

Abstract
The polymeric immunoglobulin receptor (pIgR) transports IgA antibodies across intestinal epithelial cells (IECs). Expression of pIgR is upregulated by proinflammatory signaling pathways via activation of nuclear factor-κB (NF-κB). Here, we examined the contributions of the RelA-dependent classical and RelB-dependent alternative pathways of NF-κB to pIgR regulation in the HT-29 human IEC line following stimulation with tumor necrosis factor (TNF), lipopolysaccharide (LPS; Toll-like receptor 4 (TLR4) ligand), and polyinosinic: polycytidylic acid (pIC; TLR3 ligand). Whereas induction of proinflammatory genes such as interleukin-8 (IL-8) required only RelA, pIgR expression was regulated by complex mechanisms that involved both RelA and RelB. Upregulation of pIgR expression by ligation of the lymphotoxin-β receptor suggested a direct role for the alternative NF-κB pathway. Inhibition of mitogen-activated protein kinases reduced the induction of IL-8, but enhanced the induction of pIgR by TNF and TLR signaling. Regulation of pIgR through unique signaling pathways could allow IECs to sustain high levels of IgA transport while limiting the proinflammatory responses.
Introduction
The intestinal epithelium serves as a biologically active barrier between microbes in the lumen and immune cells in the lamina propria.1, 2, 3 Secretory antibodies of the IgA class (SIgA) provide the first line of adaptive immune protection in the gut against pathogenic microbes, and prevent commensal bacteria from translocating into the lamina propria and eliciting proinflammatory responses.4 The polymeric immunoglobulin receptor (pIgR) helps to maintain mucosal barrier integrity and intestinal homeostasis by transporting polymeric IgA antibodies across intestinal epithelial cells (IECs) into gut secretions.5, 6 Proteolytic cleavage of pIgR at the apical surface of IECs releases the extracellular domain of pIgR, known as the secretory component, either in free form or as part of the SIgA complex. The secretory component enhances innate defense mechanisms by prevention of bacterial adherence to the intestinal mucous layer and neutralization of potential proinflammatory factors.7 Because one molecule of pIgR is consumed for every molecule of SIgA transported into the lumen, high expression of pIgR is necessary for a continuous supply of SIgA and secretory component.
Recent evidence suggests that nuclear factor-κB (NF-κB) signaling in IECs is critical for maintenance of epithelial barrier function, production of antimicrobial peptides, and modulation of immune responses.8 Among the genes activated by the classical NF-κB pathway are proinflammatory chemokines and cytokines, which are rapidly induced and then downregulated within hours by negative regulatory pathways.9 In contrast, the NF-κB-dependent induction of pIgR is slow and sustained,10, 11, 12 and steady-state levels of pIgR mRNA in mouse and human IECs are very high.13, 14 The NF-κB family of transcription factors comprises 5 subunits, designated RelA (p65), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2), which associate to form as many as 15 homo- and heterodimers.15 All NF-κB dimers bind to κB sites with a loosely conserved consensus sequence ( Table 1). Activation of the RelA-dependent classical NF-κB pathway by proinflammatory cytokines and Toll-like receptor (TLR) ligands leads to degradation of inhibitor of NF-κBα (IκBα), followed by phosphorylation and nuclear translocation of RelA/p50 dimers, and activation of gene transcription.15, 16, 17
Table 1. κB Sites in target genes.
| κB Sitea | Sequence compared with RelA consensus (5′-3′)b | Reference |
|---|---|---|
| RelA Consensus | GGGRNWYYCC | 18 |
| RELB promoter −247 | GGGGTTTTCC | 19 |
| RELB promoter −175 | GGGGAATTCC | |
| REL (c-Rel) promoter −578 | GGGGGTCCCC | |
| REL (c-Rel) promoter −442 | GGGAAcCaCC | |
| REL (c-Rel) promoter −278 | GGGATTTCtC | |
| REL (c-Rel) promoter +6 | GGGAAATTCC | 20 |
| NF-κB1/p50 promoter −288 | GGGGCTTCCC | |
| NF-κB1/p50 promoter −103 | GGGcTTCCCC | |
| NF-κB2/p52 promoter −110 | GGGAATTCCC | |
| NF-κB2/p52 promoter −80 | GGGcTTTCCC | 21 |
| NFKBIA (IκBα) promoter −26 | GaGACATCCC | |
| NFKBIA (IκBα) promoter −55 | GGaAATTCCC | |
| NFKBIA (IκBα) promoter −311 | GGGAAACCCC | 22 |
| IL8 promoter −82 | tGGAATTTCC | 23 |
| TNF promoter −873 | GGGACcCCCC | |
| TNF promoter −627 | GtGACTTCCC | |
| TNF promoter −598 | GGGcTgTCCC | 24 |
| A20 promoter −66 | GGaAAgTCCC | |
| A20 promoter −54 | GGaAATCCCC | 25 |
| PIGR intron 1 +4395 | GGGAAATTCC | 10 |
| NF-κB reporter plasmid | GGGAATTTCC | Stratagene |
Abbreviations: IκB, inhibitor of NF-κB; IL, interleukin; NF, nuclear factor; PIGR, polymeric immunoglobulin receptor; TNF, tumor necrosis factor.
The position of the κB site refers to bp upstream (−) or downstream (+) of the transcription start site in the corresponding human gene.
Nucleotides that do not match the κB consensus are shown in lower case (R, purine; Y, pyrimidine; W, A or T; and N, any nucleotide).
We previously demonstrated that induction of transcription of the PIGR gene in a human IEC line by tumor necrosis factor (TNF) and TLR signaling requires a κB element in the first intron.10, 12, 26 The sequence of this κB site is a perfect match to the RelA consensus sequence ( Table 1). These findings led us to hypothesize that the RelA subunit of NF-κB is the major transcription factor required for induction of pIgR by TNF and TLR signaling. To test this hypothesis, we examined the ability of TNF and TLR ligands to induce pIgR expression in a human intestinal epithelial cell line in which the expression of RelA was inhibited by stable transfection of a RelA-specific small inhibitory RNA (siRNA). For comparison, we also examined the role of RelB, which is activated by the alternative NF-κB pathway.16, 27, 28
Results
Activation of the classical NF-κB pathway by TNF and TLR signaling leads to upregulation of pIgR expression
To activate the classical RelA-dependent pathway of NF-κB activation, the human IEC line HT-29 was stimulated with TNF or ligands for TLR4 (lipopolysaccharide (LPS)) or TLR3 (polyinosinic:polycytidylic acid (pIC); Figure 1). Consistent with our previously published work,11, 12 we observed a rapid increase in the mRNA encoding the proinflammatory chemokine interleukin-8 (IL-8). Inhibition of IL-8 induction by BAY 11-7082, a small molecule that blocks the phosphorylation of IκBα,29, 30 suggested that the early proinflammatory response involved activation of the classical NF-κB pathway. The varying magnitude of IL-8 induction by TNF, LPS, and pIC suggested that there were stimulus-specific quantitative differences in NF-κB activation and/or activation of additional signaling pathways. In additional data not shown, we found that expression of IL-8 was downregulated by 24 h, consistent with our previous findings.11, 12 In contrast, induction of pIgR was delayed until 24 h, and the magnitude was similar for all three stimuli. Moreover, inhibition of pIgR induction by BAY 11-7082 suggested that early activation of the classical pathway of NF-κB activation was critical for subsequent upregulation of pIgR mRNA. Similar results were observed in another human IEC line (Supplementary Figure S1 online).
Figure 1.

Activation of the classical nuclear factor-κB (NF-κB) pathway by tumor necrosis factor (TNF), lipopolysaccharide (LPS), and polyinosinic:polycytidylic acid (pIC) in intestinal epithelial cells (IECs). HT-29 cells were stimulated with TNF (10 ng ml–1), LPS (1 μg ml–1), or pIC (100 μg ml–1) for 3 or 24 h, in the presence or absence of 10 μ BAY 11-7082, an inhibitor of IκBα phosphorylation. Concentrations of TNF, LPS, and pIC were optimized previously,11, 12 and the concentration of BAY 11-7082 was optimized in preliminary experiments (data not shown). mRNA levels for interleukin-8 (IL-8) and polymeric immunoglobulin receptor (pIgR) were quantified by quantitative reverse transcriptase-PCR (qRT-PCR) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Data from three independent experiments were combined and expressed as mean ± s.e.m. (n=9): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells (P<0.05); b, the mean for cells treated with BAY 11-7082 is significantly different from the mean for cells given the same stimulus in the absence of BAY 11-7082 (P<0.05).
Knockdown of RelA expression in HT-29 cells inhibits activation of the classical NF-κB pathway
Activation of the classical NF-κB pathway results in proteolytic degradation of the IκBα, leading to phosphorylation of RelA and translocation of active RelA/p50 dimers to the nucleus and enhanced transcription of RelA target genes.15 To inhibit this pathway, HT-29 cells were stably transfected with a plasmid encoding siRNA specific for RelA. For comparison, HT-29 cells were stably transfected with siRNA specific for RelB or a control siRNA with a random sequence. Because similar results were observed with multiple subclones of siRNA-transfected HT-29 cells (data not shown), one representative subclone for each siRNA was used in all subsequent experiments. Expression of RelA and RelB mRNA (Figure 2a) and protein (Figure 3a, b, d) was decreased in cells expressing the corresponding siRNA. RelA knockdown also caused a significant decrease in levels of RelB mRNA and protein, consistent with regulation of the human RELB gene by binding of RelA/p50 dimers to κB sites in its promoter ( Table 1). In contrast, knockdown of RelB resulted in only a minor decrease in RelA mRNA and no change in the level of RelA protein. Stimulation with TNF, LPS, or pIC did not alter levels of RelA mRNA or protein in either control cells or RelA knockdown cells (Figures 2b and 3a). Stimulation of control cells with TNF, LPS, or pIC caused a significant increase in RelB, c-Rel, p50, and p52 mRNA (Figure 2b), consistent with the presence of κB sites in the promoters of these genes ( Table 1). Knockdown of either RelA or RelB blocked the increase in RelB protein following stimulation with TLR ligands, and delayed the induction of RelB in response to TNF stimulation (Figure 3b). Taken together, these results suggest that knockdown of RelA in IECs alters the cellular pools of other NF-κB subunits, particularly RelB.
Figure 2.

Effects of RelA and RelB knockdown on mRNA levels of nuclear factor-κB (NF-κB) subunits. HT-29 cells were stably transfected with expression plasmids encoding small inhibitory RNA (siRNA) specific for RelA, RelB, or an irrelevant control sequence. (a) siRNA-mediated knockdown of RelA and RelB. mRNA levels were quantified by quantitative reverse transcriptase-PCR (qRT-PCR) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Data from four independent experiments were combined and expressed as mean ± s.e.m. (n=14): a, the mean is significantly less than the mean for cells transfected with control siRNA (P<0.05); b, the mean for cells transfected with RelB siRNA is significantly different from the mean for cells transfected with RelA siRNA (P<0.05). (b) mRNA levels for NF-κB subunits following stimulation with tumor necrosis factor (TNF; 10 ng ml–1), lipopolysaccharide (LPS; 1 μg ml–1), or polyinosinic:polycytidylic acid (pIC; 100 μg ml–1). mRNA levels were analyzed in HT-29 cells stably transfected with control, RelA, or RelB siRNAs and treated with the indicated stimulus for 3 or 24 h. Data from two independent experiments were combined and expressed as mean ± s.e.m. (n=8): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells expressing the same siRNA (P<0.05); b, the mean for cells expressing RelA or RelB siRNA is significantly different from the mean for cells expressing control siRNA given the same stimulus for the same length of time (P<0.05).
Figure 3.
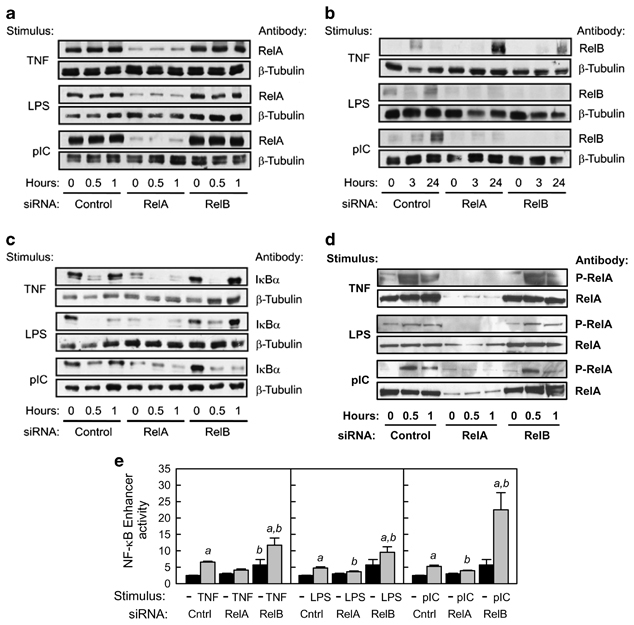
Effects of RelA and RelB knockdown on levels of RelA, RelB, and inhibitor of NF-κBα (IκBα) proteins and nuclear factor-κB (NF-κB) enhancer activity. (a–c) Whole-cell extracts were prepared from HT-29 cells stably transfected with the indicated small inhibitory RNA (siRNA) plasmid and treated with tumor necrosis factor (TNF; 10 ng ml–1), lipopolysaccharide (LPS; 1 μg ml–1), or polyinosinic:polycytidylic acid (pIC; 100 μg ml–1) for the indicated time. Approximately 40 μg of protein per lane was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and immunoblotted with rabbit polyclonal IgG antibodies to RelA, RelB, or IκBα. As a control for protein loading, parallel membranes were immunoblotted with rabbit polyclonal IgG antibody to β-tubulin. (d) Nuclear extracts were prepared from HT-29 cells stably transfected with the indicated siRNA plasmid and treated with TNF (10 ng ml–1), LPS (1 μg ml–1), or pIC (100 μg ml–1) for the indicated time. Approximately 25 μg of protein per lane was separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with rabbit polyclonal IgG antibodies to total RelA or RelA phosphorylated at Ser 536 (P-RelA). (e) HT-29 cells stably transfected with the indicated siRNA plasmids were transiently transfected with an NF-κB regulated firefly luciferase reporter plasmid (pNF-κB-TK-luc) or an enhancerless control plasmid (pTK-luc). At 24 h after transfection, cells were stimulated for 3 h with TNF (10 ng ml–1), LPS (1 μg ml–1), or pIC (100 μg ml–1). Firefly luciferase activity was analyzed in cell lysates and normalized to the activity of a co-transfected Renilla luciferase plasmid. NF-κB enhancer activity was calculated by subtracting the average luciferase activity of cells treated with pTK-luc from the luciferase activity of cells treated with pNF-κB-TK-luc. Data from two independent experiments were combined and expressed as mean ± s.e.m. (n=8): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells expressing the same siRNA (P<0.05); b, the mean for cells expressing RelA or RelB siRNA is significantly different from the mean for cells expressing control siRNA given the same stimulus (P<0.05).
To determine the consequences of RelA knockdown on activation of the classical NF-κB pathway, we analyzed cellular levels of IκBα (Figure 3c) and nuclear levels of RelA phosphorylated at Ser 536 (Figure 3d) following stimulation with TNF, LPS, or pIC. In control cells, IκBα protein was rapidly degraded and re-synthesized following treatment with all three stimuli. Basal and inducible expression of IκBα was very low in cells expressing RelA siRNA, consistent with a role for RelA in activating transcription of the IκBα gene ( Table 1). Basal expression and inducible degradation of IκBα was not affected by RelB knockdown, although re-synthesis of IκBα following pIC stimulation appeared to be delayed. Nuclear levels of phosphorylated RelA increased rapidly upon stimulation, especially with TNF and pIC (Figure 3d). No increase in phosphorylated RelA was observed in cells stably transfected with RelA siRNA, whereas knockdown of RelB did not alter stimulus-dependent RelA phosphorylation. Transcriptional activity of nuclear RelA was analyzed by transient transfection of a plasmid in which binding sites for RelA/p50 dimers regulate transcription of a luciferase reporter gene. Rapid transactivation of this reporter gene by all three stimuli was blocked by RelA knockdown (Figure 3e). Similar results were obtained at later time points poststimulation (Supplementary Figure S2 online). As we previously reported,12 pIC was a stronger stimulus of NF-κB transcriptional activity than was LPS. Interestingly, knockdown of RelB enhanced RelA-dependent transactivation, suggesting that RelB may have inhibitory activity in this context. In summary, we demonstrated that knockdown of RelA but not RelB in HT-29 cells inhibited activation of the classical NF-κB pathway by proinflammatory stimuli. Using this experimental system, we then investigated the specific contribution of this signaling pathway to regulation of pIgR expression.
Differential contributions of the classical NF-κB pathway to induction of early response genes and the polymeric immunoglobulin receptor
To compare the mechanisms underlying the early induction of proinflammatory genes and the delayed induction of pIgR, gene expression was analyzed in control, RelA, and RelB knockdown HT-29 cells stimulated with TNF, LPS, or pIC for 3 or 24 h (Figure 4). We previously reported that changes in protein levels for pIgR, IL-8, and TNF, as measured by enzyme-linked immunosorbent assay, were directly proportional to changes in mRNA levels.12 As expected, knockdown of RelA inhibited the rapid upregulation of mRNA encoding IL-8 as well as TNF, which we have previously shown to be characteristic of the early proinflammatory response in HT-29 cells.11, 12 Knockdown of RelA also blocked induction of A20, a cytoplasmic ubiquitin-editing enzyme that acts as a negative feedback regulator of the classical NF-κB pathway.31 Knockdown of RelB partially inhibited TNF-stimulated induction of early response genes, suggesting some contribution of the alternative pathway, although it did not affect the responses to TLR signaling. To rule out the possibility that knockdown of RelA had secondary effects on the mitogen-activated protein kinase (MAPK) signaling pathway, we analyzed the expression of MAPK phosphatase-1 (MKP-1), a nuclear dual-specificity phosphatase that downregulates MAPK signaling by dephosphorylating p38 MAPK and c-Jun N-terminal kinase (JNK).32 The promoter of the MKP1 gene contains multiple MAPK-responsive elements, but no κB-binding sites.33 Changes in MKP-1 expression were very minor compared with the dramatic effects on IL-8, TNF, and A20, confirming that MKP-1 is not a major target of NF-κB signaling and suggesting that RelA knockdown does not significantly impact the MAPK signaling pathway.
Figure 4.

Effect of RelA and RelB knockdown on tumor necrosis factor (TNF)-, lipopolysaccharide (LPS)-, and polyinosinic:polycytidylic acid (pIC)-stimulated gene expression. mRNA levels were analyzed by quantitative reverse transcriptase-PCR (qRT-PCR) in HT-29 cells stably transfected with control, RelA, or RelB small inhibitory RNAs (siRNAs) and treated with TNF (10 ng ml–1), LPS (1 μg ml–1), or pIC (100 μg ml–1) for 3 or 24 h. Data from two to three independent experiments were combined and expressed as mean ± s.e.m. (n=8–12): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells expressing the same siRNA (P<0.05); b, the mean for cells expressing RelA or RelB siRNA is significantly different from the mean for cells expressing control siRNA given the same stimulus for the same length of time (P<0.05).
Interestingly, the effect of RelA knockdown on upregulation of pIgR mRNA was stimulus specific, blocking the response to TNF but not the response to LPS or pIC. In contrast, knockdown of RelB did not inhibit the upregulation of pIgR mRNA for any of the stimuli. The basal levels of pIgR mRNA were significantly higher in cells expressing RelB siRNA than in those expressing either RelA or control siRNA, suggesting an inhibitory effect of RelB on RelA-dependent gene activation. Additional increases in pIgR mRNA were observed in response to TNF, LPS, and pIC stimulation in RelB knockdown cells. These findings suggest that the RelA-dependent classical pathway of NF-κB activation is required for regulation of pIgR expression by TNF, whereas other signaling pathways may compensate to allow induction of pIgR in response to TLR stimulation.
Activation of the classical NF-κB pathway is required for induction of a PIGR reporter gene by proinflammatory stimuli
We previously reported that transactivation of a reporter gene containing an 8.6-kb regulatory region from the human PIGR gene by TNF and TLR signaling requires a κB element in intron 1.10, 12, 26 To determine whether regulation of this reporter gene requires activation of the classical NF-κB pathway, HT-29 cells stably transfected with control, RelA, or RelB siRNAs were transiently transfected with the human PIGR reporter gene before stimulation with TNF, LPS, or pIC. To confirm that the classical NF-κB pathway was activated by these stimuli, we first demonstrated that the early response of a co-transfected IL8 reporter gene was inhibited by knockdown of RelA but not RelB (Figure 5a). Transcription of the IL8 reporter gene in response to LPS was lower than the response to pIC or TNF, consistent with the differential effects of these stimuli on IL-8 mRNA levels (Figure 1). We found that knockdown of RelA inhibited the induction of the PIGR reporter gene in response to all three stimuli, suggesting a requirement for the classical NF-κB pathway (Figure 5b). Surprisingly, knockdown of RelB partially inhibited transactivation of the PIGR reporter gene in response to LPS and TNF, but not pIC. These findings suggest that under some conditions RelA and RelB may cooperate to activate the κB element in intron 1 of the PIGR gene.
Figure 5.

Effect of RelA and RelB knockdown on interleukin-8 (IL8) and polymeric immunoglobulin receptor (PIGR) transcriptional activity. HT-29 cells stably transfected with the indicated small inhibitory RNA (siRNA) expression plasmid were transiently transfected with (a) a reporter plasmid containing the promoter region of the human IL8 gene (−135 to +46 bp relative to the transcription start site) fused to firefly luciferase or (b) a reporter plasmid containing an 8.6-kb fragment from the human PIGR gene (2.7 kb of 5′ flanking sequence, exon 1, intron 1 (5.7 kb) and part of exon 2 up to the ATG translation start site) fused to firefly luciferase. At 24 h after transfection, cells were stimulated for 3 h (IL8 reporter) or 24 h (PIGR reporter) with tumor necrosis factor (TNF; 10 ng ml–1), lipopolysaccharide (LPS; 1 μg ml–1), or polyinosinic:polycytidylic acid (pIC; 100 μg ml–1). Firefly luciferase activity was analyzed in cell lysates and normalized to the activity of a co-transfected Renilla luciferase plasmid. Data from two independent experiments were combined and are expressed as fold increase compared with the transcriptional activity in unstimulated cells expressing control siRNA (mean ± s.e.m., n=8): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells expressing the same siRNA (P<0.05); b, the mean for cells expressing RelA or RelB siRNA is significantly different from the mean for cells expressing control siRNA given the same stimulus (P<0.05).
The alternative NF-κB pathway may contribute to regulation of pIgR expression
Our observation of a potential contribution of RelB to transcriptional regulation of the PIGR gene (Figure 5) raised the interesting possibility that both the classical and alternative NF-κB pathways may regulate pIgR expression, depending on the stimulus. To test this hypothesis, HT-29 cells were stimulated with an agonist for the lymphotoxin β receptor (LTβR), which is known to activate the alternative NF-κB pathway.27 To confirm activation of the alternative NF-κB pathway, we first demonstrated that ligation of LTβR induced early expression of the known targets of this pathway, RelB and CCL20 (chemokine (C-C motif) ligand 20)34, 35 (Figure 6). Interestingly, ligation of LTβR induced both IL-8 and pIgR mRNA to an extent similar to that achieved by TNF, LPS, and pIC stimulation (compare Figures 6 and 1). Induction of all four genes by LTβR ligation was partially inhibited by BAY 11-7082, suggesting that either LTβR ligation activated both the classical and alternative NF-κB pathways or that BAY 11-7082 inhibits the alternative as well as the classical NF-κB pathway, as has been proposed.36
Figure 6.

Activation of the alternative nuclear factor-κB (NF-κB) pathway by lymphotoxin β receptor (LTβR) in intestinal epithelial cells (IECs). HT-29 cells were stimulated with an affinity-purified polyclonal goat IgG antibody to the LTβR (100 ng ml–1) in the presence or absence of 10 μ BAY 11-7082, an inhibitor of NF-κB activation. The optimal concentration of α-LTβR was determined in preliminary experiments (data not shown). mRNA levels for interleukin-8 (IL-8), polymeric immunoglobulin receptor (pIgR), CCL20 (chemokine (C-C motif) ligand 20), and RelB were quantified by quantitative reverse transcriptase-PCR (qRT-PCR) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Data from two independent experiments were combined and expressed as mean ± s.e.m. (n=8): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells (P<0.05); b, the mean for cells treated with BAY 11-7082 is significantly different from the mean for cells given the same stimulus in the absence of BAY 11-7082 (P<0.05).
MAP kinase signaling enhances expression of proinflammatory genes but inhibits expression of the polymeric immunoglobulin receptor
Ligation of TNF and TLRs in IECs results in multiple interactions between NF-κB and other intracellular signaling pathways, in particular those involving the MAP kinases.37 It has recently been reported that BAY11-7082 inhibits phosphorylation of the MAP kinases ERK2 (extracellular-regulated kinase 2) and JNK1,38 suggesting that some of the inhibitory effects of BAY11-7082 on TNF- and TLR-induced gene expression (Figure 1) may have been because of attenuation of MAPK signaling. To determine the contribution of MAPK signaling to the early induction of IL-8 and the delayed regulation of pIgR, HT-29 cells were stimulated with TNF, LPS, or pIC in the presence or absence of individual or combined MAPK inhibitors targeting p38, JNK, and ERK (Figure 7). Inhibition of p38 and ERK significantly reduced the induction of IL-8 by TNF and pIC but not LPS, whereas no effect was observed upon inhibition of JNK. Combined inhibition of p38, JNK, and ERK blocked the induction of IL-8 by all three stimuli, suggesting synergism among the MAPK signaling pathways. In contrast, inhibition of either p38 or ERK enhanced the induction of pIgR by TNF and inhibition of ERK enhanced the induction of pIgR expression by pIC. This finding was consistent with an earlier report that inhibition of ERK enhanced TNF-stimulated pIgR expression.39 None of the MAPK inhibitors affected the response of pIgR to LPS, suggesting that regulation of pIgR expression by MAPK signaling is stimulus specific. To examine whether inhibition of pIgR expression by MAPK signaling could have been an indirect effect of altered RelB expression, we analyzed the effects of MAPK inhibitors on TNF, LPS, and pIC-stimulated RelB mRNA expression. Neither the modest induction of RelB by pIC nor the potent induction of RelB by TNF were blocked by the MAPK inhibitors, suggesting that the RelA-dependent increase in RelB expression did not involve synergy with MAPK signaling. Furthermore, the enhanced induction of pIgR by TNF or pIC in the presence of MAPK inhibitors was not affected by siRNA-mediated RelB knockdown (data not shown). We conclude that the NF-κB and MAPK signaling pathways act synergistically to induce rapid expression of proinflammatory genes like IL-8, but may have opposing effects on the delayed expression of pIgR.
Figure 7.

Effect of mitogen-activated protein kinase (MAPK) inhibition on the response of HT-29 cells to tumor necrosis factor (TNF), lipopolysaccharide (LPS), and polyinosinic:polycytidylic acid (pIC). HT-29 cells were stimulated with TNF (10 ng ml–1), LPS (1 μg ml–1), or pIC (100 μg ml–1) for 3 or 24 h, in the presence or absence of individual MAPK inhibitors or a combination of the three inhibitors at the same concentrations (10 μ SB20358, p38 inhibitor; 10 μ SP600125, c-Jun N-terminal kinase (JNK) inhibitor; 10 μ PD98059, extracellular-regulated kinase (ERK) inhibitor). Cells cultured in the absence of MAPK inhibitors were treated with an equivalent volume of vehicle (dimethylsulfoxide (DMSO)). mRNA levels for interleukin-8 (IL-8), polymeric immunoglobulin receptor (pIgR), and RelB were quantified by quantitative reverse transcriptase-PCR (qRT-PCR) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Data from two independent experiments were combined and expressed as mean ± s.e.m. (n=8): a, the mean for stimulated cells is significantly greater than the mean for unstimulated cells (P<0.05); b, the mean for cells treated with MAPK inhibitors is significantly different from the mean for cells given the same stimulus in the absence of MAPK inhibitors (P<0.05).
Discussion
The complex functions of NF-κB in the intestine must balance pro-inflammatory innate and adaptive immune responses to potential pathogens and at the same time protect epithelial integrity.8 Mouse models involving targeted gene deletion have revealed that activation of NF-κB in IECs is critical for protection against intestinal inflammation. Mice with an IEC-specific deletion of IKK-γ, the regulatory subunit of the IκB kinase (IKK) complex, were found to develop spontaneous colitis associated with epithelial cell apoptosis and translocation of bacteria into the mucosa.40 Mice with an IEC-specific deletion of IKK-β, the catalytic subunit of IKK, had a diminished ability to clear infection with Trichuris muris associated with reduced expression of the protective cytokine thymic stromal lymphopoietin.41 IEC-targeted deletion of RelA in mice resulted in reduced expression of antimicrobial peptides and antiapoptotic and prorestitution genes, and was associated with increased rates of epithelial proliferation and apoptosis and increased susceptibility to chemically induced colitis.42 In contrast, mice with a global deletion in either RelB or NF-κB2/p100 were found to have impaired development of Peyer's patches and other secondary lymphoid structures in the intestine.43 The effects of a targeted deletion of RelB in IECs have not been reported.
Our new findings suggest that regulation of pIgR expression in IECs may be an important mechanism through which NF-κB maintains intestinal homeostasis during proinflammatory immune responses. Our initial experiments with a soluble inhibitor of IκBα phosphorylation indicated that NF-κB activation is required for TNF-stimulated upregulation of pIgR mRNA in human IEC lines (Figure 1 and Supplementary Figure S1 online). By inhibiting RelA expression in HT-29 cells, we demonstrated that induction of pIgR by TNF required the RelA subunit of NF-κB. RelA knockdown abolished both the increase in pIgR mRNA (Figure 4) and the transcriptional activity of a PIGR reporter gene (Figure 5) following TNF stimulation. These data are consistent with a mechanism in which TNF activates the classical NF-κB pathway, resulting in binding of RelA-containing dimers to the κB element in intron 1 of the PIGR gene and enhanced transcription of pIgR mRNA. The finding that TNF-induced activation of the PIGR reporter gene was also reduced in RelB knockdown cells (Figure 5) suggests that RelB may cooperate with RelA to induce PIGR gene transcription. Previous studies demonstrated that both RelA- and RelB-containing dimers were translocated to the nucleus following TNF stimulation, and electrophoretic mobility shift assays revealed that RelA- and RelB- but not c-Rel-containing dimers bound to the κB element in intron 1 of the human PIGR gene.26 However, knockdown of RelB did not inhibit the TNF-induced increase in pIgR mRNA, and basal levels of pIgR mRNA were actually higher in RelB knockdown cells (Figure 4). One possible explanation for these apparently contradictory results is that activation of the NF-κB pathway by TNF may regulate steady-state levels of pIgR mRNA by post-transcriptional as well as transcriptional mechanisms. In support of this concept, we previously reported that stimulation of HT-29 cells with TNF caused a significant increase in the stability of pIgR mRNA.11 The requirement for both transcriptional and post-transcriptional mechanisms could contribute to the delayed rise in pIgR mRNA relative to the rapid induction of proinflammatory genes like IL-8.
Regulation of pIgR expression by TLR signaling appears to involve more complex mechanisms. As was the case with TNF stimulation, we found that a soluble inhibitor of IκBα phosphorylation reduced induction of pIgR mRNA (Figure 1), and that RelA was required for the increased transcriptional activity of a PIGR reporter gene (Figure 5), following stimulation by LPS or pIC. Knockdown of RelB expression reduced LPS-stimulated but not pIC-stimulated PIGR gene transcription, suggesting that there are stimulus-specific contributions of RelA and RelB to activation of the κB site in intron 1 of the PIGR gene. Surprisingly, upregulation of pIgR mRNA by LPS or pIC was not blocked by inhibition of either RelA or RelB (Figure 4). The simplest explanation for mechanistic differences between TNF- and TLR-induced pIgR expression is that TLR ligation may induce additional signaling pathways that compensate for the loss of RelA or RelB. Such a mechanism could involve regulatory elements in the endogenous PIGR gene, outside the 8-kb regulatory region in the PIGR reporter gene. The observation that TLR signaling may utilize different mechanisms than does TNF signaling for induction of pIgR is consistent with the concept of homeostatic cross-talk between IECs and elements of the intestinal microbiota that maintains the “tone” of pIgR expression in the absence of overt inflammation. This would be particularly relevant in the case of TLR4 signaling, given the constant presence at the epithelial surface of soluble LPS shed by Gram-negative commensal bacteria. In support of this concept, we recently reported that mice deficient in MyD88 (myeloid differentiation primary response gene (88)), a cytoplasmic adaptor in the TLR4 signaling pathway, have significantly reduced expression of pIgR in IECs.14
Our finding that ligation of LTβR can upregulate pIgR mRNA (Figure 6) reinforces the concept that both the RelA-dependent classical and RelB-dependent alternative NF-κB pathways may contribute to the maintenance of pIgR expression. We suggest that induction of pIgR expression in IECs may be part of the developmental program of the intestinal immune system, which is known to require the RelB-dependent alternative pathway.43 Because ligands that activate the alternative NF-κB pathway have been shown to induce class switching to IgA in mucosal B cells,44 a similar mechanism for induction of pIgR expression in IECs would coordinately enhance the production and transport of secretory IgA.
Our results suggested that additional signaling pathways could potentially modulate the effects of NF-κB signaling on regulation of pIgR expression. However, we found that inhibition of MAP kinases, known to be activated by TNF and TLR signaling, did not block induction of pIgR (Figure 7). In fact, inhibition of ERK enhanced TNF- and pIC-induced pIgR expression, suggesting that this pathway could antagonize rather than synergize with NF-κB signaling. These effects were in contrast to the positive effects of MAPK signaling on early expression of the proinflammatory gene IL-8, illustrating the differences in regulatory mechanisms.
We conclude that maintenance of high levels of pIgR expression by NF-κB signaling may be an important mechanism through which products of the gut microbiota and host cytokines promote intestinal epithelial integrity and barrier function. Our results suggest that pIgR expression is regulated through multiple transcriptional and post-transcriptional mechanisms that involve both the RelA and RelB subunits of NF-κB, as well as other signaling pathways. By utilizing signaling pathways that are distinct from those involved in the early proinflammatory response, IEC are able to upregulate pIgR expression in response to a complex mix of microbial and host stimuli while suppressing a potentially damaging proinflammatory response.
Methods
Cell culture. The HT-29v20 subclone of the human colon adenocarcinoma cell line HT-2945 was cultured as described.14 Where indicated, cells were stimulated with human recombinant TNF (R&D Systems, Minneapolis, MN) at 10 ng ml–1, purified Escherichia coli O26:B6 LPS (Sigma-Aldrich, St Louis, MO) at 1 μg ml–1, pIC (Sigma-Aldrich) at 100 μg ml–1, or an affinity-purified polyclonal goat IgG antibody to the LTβR (R&D Systems) at 100 ng ml–1. In some experiments, cells were treated 30 min before stimulation with the NF-κB inhibitor BAY 11-7082 (EMD Chemicals, Gibbstown, NJ) at a final concentration of 10 μ, or with specific MAPK kinase inhibitors, added individually or combined (SB20358, p38 inhibitor; SP600125, JNK inhibitor PD98059, ERK inhibitor; Sigma-Aldrich) at a final concentration of 10 μ each.
RNA isolation, complementary DNA synthesis, and real-time quantitative reverse transcriptase-PCR. Total cellular RNA was extracted, reverse transcribed to generate complementary DNA, and specific mRNA levels were quantified by real-time quantitative reverse transcriptase-PCR using the ABI Prism 7700 Sequence Detection System (Life Technologies, Carlsbad, CA) as described.11 The sequences of primers and fluorescent probes for human GAPDH (glyceraldehyde 3-phosphate dehydrogenase), pIgR, TNF, and IL-8 mRNAs were previously reported;11 for all other mRNAs, predesigned primers and probes were purchased from Applied Biosystems. The mRNA levels were normalized to GAPDH mRNA according to the formula: (2−(CT test−CT GAPDH)) × 100%.
Stable transfection of HT-29 cells with siRNA expression plasmids. To knockdown expression of RelA or RelB, HT-29 cells were transfected with the pSIREN-RetroQ expression plasmid (BD Biosciences, San Jose, CA) encoding RelA, RelB, or an irrelevant control siRNA, selected for stable integration of the plasmid by resistance to puromycin, and cloned by limiting dilution. The sequence of the RelB siRNA was previously reported.26 The sequence of the RelA siRNA (5′–3′) was GATCCGATCAATGGCTACACAGGATTCAAGAGATCCTGTGTAGCCATTGATCTTTTTTG and the sequence of the control siRNA (5′–3′) was GATCCGCGGGCTGATGCTGCACCAATTCAAGAGATTGGTGCAGCATCAGCCCGTTTTTTG.
Immunoblot analysis of NF-κB subunits. For the preparation of whole-cell lysates, HT-29 cells were disrupted by addition of 700 μl cell lysis buffer (50 m Tris, pH 8.0, 150 m NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 1 m dithiothreitol)46 supplemented with protease inhibitors (2.8 μg ml–1 aprotinin, 1 μg ml–1 pepstatin A, 1 μg ml–1 leupeptin, 150 m benzamidine, and 1 m phenylmethylsulfonyl fluoride; Sigma-Aldrich) and incubated on ice for 15 min with agitation. Supernatants were collected following centrifugation for 10 min at 15,800 g, and protein concentrations were analyzed by the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Nuclear extracts were prepared according to Dignam et al.47 as previously described.48 Protein concentrations were analyzed using the Pierce BCA Protein assay (Thermo Fisher Scientific, Fairland, NJ). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting were performed as described,48 using ∼40 μg protein per lane for experiments using whole-cell lysates and 20–25 μg protein per lane for nuclear extracts. Rabbit polyclonal IgG antibodies to RelA, phosphorylated RelA (at Ser 536), RelB, IκBα, and goat anti-rabbit IgG-horseradish peroxidase were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA). For whole-cell lysates, duplicate membranes were probed with affinity-purified rabbit polyclonal IgG to β-tubulin (Thermo Fisher Scientific) to control for protein loading.
Transient transfection of HT-29 cells and gene reporter assays. The NF-κB reporter plasmid (pNF-κB-TK-luc) (BD Biosciences Clontech, Mountain View, CA) contains four copies of a RelA consensus site (5′-GGGAATTTCC-3′) upstream of a minimal thymidine kinase (TK) promoter and the coding region of the firefly luciferase gene. The corresponding control plasmid (pTK-luc) contains the same minimal TK promoter without the upstream NF-κB sites. The human PIGR-luciferase reporter plasmid includes 2684 bp of PIGR 5′-flanking sequence, exon 1 (132 bp), intron 1 (5751 bp), and the first 56 bp of exon 2, up to and including the translation start site.49 The human IL8-luciferase reporter plasmid, which contains the IL8 promoter region (nt −135 to + 46), was generously provided by Dr Gary Wu, University of Pennsylvania.50 HT-29 cells at 50% confluence were transfected for 2 h with 1 μg of the indicated gene reporter plasmid and 0.02 μg of a control Renilla luciferase plasmid (pRL-CMV; Promega, Madison, WI) with 3.1 μl Tfx-50 transfection reagent (Promega). At 24 h following transfection, cells were treated with TNF, LPS, or pIC as indicated. Cell lysates were analyzed for firefly and Renilla luciferase activities using the Dual Luciferase Reporter Assay System (Promega). NF-κB enhancer activity was calculated by subtracting the normalized luciferase activity of cells treated with pTK-luc from the normalized luciferase activity of cells treated with pNF-κB-TK-luc. PIGR and IL8 promoter activities were calculated as the ratio of firefly to Renilla luciferase activity.
Statistical analyses. Statistical differences among treatment groups were determined by analysis of variance and Fisher's protected least significant difference test.
Acknowledgments
This work was supported by the NIH grant R21AI069027 (to C.S.K.), a Senior Research Award from the Crohn's & Colitis Foundation of America (to C.S.K.), and Research Council of Norway and University of Oslo (to F.E.J.).
The authors declared no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
Supplementary Material
References
- Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008;8,:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- Duerkop B.A., Vaishnava S., Hooper L.V. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31,:368–376. doi: 10.1016/j.immuni.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Sansonetti P.J., Medzhitov R. Learning tolerance while fighting ignorance. Cell. 2009;138,:416–420. doi: 10.1016/j.cell.2009.07.024. [DOI] [PubMed] [Google Scholar]
- Macpherson A.J., McCoy K.D., Johansen F.E., Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol. 2008;1,:11–22. doi: 10.1038/mi.2007.6. [DOI] [PubMed] [Google Scholar]
- Norderhaug I.N., Johansen F.E., Schjerven H., Brandtzaeg P. Regulation of the formation and external transport of secretory immunoglobulins. Crit. Rev. Immunol. 1999;19,:481–508. [PubMed] [Google Scholar]
- Kaetzel C.S., Bruno M.E.C.Epithelial transport of IgA by the polymeric immunoglobulin receptor Mucosal Immune Defense: Immunoglobulin A(Kaetzel, C.S., ed)43–89.(Springer, New York; 2007). [Google Scholar]
- Phalipon A., Corthésy B. Novel functions of the polymeric Ig receptor: well beyond transport of immunoglobulins. Trends Immunol. 2003;24,:55–58. doi: 10.1016/s1471-4906(02)00031-5. [DOI] [PubMed] [Google Scholar]
- Spehlmann M.E., Eckmann l. Nuclear factor-κB in intestinal protection and destruction. Curr. Opin. Gastroenterol. 2009;25,:92–99. doi: 10.1097/MOG.0b013e328324f857. [DOI] [PubMed] [Google Scholar]
- Shibolet O., Podolsky D.K. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292,:G1469–G1473. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- Schjerven H., Brandtzaeg P., Johansen F.E. A novel NF-κB/Rel site in intron 1 cooperates with proximal promoter elements to mediate TNF-α-induced transcription of the human polymeric Ig receptor. J. Immunol. 2001;167,:6412–6420. doi: 10.4049/jimmunol.167.11.6412. [DOI] [PubMed] [Google Scholar]
- Bruno M.E.C., Kaetzel C.S. Long-term exposure of the HT-29 human intestinal epithelial cell-line to TNF causes sustained up-regulation of the polymeric Ig receptor and pro-inflammatory genes through transcriptional and post-transcriptional mechanisms. J. Immunol. 2005;174,:7278–7284. doi: 10.4049/jimmunol.174.11.7278. [DOI] [PubMed] [Google Scholar]
- Schneeman T.A., et al. Regulation of the polymeric Ig receptor by signaling through Toll-like receptors 3 and 4: linking innate and adaptive immune responses. J. Immunol. 2005;175,:376–384. doi: 10.4049/jimmunol.175.1.376. [DOI] [PubMed] [Google Scholar]
- Arsenescu R., et al. Signature biomarkers in Crohn's disease: toward a molecular classification. Mucosal Immunol. 2008;1,:399–411. doi: 10.1038/mi.2008.32. [DOI] [PubMed] [Google Scholar]
- Bruno M.E.C., et al. Regulation of the polymeric immunoglobulin receptor in intestinal epithelial cells by Enterobacteriaceae: implications for mucosal homeostasis. Immunol. Invest. 2010;39,:356–382. doi: 10.3109/08820131003622809. [DOI] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132,:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Bonizzi G., Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25,:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Xiao G., Rabson A.B., Young W., Qing G., Qu Z. Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17,:281–293. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Fusco A.J., et al. NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep. 2009;10,:152–159. doi: 10.1038/embor.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bren G.D., et al. Transcription of the RelB gene is regulated by NF-κB. Oncogene. 2001;20,:7722–7733. doi: 10.1038/sj.onc.1204868. [DOI] [PubMed] [Google Scholar]
- Viswanathan M., Yu M., Mendoza L., Yunis J.J. Cloning and transcription factor-binding sites of the human c-rel proto-oncogene promoter. Gene. 1996;170,:271–276. doi: 10.1016/0378-1119(95)00773-3. [DOI] [PubMed] [Google Scholar]
- Cogswell P.C., Scheinman R.I., Baldwin A.S., Jr Promoter of the human NF-κB p50/p105 gene. Regulation by NF-κB subunits and by c-REL. J. Immunol. 1993;150,:2794–2804. [PubMed] [Google Scholar]
- Ito C.Y., Kazantsev A.G., Baldwin A.S., Jr Three NF-(B sites in the I(B( promoter are required for induction of gene expression by TNF-α. Nucleic Acids Res. 1994;22,:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein B., Baldwin A.S. Distinct mechanisms for regulation of the interleukin-8 gene involve synergism and cooperativity between C/EBP and NF-κB. Mol. Cell Biol. 1993;13,:7191–7198. doi: 10.1128/mcb.13.11.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udalova I.A., Knight J.C., Vidal V., Nedospasov S.A., Kwiatkowski D. Complex NF-κB interactions at the distal tumor necrosis factor promoter region in human monocytes. J. Biol. Chem. 1998;273,:21178–21186. doi: 10.1074/jbc.273.33.21178. [DOI] [PubMed] [Google Scholar]
- Ainbinder E., et al. Mechanism of rapid transcriptional induction of tumor necrosis factor-α-responsive genes by NF-κB. Mol. Cell Biol. 2002;22,:6354–6362. doi: 10.1128/MCB.22.18.6354-6362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjerven H., Tran T.N., Brandtzaeg P., Johansen F.E. De novo synthesized RelB mediates TNF-induced up-regulation of the human polymeric Ig receptor. J. Immunol. 2004;173,:1849–1857. doi: 10.4049/jimmunol.173.3.1849. [DOI] [PubMed] [Google Scholar]
- Bonizzi G., et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J. 2004;23,:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E. The alternative NF-κB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem. Pharmacol. 2006;72,:1161–1179. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Pierce J.W., et al. Novel inhibitors of cytokine-induced IκBαphosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 1997;272,:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Sakurai H., et al. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 2003;278,:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- Coornaert B., Carpentier I., Beyaert R. A20: central gatekeeper in inflammation and immunity. J. Biol. Chem. 2009;284,:8217–8221. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros T., Chevet E., Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol. Rev. 2008;60,:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- Ryser S., Massiha A., Piuz I., Schlegel W. Stimulated initiation of mitogen-activated protein kinase phosphatase-1 (MKP-1) gene transcription involves the synergistic action of multiple cis-acting elements in the proximal promoter. Biochem. J. 2004;378,:473–484. doi: 10.1042/BJ20031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbo M., Sierro F., Debard N., Kraehenbuhl J.P., Finke D. Lymphotoxin-β receptor signaling induces the chemokine CCL20 in intestinal epithelium. Gastroenterology. 2004;127,:213–223. doi: 10.1053/j.gastro.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Sirard J.C., Didierlaurent A., Cayet D., Sierro F., Rumbo M. Toll-like receptor 5- and lymphotoxin-β receptor-dependent epithelial Ccl20 expression involves the same NF-κB binding site but distinct NF-kB pathways and dynamics. Biochim. Biophys. Acta. 2009;1789,:386–394. doi: 10.1016/j.bbagrm.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Travert M., et al. CD40 ligand protects from TRAIL-induced apoptosis in follicular lymphomas through NF-kappaB activation and up-regulation of c-FLIP and Bcl-xL. J. Immunol. 2008;181,:1001–1011. doi: 10.4049/jimmunol.181.2.1001. [DOI] [PubMed] [Google Scholar]
- Haller D. Intestinal epithelial cell signalling and host-derived negative regulators under chronic inflammation: to be or not to be activated determines the balance towards commensal bacteria. Neurogastroenterol. Motil. 2006;18,:184–199. doi: 10.1111/j.1365-2982.2006.00762.x. [DOI] [PubMed] [Google Scholar]
- Lee H.S., et al. A noble function of BAY 11-7082: inhibition of platelet aggregation mediated by an elevated cAMP-induced VASP, and decreased ERK2/JNK1 phosphorylations. Eur. J. Pharmacol. 2010;627,:85–91. doi: 10.1016/j.ejphar.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Takenouchi-Ohkubo N., Moro I., Mukae S., Kaneko Y., Komiyama K. Tumour necrosis factor-αmediated human polymeric immunoglobulin receptor expression is regulated by both mitogen-activated protein kinase and phosphatidylinositol-3-kinase in HT-29 cell line. Immunology. 2008;123,:500–507. doi: 10.1111/j.1365-2567.2007.02716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenci A., et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446,:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- Zaph C., et al. Epithelial-cell-intrinsic IKK-β expression regulates intestinal immune homeostasis. Nature. 2007;446,:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- Steinbrecher K.A., Harmel-Laws E., Sitcheran R., Baldwin A.S. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J. Immunol. 2008;180,:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- Yilmaz Z.B., Weih D.S., Sivakumar V., Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22,:121–130. doi: 10.1093/emboj/cdg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorny A., Puga I., Cerutti A. Innate signaling networks in mucosal IgA class switching. Adv. Immunol. 2010;107,:31–69. doi: 10.1016/B978-0-12-381300-8.00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanch V.J., Piskurich J.F., Kaetzel C.S. Cutting edge: coordinate regulation of IFN regulatory factor-1 and the polymeric Ig receptor by proinflammatory cytokines. J. Immunol. 1999;162,:1232–1235. [PubMed] [Google Scholar]
- Greer S.F., Zika E., Conti B., Zhu X.S., Ting J.P. Enhancement of CIITA transcriptional function by ubiquitin. Nat. Immunol. 2003;4,:1074–1082. doi: 10.1038/ni985. [DOI] [PubMed] [Google Scholar]
- Dignam J.D., Lebovitz R.M., Roeder R.G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11,:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno M.E.C., West R.B., Schneeman T.A., Bresnick E.H., Kaetzel C.S. Upstream stimulatory factor but not c-Myc enhances transcription of the human polymeric immunoglobulin receptor gene. Mol. Immunol. 2004;40,:695–708. doi: 10.1016/j.molimm.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Schjerven H., Brandtzaeg P., Johansen F.E. Mechanism of IL-4-mediated up-regulation of the polymeric Ig receptor: role of STAT6 in cell type-specific delayed transcriptional response. J. Immunol. 2000;165,:3898–3906. doi: 10.4049/jimmunol.165.7.3898. [DOI] [PubMed] [Google Scholar]
- Wu G.D., Lai E.J., Huang N., Wen X. Oct-1 and CCAAT/enhancer-binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. The role of Oct-1 as a transcriptional repressor. J. Biol. Chem. 1997;272,:2396–2403. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.