AHPC treatment (Fig. 7, A and B). These data suggest that treatment with 3Cl-AHPC may inhibit expression of known SHP target genes in a gene-specific manner, although the underlying mechanism for the gene-specific regulation is unclear.
Fig. 7.

3Cl-AHPC inhibits expression of metabolic SHP target genes in a gene-specific manner. A and B, PHH (A) or HepG2 cells (B) were treated with 25, 100, or 200 nm 3Cl-AHPC or vehicle overnight, and cells were collected for gene expression studies. The mRNA levels of representative genes, as indicated, involved in bile acid biosynthesis, bile acid transport, cholesterol efflux, hepatic lipogenesis, and gluconeogenesis were measured by q-RT-PCR analysis as described in Materials and Methods. The mean and sem are plotted (n = 4). Statistical significance was measured using the Student's t test. *, P < 0.05. NS, Nonsignificant; DMSO, dimethylsulfoxide; BSEP, bile salt excretion pump.
Discussion
In this study, we present evidence suggesting that the activity of an orphan nuclear receptor, SHP, can be regulated by ligands in the repression of the hepatic bile acid biosynthetic CYP7A1 and CYP8B1 genes. In vitro and in-cell protein interaction studies revealed that 3Cl-AHPC binds to SHP, dramatically increases interaction of SHP with LRH-1, an orphan nuclear receptor and hepatic activator of CYP7A1 and CYP8B1 genes, and also increases selectively the interaction with chromatin modifying repressive cofactors, Brm and mSin3A/HDAC1, but not with G9a. Consistent with the increased interaction with LRH-1, a docking protein for SHP in many metabolic genes, including CYP7A1 and CYP8B1, occupancy of SHP was increased at the promoters of the CYP7A1 and CYP8B1 genes in HepG2 cells. Three lines of evidence suggest that 3Cl-AHPC binds as a ligand to SHP. First, protein interaction and functional studies combined with molecular modeling have revealed that mutation of Leu-100, a putative 3Cl-AHPC-interacting amino acid residue, reduced the effects of 3Cl-AHPC on SHP. Second, addition of 3Cl-AHPC to cell-free extracts in vitro enhanced SHP interactions with LRH-1 and the cofactors. Third, 3A-AHPC competes for binding of 3Cl-AHPC to SHP and antagonized the effect of 3Cl-AHPC on SHP repression. These results suggest that 3Cl-AHPC directly binds to the LBD of SHP and alters the interaction of SHP with its cofactors and with LRH-1.
An interesting finding in this study is that the dose-dependent effects of 3Cl-AHPC on regulation of CYP7A1 gene expression are biphasic. At micromolar concentrations that were used in the previous apoptotic studies in tumor cells (23, 24, 26, 28), 3Cl-AHPC increased CYP7A1 expression in HepG2 cells, which is the opposite of expected results. Although the binding affinity of 3Cl-AHPC to SHP was not directly measured, a related analog AHPN was reported to bind to SHP LBD with high affinity (dissociation constant of 2.8 nm), and 3Cl-AHPC efficiently competed for binding of AHPN to SHP (23). At nanomolar concentrations of 3Cl-AHPC, the activity of SHP and repression of CYP7A1 were enhanced in contrast to the opposite effects at micromolar concentrations. At the higher concentrations, p38 kinase was activated, and inhibition of p38 kinase blocked the increased expression of CYP7A1 in response to 3Cl-AHPC. In line with these findings, it had been shown that p38 kinase increases CYP7A1 expression by phosphorylating and subsequently by increasing nuclear abundance of a key CYP7A1 activator, HNF-4 (32). These results are most consistent with 3Cl-AHPC binding directly to SHP as an agonist to enhance CYP7A1 repression at nanomolar concentrations, whereas at micromolar concentrations, 3Cl-AHPC activates the p38 kinase and thereby indirectly increases CYP7A1 expression.
Based on the effects of nanomolar concentrations of 3Cl-AHPC, we propose that 3Cl-AHPC directly binds to SHP and dramatically increases SHP interaction with LRH-1, which results in increased occupancy of SHP at the promoter of the CYP7A1 and CYP8B1 genes and possibly other SHP/LRH-1 target genes, including ApoA1 and SREBP-1c genes (Fig. 8). Furthermore, 3Cl-AHPC selectively increases the interaction of SHP with the mSin3A/HDAC1 corepressors and Brm of the Swi/Snf chromatin remodeling complexes but not with G9a methyltransferase. Increased occupancy of SHP and repressive cofactors results in repression of CYP7A1, which is consistent with decreased occupancy of RNA polymerase II at the CYP7A1 promoter after 3Cl-AHPC treatment. Mutation of Leu-100, a residue that is predicted to contact 3Cl-AHPC within the ligand binding pocket based on molecular modeling, dramatically decreased SHP interaction with LRH-1 and Brm, resulting in impaired SHP repression of LRH-1 transactivation. Our data from biochemical, functional studies, and molecular modeling, taken together with previous findings (23, 24, 26), strongly support the conclusion that 3Cl-AHPC binds to the LBD of SHP and increases SHP interaction with LRH-1.
Fig. 8.

Model of the mechanism of the enhancement of SHP activity by 3Cl-AHPC. In this model, 3Cl-AHPC binds to the ligand binding pocket of SHP, which leads to a conformational change of SHP that allows increased interaction of SHP with LRH-1, a DNA binding activator and SHP-docking protein for CYP7A1 and other metabolic genes, such as CYP8B1, SREBP-1c, and ApoA1. Binding of 3Cl-AHPC also selectively increases interaction of SHP with chromatin modifying repressive cofactors, Brm, an ATPase of Swi/Snf chromatin remodeling complexes, and mSin3A/HDAC1 corepressors, but not with G9a lysine methyltransferase. Increased interaction between SHP and LRH-1 leads to increased occupancy of SHP, followed by increased occupancy of repressive cofactors of SHP and decreased occupancy of RNA polymerase II at the promoter of the CYP7A1 and CYP8B1 genes (and possibly other SHP/LRH-1 target genes, such as ApoA1 and SREBP-1c genes), resulting in gene repression. In contrast to this model for nanomolar concentrations of 3Cl-AHPC, micromolar concentrations results in increased CYP7A1 gene expression due to activation of the cellular p38 kinase.
The demonstration that SHP activity can be modulated by ligands raises an important question of whether SHP can be regulated physiologically by unidentified endogenous ligands. Endogenous ligands could potentially modulate SHP function in a gene-specific or metabolic pathway-specific manner or also in a tissue-specific manner, so that efforts to identify such endogenous ligands in target tissues are warranted. SHP function is known to be regulated at the level of gene expression, protein stability, and activity by posttranslational modification (8, 12, 13, 18, 20). Direct regulation of activity by ligand binding would provide an additional mechanism for the tight regulation of this important transcriptional regulatory protein.
SHP has been shown to be an important regulator of cellular metabolism, including bile acid and cholesterol homeostasis, hepatic lipogenesis, and gluconeogenesis (7–11). Mutations of SHP in humans are associated with metabolic abnormalities, such as early onset of diabetes, fatty liver, and insulin resistance (40–42), further demonstrating the important role of SHP in metabolic regulation. The demonstration that SHP activity can be regulated by retinoid-related compounds suggests that SHP may be a potential therapeutic target for the development of small molecules to treat metabolic disease. From gene expression studies, we observed that repression of CYP7A1 and other SHP target genes involved in metabolic pathways are selectively modulated by 3Cl-AHPC, which raised the intriguing possibility of therapeutic agents that target SHP and selectively affect different metabolic pathways. Although the underlying mechanism for the gene-specific effects by 3Cl-AHPC was not delineated in this study, our data suggest that synthetic ligands of SHP may be developed to target specific metabolic pathways and, thus, to selectively treat SHP-related human diseases, such as metabolic syndrome, cholestasis, cancer, and infertility. Furthermore, the demonstration that high concentrations of 3Cl-AHPC activate p38 kinase signaling illustrates the need to develop synthetic ligands selective for binding to SHP with diminished effects on cellular kinase activity to reduce side effects of the drugs.
Materials and Methods
Reagents
Antibodies for SHP (sc30169), LRH-1 (sc-5995 X), HDAC1 (sc-7872), mSin3A (sc-994), Brm (sc6450), tubulin (sc-8035), and RNA polymerase II (sc-9001) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). M2 and G9a antibodies were purchased from Sigma (St. Louis, MO) and Upstate Biotechnology (Waltham, MA), respectively. Antibodies for total and phosphorylated p38 and JNK were purchased from Cell Signaling (Beverly, MA). Kinase inhibitors were purchased from Sigma. 3Cl-AHPC was synthesized as previously described (23, 24, 26).
Plasmid and adenoviral vector constructs
Flag-mouse SHP mutants were constructed by site-directed mutagenesis (Stratagene, Inc., La Jolla, CA) and confirmed by sequencing. Adenovirus (Ad)-flag-SHP wild type and Ad-flag-SHP L100E mutant were constructed with the Ad-CMV-Track system, and recombinant Ad vectors were amplified in Ad-293 cells. The viruses were isolated by CsCl step gradient centrifugation, and total viral particles were determined by absorbance at A260 (one A260 unit is approximately 1 × 1012 particles), and the number of active viral particles was determined by titering and determining the number of cells expressing green fluorescent protein by confocal microscopy.
Cell culture and cell-based reporter assays
HepG2 cells (HB8065; American Type Culture Collection, Manassas, VA) were maintained in DMEM/F12 (1:1) media. PHH were obtained from the Liver Tissue Procurement and Distribution System of the National Institutes of Health (S. Strom, University of Pittsburgh, Pittsburgh, PA). PHH cells were maintained in modified William's E medium (Clonetics, Inc., San Diego, CA) as previously described (43). For reporter assays, HepG2 cells were cotransfected with 200-ng −1887 human CYP7A1-luc reporter or Gal4-TATA-luc along with 300 ng of expression vectors of CMV β-galactosidase as an internal control, 50–100 ng of expression plasmids for HNF-4, LRH-1, and PGC-1α, 50 ng of Gal4 DNA-binding domain-LRH-1, and 10–25 ng of pcDNA3-flag-mouse SHP. Transfection was carried out using lipofectamine 2000 in 24-well plates. Cells were treated with 100–200 nm 3Cl-AHPC overnight, and firefly luciferase activities were divided by β-galactosidase activities to normalize for transfection efficiency. Consistent results were observed in two (CYP7A1-luc) or four (Gal4-luc system) independent triplicate assays.
Measurement of bile acid levels in HepG2 cells
HepG2 cells (three 15-cm plates for each group) were maintained in serum-free media overnight followed by the treatment with dimethylsulfoxide, 200 nm 3Cl-AHPC, or 200 nm GW4064, as a control, for 24 h. Cells were collected and sonicated in 70% ethanol. Cell lysates were incubated at 50 C overnight and centrifuged (13,000 × g, 5 min). The supernatants from lysates were used for bile acid assays. Total bile acids were analyzed using the Total Bile Acid Assay kit (Trinity Biotechnology, Jamestown, NY) according to the manufacturer's instruction.
Real-time RT-PCR
HepG2 cells were incubated with serum-free media overnight and then treated with different concentrations of 3Cl-AHPC for 8 h to overnight as indicated in figure legends. Total RNA was isolated from HepG2 cells or PHH using Trizol reagent, and cDNA was synthesized using a reverse transcriptase kit (Promega, Madison, WI). Real-time RT-PCR was performed with an iCycler iQ (Bio-Rad, Inc., Hercules, CA) following the manufacturer's instructions. The mRNA levels of each gene were normalized by dividing by those of 36B4. Sequences of the primers are available upon request.
Protein structure modeling
An active SHP model was built by using the Prime program. The crystal structure of COUP-TFII (Protein Data Bank code, 3CJW) as described previously (37) and RXRα (Protein Data Bank code, 1FM9) were retrieved from the Research Collaboratory for Structural Bioinformatics website (www.rcsb.org) and used as a template for this active model. The structure of 3Cl-AHPC was built and minimized with OPLS 2005 force field in Maestro (Maestro, version 8.5; Schrödinger, LLC, New York, NY) interface. The grid-enclosing box was centered in the LBD with approximate dimensions of 25 Å × 25 Å × 25 Å. A scaling factor of 1.0 was set to van der Waals radii of those receptor atoms with the partial atomic charge less than 0.25. The minimized 3Cl-AHPC was docked into the ligand binding pocket of SHP using the Glide (version 5.0; Schrödinger, LLC) software package with standard parameters.
CoIP assay
For in vitro CoIP assays, cell extracts were prepared first and then treated with 3Cl-AHPC or vehicle on ice for 30 min. For in-cell CoIP assays, HepG2 cells were treated with 3Cl-AHPC or vehicle for 2 h, and cell extracts were prepared. Cell extracts were incubated in lysis buffer [20 mm Tris-HCl (pH 8.0), 0.5 mm EDTA, 5% glycerol, 150 mm NaCl, 0.3% Nonidet P-40, 1 mm dithiothreitol, and protease inhibitors] with antibodies against chromatin modifying cofactors or SHP or rabbit IgG at 4 C for 4 h to overnight, and the immune complex was collected by incubation with 30 μl of a 25% slurry of protein G agarose for 2 h. Immunoprecipitates were washed four times with lysis buffer supplemented with NaCl to 250 mm and subjected to Western blotting.
ChIP assays
Confluent HepG2 cells in 15-cm plates (2 × 107) were infected at a multipicity of infection of 5–10 with adenoviruses, and 24 h after infection, cells were treated with 3Cl-AHPC or vehicle for 2 h and collected for ChIP assays. ChIP assays in HepG2 cells were carried out essentially as described in previous studies (19, 44, 45). Experiments were repeated multiple times as indicated in figure legends with consistent results. Sequences of the primers are available upon request.
Supplementary Material
Acknowledgments
We thank Yoon Kwang Lee and David Moore for expression plasmids for SHP and G4-LRH-1 and Gal4-TATA-Luc, Bryan Goodwin for pcDNA3 LRH-1, John Chiang for −1887 human CYP7A1-luc, Bruce Spiegelman for pcDNA3 PGC-1α, Anthony Imbalzano and Said Sif for the Brm expression vector, Tiangang Li and John Chiang for kindly providing a protocol for measuring bile acid levels in HepG2 cells, the Liver Tissue Procurement and Distribution System funded by the National Institutes of Health Contract N01-DK-7-0004 (Stephen Strom at University of Pittsburgh, Pittsburgh, PA) for providing human primary hepatocytes, and Byron Kemper for critical comments on the manuscript.
This work was supported by National Institutes of Health Grants DK062777 and DK080032, and by an American Diabetes Association Basic Science award (J.K.K.).
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: SHP | LRH-1;
Coregulators: BRM | Sin3A | HDAC1;
Ligands: GW4064.
Footnotes
- Ad
- Adenoviral
- AHPN
- 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid
- ApoA1
- apolipoprotein A1
- Brm
- Brahma
- ChIP
- chromatin immunoprecipitation
- 3Cl-AHPC
- 4-(3-(1-adamantyl)-4-hydroxyphenyl)-3-chlorocinnamic acid
- CoIP
- coimmunoprecipitation
- CYP7A1
- cholesterol 7α hydroxylase
- CYP8B1
- sterol 12α hydroxylase
- Fox
- forkhead box
- GPS2
- G-protein pathway suppressor-2
- GST
- glutathione S-transferase
- HDAC
- histone deacetylase
- HNF
- hepatic nuclear factor
- JNK
- c-Jun N-terminal kinase
- LRH
- liver receptor homologue
- mSin3a
- mammalian mSin3a
- N-CoR
- nuclear receptor corepressor
- PGC
- peroxisome proliferator-activated receptor γ coactivator
- PHH
- primary human hepatocyte
- q-RT-PCR
- quantitative RT-PCR
- SHP
- small heterodimer partner
- SREBP
- sterol regulatory element-binding protein
- Swi/Snf
- switch/sucrose nonfermentable.
References
- 1. Seol W, Choi HS, Moore DD. 1996. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science 272:1336–1339 [DOI] [PubMed] [Google Scholar]
- 2. Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. 2000. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol Cell Biol 20:187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee YK, Moore DD. 2002. Dual mechanism for repression of the monomeric orphan receptor liver receptor homologous protein-1 (LRH-1) by the orphan small heterodimer partner (SHP). J Biol Chem 277:2463–2467 [DOI] [PubMed] [Google Scholar]
- 4. Johansson L, Båvner A, Thomsen JS, Färnegårdh M, Gustafsson JA, Treuter E. 2000. The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors. Mol Cell Biol 20:1124–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim JY, Kim HJ, Kim KT, Park YY, Seong HA, Park KC, Lee IK, Ha H, Shong M, Park SC, Choi HS. 2004. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding. Mol Endocrinol 18:2880–2894 [DOI] [PubMed] [Google Scholar]
- 6. Lee J, Padhye A, Sharma A, Song G, Miao J, Mo YY, Wang L, Kemper JK. 2010. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J Biol Chem 285:12604–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Båvner A, Sanyal S, Gustafsson JA, Treuter E. 2005. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab 16:478–488 [DOI] [PubMed] [Google Scholar]
- 8. Chanda D, Park JH, Choi HS. 2008. Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocr J 55:253–268 [DOI] [PubMed] [Google Scholar]
- 9. Schoonjans K, Auwerx J. 2002. A sharper image of SHP. Nat Med 8:789–791 [DOI] [PubMed] [Google Scholar]
- 10. Volle DH, Decourteix M, Garo E, McNeilly J, Fenichel P, Auwerx J, McNeilly AS, Schoonjans K, Benahmed M. 2009. The orphan nuclear receptor small heterodimer partner mediates male infertility induced by diethylstilbestrol in mice. J Clin Invest 119:3752–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I. 2005. Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J 24:2624–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515 [DOI] [PubMed] [Google Scholar]
- 13. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526 [DOI] [PubMed] [Google Scholar]
- 14. Moore DD. 2002. Does loss of bile acid homeostasis make mice melancholy? J Clin Invest 110:1067–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiang JYL. 2002. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr Rev 23:443–463 [DOI] [PubMed] [Google Scholar]
- 16. Fayard E, Auwerx J, Schoonjans K. 2004. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol 14:250–260 [DOI] [PubMed] [Google Scholar]
- 17. Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. 2004. Role of a mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol 24:7707–7719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miao J, Xiao Z, Kanamaluru D, Min G, Yau PM, Veenstra TD, Ellis E, Strom S, Suino-Powell K, Xu HE, Kemper JK. 2009. Bile acid signaling pathways increase stability of small heterodimer partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev 23:986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. 2007. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol 27:1407–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanamaluru D, Xiao Z, Fang S, Choi SE, Kim DH, Veenstra TD, Kemper JK. 2011. Arginine methylation by PRMT5 at a naturally-occurring mutation site is critical for liver metabolic regulation by small heterodimer partner. Mol Cell Biol 31:1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanyal S, Båvner A, Haroniti A, Nilsson LM, Lundåsen T, Rehnmark S, Witt MR, Einarsson C, Talianidis I, Gustafsson JA, Treuter E. 2007. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc Natl Acad Sci USA 104:15665–15670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miao J, Fang S, Lee J, Comstock C, Knudsen KE, Kemper JK. 2009. Functional specificities of Brm and Brg-1 Swi/Snf ATPases in the feedback regulation of hepatic bile acid biosynthesis. Mol Cell Biol 29:6170–6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Farhana L, Dawson MI, Leid M, Wang L, Moore DD, Liu G, Xia Z, Fontana JA. 2007. Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor. Cancer Res 67:318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dawson MI, Xia Z, Liu G, Ye M, Fontana JA, Farhana L, Patel BB, Arumugarajah S, Bhuiyan M, Zhang XK, Han YH, Stallcup WB, Fukushi J, Mustelin T, Tautz L, Su Y, Harris DL, Waleh N, Hobbs PD, Jong L, Chao WR, Schiff LJ, Sani BP. 2007. An adamantyl-substituted retinoid-derived molecule that inhibits cancer cell growth and angiogenesis by inducing apoptosis and binds to small heterodimer partner nuclear receptor: effects of modifying its carboxylate group on apoptosis, proliferation, and protein-tyrosine phosphatase activity. J Med Chem 50:2622–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dawson MI, Xia Z, Jiang T, Ye M, Fontana JA, Farhana L, Patel B, Xue LP, Bhuiyan M, Pellicciari R, Macchiarulo A, Nuti R, Zhang XK, Han YH, Tautz L, Hobbs PD, Jong L, Waleh N, Chao WR, Feng GS, Pang Y, Su Y. 2008. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis, and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J Med Chem 51:5650–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farhana L, Dawson MI, Dannenberg JH, Xu L, Fontana JA. 2009. SHP and Sin3A expression are essential for adamantyl-substituted retinoid-related molecule-mediated nuclear factor-κB activation, c-Fos/c-Jun expression, and cellular apoptosis. Mol Cancer Ther 8:1625–1635 [DOI] [PubMed] [Google Scholar]
- 27. Cellanetti M, Gunda V, Wang L, Macchiarulo A, Pellicciari R. 2010. Insights into the binding mode and mechanism of action of some atypical retinoids as ligands of the small heterodimer partner (SHP). J Comput Aided Mol Des 24:943–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y, Soto J, Park K, Viswanath G, Kuwada S, Abel ED, Wang L. 2010. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol Cell Biol 30:1341–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pfahl M, Piedrafita FJ. 2003. Retinoid targets for apoptosis induction. Oncogene 22:9058–9062 [DOI] [PubMed] [Google Scholar]
- 30. Olson JM, Hallahan AR. 2004. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med 10:125–129 [DOI] [PubMed] [Google Scholar]
- 31. Li T, Jahan A, Chiang JY. 2006. Bile acids and cytokines inhibit the human cholesterol 7α-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology 43:1202–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu Z, Tavares-Sanchez OL, Li Q, Fernando J, Rodriguez CM, Studer EJ, Pandak WM, Hylemon PB, Gil G. 2007. Activation of bile acid biosynthesis by the p38 mitogen-activated protein kinase (MAPK): hepatocyte nuclear factor-4α phosphorylation by the p38 MAPK is required for cholesterol 7α-hydroxylase expression. J Biol Chem 282:24607–24614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA. 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 17:1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gupta S, Stravitz RT, Dent P, Hylemon PB. 2001. Down-regulation of cholesterol 7 a-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-jun N-terminal kinase pathway. J Biol Chem 276:15816–15822 [DOI] [PubMed] [Google Scholar]
- 35. Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, Wei P, Heyman RA, Karin M, Moore DD. 2002. Redundant pathways for negative feedback regulation of bile acid production. Developmental Cell 2:721–731 [DOI] [PubMed] [Google Scholar]
- 36. Boulias K, Talianidis I. 2004. Functional role of G9a-induced histone methylation in small heterodimer partner-mediated transcriptional repression. Nucleic Acids Res 32:6096–6103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kruse SW, Suino-Powell K, Zhou XE, Kretschman JE, Reynolds R, Vonrhein C, Xu Y, Wang L, Tsai SY, Tsai MJ, Xu HE. 2008. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol 6:e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. 2004. Identification of liver receptor homolog-1 as a novel regulator of apolipoprotein AI gene transcription. Mol Endocrinol 18:2378–2387 [DOI] [PubMed] [Google Scholar]
- 39. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. 2004. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishigori H, Tomura H, Tonooka N, Kanamori M, Yamada S, Sho K, Inoue I, Kikuchi N, Onigata K, Kojima I, Kohama T, Yamagata K, Yang Q, Matsuzawa Y, Miki T, Seino S, Kim MY, Choi HS, Lee YK, Moore DD, Takeda J. 2001. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc Natl Acad Sci USA 98:575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Echwald SM, Andersen KL, Sørensen TI, Larsen LH, Andersen T, Tonooka N, Tomura H, Takeda J, Pedersen O. 2004. Mutation analysis of NR0B2 among 1545 Danish men identifies a novel c. 278G>A (p.G93D) variant with reduced functional activity. Hum Mutat 24:381–387 [DOI] [PubMed] [Google Scholar]
- 42. Enya M, Horikawa Y, Kuroda E, Yonemaru K, Tonooka N, Tomura H, Oda N, Yokoi N, Yamagata K, Shihara N, Iizuka K, Saibara T, Seino S, Takeda J. 2008. Mutations in the small heterodimer partner gene increase morbidity risk in Japanese type 2 diabetes patients. Hum Mutat 29:E271–E277 [DOI] [PubMed] [Google Scholar]
- 43. Song KH, Ellis E, Strom S, Chiang JY. 2007. Hepatocyte growth factor signaling pathway inhibits cholesterol 7α-hydroxylase and bile acid synthesis in human hepatocytes. Hepatology 46:1993–2002 [DOI] [PubMed] [Google Scholar]
- 44. Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. 2004. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1α. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem 279:45139–45147 [DOI] [PubMed] [Google Scholar]
- 45. Fang S, Tsang S, Jones R, Ponugoti B, Yoon H, Wu SY, Chiang CM, Willson TM, Kemper JK. 2008. The p300 acetylase is critical for ligand-activated farnesoid X receptor (FXR) induction of SHP. J Biol Chem 283:35086–35095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.