

Abstract
Migraine and its transformation to chronic migraine are healthcare burdens in need of improved treatment options. We seek to define how neural immune signaling modulates the susceptibility to migraine, modeled in vitro using spreading depression (SD), as a means to develop novel therapeutic targets for episodic and chronic migraine. SD is the likely cause of migraine aura and migraine pain. It is a paroxysmal loss of neuronal function triggered by initially increased neuronal activity, which slowly propagates within susceptible brain regions. Normal brain function is exquisitely sensitive to, and relies on, coincident low-level immune signaling. Thus, neural immune signaling likely affects electrical activity of SD, and therefore migraine. Pain perception studies of SD in whole animals are fraught with difficulties, but whole animals are well suited to examine systems biology aspects of migraine since SD activates trigeminal nociceptive pathways. However, whole animal studies alone cannot be used to decipher the cellular and neural circuit mechanisms of SD. Instead, in vitro preparations where environmental conditions can be controlled are necessary. Here, it is important to recognize limitations of acute slices and distinct advantages of hippocampal slice cultures. Acute brain slices cannot reveal subtle changes in immune signaling since preparing the slices alone triggers: pro-inflammatory changes that last days, epileptiform behavior due to high levels of oxygen tension needed to vitalize the slices, and irreversible cell injury at anoxic slice centers.
In contrast, we examine immune signaling in mature hippocampal slice cultures since the cultures closely parallel their in vivo counterpart with mature trisynaptic function; show quiescent astrocytes, microglia, and cytokine levels; and SD is easily induced in an unanesthetized preparation. Furthermore, the slices are long-lived and SD can be induced on consecutive days without injury, making this preparation the sole means to-date capable of modeling the neuroimmune consequences of chronic SD, and thus perhaps chronic migraine. We use electrophysiological techniques and non-invasive imaging to measure neuronal cell and circuit functions coincident with SD. Neural immune gene expression variables are measured with qPCR screening, qPCR arrays, and, importantly, use of cDNA preamplification for detection of ultra-low level targets such as interferon-gamma using whole, regional, or specific cell enhanced (via laser dissection microscopy) sampling. Cytokine cascade signaling is further assessed with multiplexed phosphoprotein related targets with gene expression and phosphoprotein changes confirmed via cell-specific immunostaining. Pharmacological and siRNA strategies are used to mimic and modulate SD immune signaling.
Keywords: Neuroscience, Issue 52, innate immunity, hormesis, microglia, T-cells, hippocampus, slice culture, gene expression, laser dissection microscopy, real-time qPCR, interferon-gamma
Protocol
Several points are worth emphasizing for successful study of immune-related SD signaling in brain slices. First, initiating stimuli must synchronously depolarize a sufficient volume of gray matter brain to initiate SD. This means an interface configuration (i.e., fluid exposure only below and gas exchange above slice culture insert) is required.1,2 Second, acute brain slices sustain SD but trauma from their preparation, as well as the use of 95%-oxygen aerated Ringer's solution generate pro-inflammatory changes and epileptiform behavior respectively, which can alter neural circuit function and immune homeostasis.3 Third, while hippocampal slice cultures overcome these acute slice limitations, it is important to note that slice cultures should be maintained for adequate periods before use (i.e., greater than 10 days in vitro) so that microglia and astrocytes become quiescent.3-5 Finally, SD should be induced using sterile and aseptic techniques. The techniques outlined below are designed to fulfill this need.
1. Spreading Depression in Slice Cultures
- Culture preparation and maintenance.
- Slice cultures are prepared and maintained in horse-serum-based medium or a serum-free medium (SFM)6,7 as previously described8 with minor modifications to the medium. Incubation parameters are 36°C, 5% CO2, 95% humidity-balance air.
- We find that cultures can be switched to a SFM after 18 days in vitro and maintained to at least 35 days in vitro by use of our previously defined SFM that includes extra calcium and magnesium.
- In addition, antibiotics and an antifungicide are added to prevent infectious contamination associated with multiple recording episodes.
- This long-term (or recording) medium formulation contains (per 100 mL): Neurobosal medium, 97 mL; Gem-21, 2.0 mL; Glutamax (200 mM), 0.5 mL; Gentamycin (10 mg/mL), 10 μL; D-glucose (45%), 680 μL; ascorbic acid (0.5 M), 100 μL; Fungizone, (250 mg/mL), 400 μL; NaCl (5.0 M), 820 μL, Mg2Cl2 (1.0 M), 80 μL; CaCl2 (1.0 M), 160-240 μL.
- Cultures are used for SD from 21-35 days in vitro.
- Vitality verification.3,6,7
- Cultures are screened for evidence of pyramidal neuron death before use.
- At 18 days in vitro, inserts are incubated for 20 min in medium (under normal incubation conditions) containing 5 μM Sytox Green, a marker which becomes fluorescent when it binds to DNA (i.e., by penetrating into dead cells).
- Inserts are then transferred back to their previous medium and examined with fluorescence microscopy (504 excitation and 523 emission) for evidence of CA3 or CA1 pyramidal neuron layer injury.
- Slices with more than 20 cells or a standardized level of fluorescence greater than 250 IU were excluded from further use.
- Materials fabrication.
- Ground electrodes are made with glass tubes (2.0 mm OD / 1.16 mm ID) cut to 4.5 mm lengths and briefly fire polished at both ends.
- We make about 20 electrodes at a time, which can last for more than a year.
- Bring 100 mL of 1M KCl to boil and add, with stirring, 3.5 g agar and 0.5 g EDTA (ethylenediaminetetraacetic acid disodium salt dihydrate) to produce a clear yellow solution.
- Use a short length of flexible tubing fitted over each glass tube to draw warm KCl/agar solution into the glass tubes as well as a short distance into the flexible tubing.
- Release suction and allow the KCl/agar to drip slowly out of the open tip of a glass tube; then hold the open glass tube tip against a piece of ice.
- The latter maneuver will prompt the agar to gel at the electrode tip and not gel after receding back up into the glass tube.
- Once the agar has gelled in the cold running water, the flexible tube can be removed without altering the agar positioned in the glass tubes.
- Place 0.5 mL of 1 M KCl into each well of a 1.5 mL centrifuge tube rack. Then place one tip of each KCl/agar filled glass tube in individual wells.
- Next, hold an 80 cm length of 15 gauge silver wire with a hemostat and clean each of four sides by scraping the wire through an Emery board held to the counter top.
- Cut the wire into 4 cm lengths and place them into a scintillation vial filled with bleach for 20-30 minutes to place a firm Ag-AgCl coat over the cleaned silver surface.
- Bend each wire in half and insert the free ends into one end of the glass tube filled with KCl/agar, leaving about 4-6 mm exposed.
- Finally, coat the glass tube shank containing the silver wire and a small extent of the wire with Goop, a water-resistant and flexible glue, to prevent the KCl/agar from drying. Repeat for all electrodes. Let the glue dry overnight.
- Store the electrodes at 4°C in scintillation vials (5-6 electrodes/vial) containing ~ 5 mL 1 M KCl and 25 mg of EDTA, which markedly retards bacterial growth.
- Recording electrodes are made from 1.5 mm diameter (0.86 mm ID; 10 cm length) borosilicate glass tubes with filaments (Video Figure 1).
- Pull microelectrodes to a fine tip with short glass tubes pulled to a sharp point.
- We typically pull electrodes ~ 7.5 cm in length with a 1 cm taper. This allows adequate positioning and electrode tip visualization.
- Electrodes are pulled with a Narishige PE-2 puller.
- Microelectrode tips are broken back to 2-4 μm under visualization using a compound microscope fitted with a calibrated optical micrometer. We use the edge of a standard glass histology slide (25 x 75 x 1 mm) attached to a hydraulic one-dimensional micromanipulator held by another 3-dimensional manipulator to gently break the microelectrode tips.
- First, the electrode is mounted attached to a microscope slide using clay.
- Then the slide is placed on the microscope slide holder which is used to move the microelectrode tip near the edge of the glass slid attached to the hydraulic manipulator.
- Finally, the tip of the microelectrode is gently broken by pressing it up against the hydraulically driven microscope slide edge.
- Stimulating electrodes are twisted wire bipolar electrodes because:
- They allow recording of evoked field potentials, which otherwise would be obscured by the stimulus artifact from monopolar stimulation.
- A bipolar stimulating electrode can be gently applied to the dentate gyrus surface without injury, unlike sharp-tipped bipolar electrodes.
- Finally, applied current is localized to the transverse (bare) circular surfaces of the Teflon insulated wires used for the electrode.
- Stimulating electrode fabrication begins with cutting ~ 20 cm length of platinum-iridium (125 μm bare diameter; 200 μm coated diameter) and Teflon coated wire. Then, bend the length in half and tie the loose ends into a loose square knot, leaving ~ 2 cm from the knot to the ends of the wire.
- Secure the knot in the chuck of an electric hand drill placed on a table.
- While holding the other end of the wire with a hemostat, briefly turn the drill on and off until the wire is tightly twisted (i.e., each cut surface would be an equal distance from the other when the wire is cut in the transverse direction without unraveling).
- Next, the bare ends of the platinum-iridium twisted wires need to be attached to lead wires used to connect the stimulating electrode to a stimulus isolator.
- This is done by soldering the free ends of the platinum-iridium wire to ~50 cm 30 gauge wires.
- First, tape the platinum-iridium twisted wire to a bench top and drop a puddle of concentrated HCl over one free end of the twisted wire.
- Next, strip off about 1 cm of insulation from each wire using a wire cutter.
- Place the stripped end on a lead wire adjacent to the twisted wire end and apply heat from a fine-tipped soldering iron, then solder until the two wires are firmly attached.
- Slip a small length of heat shrink tubing over the exposed wire connection and shrink it fit with heat.
- Repeat for the second wire.
- Fire polish the tip of a 15 cm Pasteur pipette and guide the twisted platinum -iridium wire down the pipette until about 6 cm of the twisted protrudes out.
- Use a water repellent epoxy (e.g., J-B Weld) to seal both ends of the electrode.
- Finally, attach banana plugs to the lead wire ends for connection to the stimulus isolator.
- Under stereoscopic observation, place the electrode tip in PBS and look for electrolytically produced bubbles coming from only the cut end of the electrode tips. If any bubbles come from the sides of the twisted wires, cut the electrode with a transverse cut using a fresh single edge razor blade to re-establish intact insulation to the wire long axes.
- When troubleshooting to resolve aberrant stimulus effects, try making a freshly cut electrode tip.
- Recording dishes consist of a slice culture insert in a 35 mm culture dish that sits on two 1.0 x 12 mm glass rods spaced about 1.0 cm apart. Attach glass rods to the dish base by heating the glass tubes and then pressing them into the base with forceps.
- For static recording conditions, add 1.5 mL of medium to the dish.
- Next, soak a ~ 10 mm wide and 3-4 cm long piece of sterile cotton in 1.0 mL of medium. Fold the cotton in half along the long axis and place it along the inner well on the insert with sterile forceps. The wet cotton helps maintain dish humidity.
- Three compressible 4 mm lengths of tubing are equally spaced around the insert.
- Cover the dish tightly with polyvinyl chloride film, which allows gas exchange.
- Cut around its mid-vertical wall with a single edge razor blade to remove excess polyvinyl chloride film.
- Electrophysiological setup
- A slice culture insert assembly is placed into the PDMI recording chamber for electrophysiologic recordings.
- The insert/culture dish assembly in the PDMI chamber is covered by a 2 mm thick plastic disc supplied with the PDMI chamber, with various small holes positioned as needed for recordings electrodes, medium inflow/outflow tubes, etc.
- We periodically monitor medium pH with a miniature combination pH electrode and temperature with a miniature Type T thermocouple probe mounted into a sealed semi-microelectrode to ensure 7.3 pH and 36.0°C conditions.
- The insert/chamber is aerated with 5% carbon dioxide-balance air and medium is either held static or in a flow (1.2 mL/minute) configuration with the insert center held at 35-36°C.
- For flow conditions, medium is warmed with an inline heater, again to keep the center of the recording chamber at 36°C.
- A peristaltic pump system with a pressure relief brings medium into to the slice cultures without pulsations from the pump. An aspirator supplied with the PDMI chamber is used to remove medium from the insert via gentle suction.
- For stability, the chamber is mounted on a specially designed Burleigh-Gibraltar inverted microscope stage that holds an inverted microscope and sits on a vibration free table.
- Small holes are opened through the polyvinyl chloride insert wrap with a small, battery-operated cautery tool.
- Next, if a flow configuration is to be used, inlet and outlet flow is begun, followed by placement of a ground electrode. Otherwise, simply put the ground electrode in place.
- Electrodes, held in place with micromanipulators, are positioned just above a slice visualized with an inverted microscope at 5x magnification (Fig. 1).
- Begin with the recording electrode followed by the stimulating electrode.
- We use an audio monitor to note when the microelectrode makes contact with the slice. The tip is positioned at the midpoint along CA3 pyramidal cell body layer.
- Recordings are started, and then the stimulating electrode is gently touched to the midpoint surface of the dentate gyrus.
- In both instances, initial electrode movements are accomplished with coarse controls, and final positioning only with the hydraulic, fine controls using phase contrast illumination so that cell body layers can be easily distinguished.
- Advance electrodes in ~25 μm increments under direct microscopic visualization.
- Once electrodes touch the tissue, advance the electrodes another 20-30 μm.
- Recordings are begun before the stimulating electrode is put in place to be certain this does not trigger SD (i.e., via a mechanical stimulus).
- ~10 minutes are allowed to elapse before experiments are started.
- Transynaptically induced SD.
- Optimize evoked CA3 field potentials as follows (Fig. 2).
- CA3 field potential are evoked with 100 μs pulses (0.2 Hz) beginning with a current that is sufficient to trigger a response (e.g., ~ 1-5 μA).
- Move the recording electrode in increments along the pyramidal neuron long axis until the field potential is maximal.
- Then, increase the current with the recording electrode at this position and note the maximal current response (e.g., ~ 10-20 μA).
- Finally, use half-maximal stimulus intensity for experiments (e.g., sham control periodic stimulation).
- Determine the threshold for synaptically evoked SD (Fig. 2).
- With the electrodes configured as outlined above for evoked field potentials, switch the stimulation paradigm to single, manually evoked bursts of 10 pulses (10 Hz @ 100 μs/pulse), a paradigm previously applied in whole animal studies.
- Progressively increase current intensity per stimulus burst until SD is triggered. Wait at least 60-120 sec between stimuli.
- Report SD threshold in coulombs (i.e., current x time).
- In other experiments that require measuring responses to the induction of multiple SDs, use a stimulus strength likely to evoke SD (e.g., ~100-500 nC).
- Trigger SD every 9 min for an hour.
- Be sure to use aseptic techniques throughout experiments.
- After the last SD, return the culture insert to normal incubation conditions.
- Mark the culture dish to note the culture that experienced SD.
- Our habit is to place a single mark to the left and two marks to the right of the slice culture insert, noting each of the three cultures as #1, #2, and #3 from left to right.
- Change medium every 3-4 days until culture harvest.
- For recurrent SD, follow procedures outlined above.
- We typically test for a change in SD threshold 1, 3, 7, and 14 days after an initial hour of multiple SDs.
- CA3 area fast potential and slow potential changes are recorded via separate digital signal processing systems.
2. Exemplary Vital Imaging in Hippocampal Slice Cultures
- Dynamic functional behavior of resident immune cells (i.e., microglia) can similarly be monitored in slice cultures without cell injury or immune activation (Fig. 3).9
- For example, to label microglia to assess their movement associated with synaptic activity changes of SD, we used fluorophore-tagged isolectin-IB4.
- "Lectin PBS" is PBS with additional cations (0.1 mM each CaCl2, MgCl2, MnCl2) since divalent cations (0.1-1.0 mM) are required to bind lectin to α-D-galactosyl moieties on microglial cell membranes.
- Make a solution of "Lectin PBS":
- For 1 L PBS, using aseptic technique and sterile filtration, add:
- 100 μL of 1M MgCl2
- 100 μL of 1M MnCl2
- 100 μL of 1M CaCl2
- Mix thoroughly to combine and keep refrigerated at 4°C.
- Only 500 μL of "lectin PBS" is needed per vial of isolectin, but since it can be refrigerated and kept for months at a time, it may be useful to make up larger volumes.
- Reconstitute AlexaFluor 594-tagged isolectin-IB4 (isolectin) lyophilized powder to1 mg/mL in "lectin PBS".
- In order to minimize photobleaching, perform this step as quickly as possible, minimizing light exposure.
- Isolectin can be aliquoted to 20 μL once reconstituted at 1mg/ml and kept at -20°C for up to one year.
- Dilute isolectin to 20 μg/mL in growth medium and incubate slice cultures at normal incubation conditions 4-10 hours before imaging.
- Imaging set-up.
- Imaging set-up consists of: microscope, shutter, camera, light, 2.0 neutral density filter, 36°C, gas: 5% CO2, air (or 20% oxygen, balance nitrogen).
- Turn on the microscope, camera, UV light, temperature control, and gases at least one hour before imaging.
- Set up MetaMorph Imaging Protocol.
- From the "Journal" drop-down menu, choose "Run Journal...".
- This should prompt the opening of the JOURNALS folder, from which you should select "Motion w-var afs (journal previously established that follows the steps below)", click "Open".
- The "Autofocus Frequency" window will pop up next, keep Number at 1, click "OK".
- Next, the Setup "Sequential File Na..." window will pop up, keep everything as-is:
- Base Name: Image;
- Image Number: 1;
- Number Width: 3;
- Save As Type: MetaMorph TIFF;
- if image already exists -> Overwrite automatically;
- Click "Select Directory...", this will cause the "Browse for Folder" window to pop up. Here, select the folder (or create a new folder) where the movie frames should be saved.
- Then, when the correct folder (e.g. C:DataNew Folder) is displayed in the bottom of the "Setup Sequential File Na..." window, click "OK".
- In the Acquire window:
- Select the Setting in the bottom left corner to be DiIMGBIN2;
- "Exposure Time": 20ms; "Binning": 1.
- Then, in the Special tab:
- "Sensor Mode": FT;
- "Digitizer": 10MHz (EM Gain);
- "Gain": Gain3 (3x);
- Select "EM Gain: 45"; Camera Shutter: Open for Expose; Clear Mode: CLEAR PRE EXP; Clear Count: 2, Trigger Mode & Live Trigger Mode: Normal (TIMED).
- "Frames To Avg": 3.
- Click "Close."
- After setting up the MetaMorph imaging, place the insert in a 35mm culture dish with two 1 mm glass rods in the bottom.
- Place three 2 mm pieces of rubber tubing fitted between the walls of the insert and the culture dish to further stabilize the insert.
- Place a ~10 mm wide piece of cotton soaked in an additional 1 mL medium inside the insert to maintain a humidified environment. Make sure that it is flush with the walls and away from the hippocampal slices.
- Cover the top of the culture dish tightly with polyvinyl chloride film to prevent fluid loss.
- Make sure that the focus of the imaging is the CA3 pyramidal cell layer. Note the slice's orientation by taking phase pictures at 5x, 10x, and 20x.
- In the "Find Focus" window that appears on the screen, set Range, current +/- to be "8", and Accuracy, in micron(s), to be "1" (both boxes at the bottom should be checked).
- Click "Find Focus" to ensure an in-focus image.
- In the "Acquire Timelapse" window, select Time interval to be "1 minutes", and Duration to be "6 hours" (these are our preferred acquisition parameters).
- To accurately capture microglial movements, imaging should be no less frequent than once per minute.
- In Image Storage, select Stack.
- The Update Image Window box should be checked, but not the other two boxes underneath.
- Select "Illum" to be "Lambda".
- Click "OK".
- The movie will now begin to run. This will mean that nothing else can be performed within the MetaMorph program until the movie finishes or until you stop the movie early by clicking Esc, after which the movie will then stop at the next timed acquisition step.
- At the end of the movie, check for cell injury by Sytox screening as noted above (A).
3. RNA Extraction for Low-Level Cytokine Signaling11-15
We previously presented detailed instructions for detection of low-level cytokine signaling in slice cultures using qPCR and PCR array techniques.6,7
We have markedly improved the sensitivity of our approaches for cytokine mRNA detection in slice cultures and present these improvements here.
The improvements include our approach to tissue harvest and RNA isolation, prescreening of samples for tumor necrosis factor alpha (TNF-α) change, and cDNA preamplfication, which are ~ 6 times more sensitive than previous techniques and, for example, allow detection for the first time of slice culture interferon-gamma (IFN-λ) detection.15
- PCR preparation.
- Slice culture harvest.
- Following experiments, slice culture inserts are submerged in 2-3 mL RNAlater and stored at 4°C for up to 3 days until further processing.
- To harvest, remove extraneous (i.e., any tissues adjacent to the hippocampus proper) tissue using a diamond knife and lift the slices into individual RNase/DNase free 1.5 mL centrifuge tubes containing 0.5 mL sterile phosphate buffered saline (PBS) using a fine tipped paint brush.
- Spin samples (10,000 rpm x 1 min) in a stereotypic fashion so all slices adhere to the same side of their tubes.
- Remove PBS supernatant and resuspend sample in 500 μL TRIzol.
- Store samples at -80°C or keep on ice until RNA extraction.
- RNA extraction usingTRIzol with the RNeasy Micro kit results in greater RNA yield (Fig. 4) than use of the kit alone.
- Thaw TRIzol-samples and incubate at room temperature for 5 min.
- Dissociate tissue by drawing samples up and down with 1 mL tipped pippetor and/or vortexing until solid tissue is no longer visible.
- Add 100 μL RNase free (RF) chloroform and invert tube 2-3 times to mix.
- Incubate 3 min at room temp, vortexing every min.
- Centrifuge at 13,000 rpm for 15 min at 4°C.
- Transfer supernatant to a clean 1.5 mL microcentrifuge tube. Be careful not to disturb interface.
- Add an equal volume of room temperature RF 70% molecular biology grade ethanol (~300 μL).
- Mix gently by pipetting up and down a few times.
- Continue with the following steps per RNeasy Micro kit directions:
- Apply sample to an RNeasy micro column placed in a 2mL collection tube.
- Centrifuge at 10,000 rpm for 15 sec, discard flow-through.
- Wash RNeasy column with 350 μL buffer RW1.
- Centrifuge at 10,000 rpm for 15 sec, discard flow-through.
- Add 10 uL DNase 1 stock solution to 70 μL buffer RDD, mix gently.
- Apply 80 μL of DNase solution directly onto membrane and incubate at room temperature for 20 min.
- Wash RNeasy column with 350 μL buffer RW1.
- Centrifuge at 10,000 rpm for 15 sec, discard flow-through and tube.
- Add 500 μL buffer RPE to RNeasy column.
- Centrifuge at 10,000 rpm for 15 sec, discard flow-through.
- Add 500 μL RF 80% molecular biology grade ethanol to RNeasy column.
- Centrifuge at 10,000 rpm for 2 min, discard flow-through and tube.
- Place RNeasy column in new tubes with the caps open.
- Centrifuge at full speed (i.e., 14,000 rpm) for 5 min.
- Transfer RNeasy column to a RF microcentrifuge tube.
- Apply 14 μL RF water directly to the membrane.
- Centrifuge at 10,000 rpm for 1 min to elute.
- Store samples at -80°C or continue to next steps.
- RNA quantification.
- Dilute RiboGreen 1:200 in RF 1x Tris-EDTA (TE) buffer.
- Prepare yeast tRNA to be used as the RNA standard.
- Working stock is stored in -20°C at 100 μg/mL
- Dilute to 1 μg/mL (10 μL in 990 μL TE).
- Create standards as follows: For 100 ng/ μL, add 100 μL, for 80 ng/ μL, add 80 μL tRNA and 20 μL TE, for 60 ng/μL, add 60 μL tRNA and 40 μL TE, for 40 ng/μL add 40 μL tRNA and 60 μL TE, for 20 ng/μL add 20 μL tRNA and 80 μL TE, and for 0 ng/μL add 100 μL TE.
- Prepare samples:
- Combine 99 μL TE + 1 μL sample.
- Run each sample in duplicate.
- Using a multi-channel pipettor, add 100 μL of diluted RiboGreen per well.
- Pipette up and down to mix.
- Measure fluorescence on a plate reader.
- Fluorescein (485 nm/535 nm, 0.1 sec) program.
- Export results into an Excel spreadsheet.
- Determine RNA concentration.
- Graph absorbency versus RNA concentration and create a best-fit linear regression line in Excel.
- Determine sample RNA concentrations from the regression equation associated with the best-fit line.
- Determine RNA quality.
- Ribosomal RNA band integrity is measured via agarose gel (Fig. 4).
- Though it is impractical to run a fraction of each RNA sample obtained (since = 1 μg of RNA is required and our yields are small), it is a good idea to periodically run a denaturing agarose gel on an exemplary sample as proof that the method, when done properly, yields high quality RNA (i.e., without degradation).
- Prepare a 1% agarose gel (for a 100 mL volume).
- Clean the electrophoresis tank, casting tray and gel combs thoroughly with RNase AWAY.
- Weigh out 1 g of ultrapure agarose into a flask.
- Add 83 mL of RNase free water and 10 mL of 10x MOPS [3-(N-Morpholino)propanesulfonic acid] buffer and microwave until agarose is dissolved and solution is clear (~3 min).
- To make 10x MOPS buffer
- Combine 83.7 g MOPS, 13.6 g sodium acetate, and 3.7 g EDTA.
- Add 800 mL of RNase free water, mix to dissolve.
- Adjust pH to 7.0 and fill to 1 L.
- Filter sterilize or autoclave.
- Store at room temperature, protected from light.
- Allow gel to cool to 55°C and add 7 mL 37% formaldehyde (this step should be done in a chemical fume hood).
- Pour gel into casting tray and allow gel to solidify in the hood.
- Prepare the RNA samples.
- RNA sample buffer (500 μL, make fresh).
- Combine 50 μL 10x MOPS, 250 μL formamide, 90 μL 37% formaldehyde, 108 μL RF H2O and 2 μL ethidium bromide (stock solution 10 mg/mL).
- To 1-10 μg of RNA, add double its volume using sample buffer, for a three-fold dilution.
- Denature at 95°C for 5 min, and then chill on ice for 5 min.
- Spin briefly and load the full sample into wells alongside a RNA ladder (add loading dye if desired).
- Electrophorese
- Fill chamber with 1x MOPS buffer.
- Electrophorese at 50-75 volts until markers have migrated about 2/3rd the length of the gel.
- Visualize the gel on a UV transilluminator.
- Verify that there are sharp 18S and 28S ribosomal RNA (rRNA) bands, and that the 28S band is twice as intense as the 18S band (Fig. 4).
- Degraded RNA will have a smeared appearance, fuzzy bands, or will not exhibit a 2:1 ratio.
- Typically we use optical density to assess total RNA quality.
- Though not as sensitive, UV spectrophotometry via a Nanodrop is a quick and cost efficient means of checking RNA quality.
- Look for optical density (OD) 260/280 ratio of 1.5-2.0.
- Keep in mind that the solution in which RNA is resuspended (TE buffer versus water) will affect the OD ratio.
- PCR activities.
- Pre-PCR setup
- Design specific primers
- Using NCBI's Primerblast http://www.ncbi.nlm.nih.gov/tools/primer-blast/:
- Enter the target gene's Refseq accession number.
- Specify PCR product size of 70 to 200 bp.
- Specify a Tm of 55-72°, with less than 5° difference between forward and reverse primers.
- Select "Primer must span an exon-exon junction" to reduce genomic background.
- Enable specificity check for Rat Refseq RNA database to ensure that primers don't recognize additional targets.
- Choose a primer pair from results that fits the following criteria:
- 18-30 bases long.
- Less than 2000 bases apart on your template.
- 40-60% guanine-cytosine content to ensure stable binding of primer to template.
- Choosing a reference gene.
- Not every reference gene will be suitable for your experimental set up. Each time a new paradigm is used, the stability of your reference should be verified. For consistency, we opted to use one of the four reference genes provided on the RT2 Profiler PCR array.
- A good reference gene should meet the following criteria:
- Its expression level is not altered by the experimental paradigm.
- It is expressed at levels similar to that of the target gene.
- To select a suitable reference:
- Review the literature to identify likely candidates for a stable reference gene in your system. For example, we tested Rpl13a because it was shown to be stable in studies of ischemia.11,12
- Design primers as discussed above (or find already-validated primers for common reference genes in a database like RTPrimerDB http://www.rtprimerdb.org/).
- Optimize primers alongside target gene primers (see below).
- Determine which candidate reference is most stable using software like Normfinder (http://www.mdl.dk/publicationsnormfinder.htm) which calculates a stability value for each gene based on the intragroup variance and selects the gene(s) with the least expression variation over the samples. For example, Rpl13a is an optimal reference gene for SD (Fig. 4).
- Optimize primers.
- To obtain consistent and reliable results from qPCR, primers have to meet certain criteria of reproducibility, sensitivity, and specificity.
- It is best to test these parameters for each new primer pair.
- Reproducibility is tested for by running technical replicates.
- Sensitivity can be assessed by performance with a standard curve of successive 4-fold dilutions.
- Specificity is determined by confirming a single product by melt curve analysis and gel electrophoresis (Fig. 5).
- Run a qPCR plate to test the quality of your primers, and the concentration of primers to be used.
- Using cDNA synthesized from whole-brain RNA, create a standard curve (series of consecutive 4-fold dilutions) as follows: 1, 1:4, 1:16, 1:64, 1:256, and no template control (NTC).
- For each dilution, run 1 μL cDNA in duplicate for each concentration of primers, (i.e., 100 nM, 200 nM, 300 nM).
- Confirm that technical replicates give consistent Ct values.
- Examine Ct values for the dilution curve.
- Plot the Ct values against concentration for each of the dilutions. The resultant graph should be linear with a good correlation coefficient (i.e., >0.95).
- Examine melt curve to confirm the presence of a single amplification product (i.e., single peak).
- If there are multiple peaks, or the peaks are not similar, this might suggest contamination, mispriming, primer-dimer artifact, etc. (Fig. 5).
- A peak in the NTC will likely correspond to primer dimers which form more readily in the absence of template cDNA, and shouldn't be a concern.
- Confirm melt-curve data by resolving amplified products on a 1% agarose gel.
- Prepare a 1% agarose gel (for a 100 mL volume)
- Weigh out 1 g of agarose into a flask.
- Add 100 mL of 1x Tris-Acetate EDTA (TAE) buffer and microwave until agarose is dissolved and solution is clear.
- 1 L 50x TAE buffer consists of 2 M Tris base, 57 mL glacial acetic acid, and 0.05 M EDTA (pH 8.0).
- Dilute TAE buffer to 1x with distilled water (final concentrations: 40 mM Tris-acetate and 1.0 mM EDTA).
- Allow gel to cool to 55°C before adding ethidium bromide to a concentration of 0.5 μg/mL.
- Pour gel into casting tray and allow it to solidify at room temperature.
- To run, place gel in electrophoresis chamber filled with 1x TAE, combine 10 μL of sample from the qPCR plate with 2 μL of 6x loading dye, mix well and load into wells alongside a molecular weight ladder.
- Electrophorese at 50-150 volts until markers have migrated an appropriate distance, depending on the expected size of your cDNA product.
- Visualize with a UV transilluminator.
- Confirm that the amplified PCR product is of appropriate size for your target.
- Primer dimers will be around 50 bp.
- Pre-screen for increased TNF-α.
- Since TNF-α initiates inflammation reactions in the periphery, we suspect this is also true for brain and use initial screening for TNF-α mRNA as a marker of the slice culture immune status (i.e., normal, sham, and experimental groups) before proceeding to full PCR array analyses.
- If normal and sham control TNF-α mRNA levels are not within an interassay range found within our laboratory, experiments (i.e., normal, sham, and experimental groups) are discarded and do not proceed to full PCR array analyses.
- iScript cDNA synthesis.
- For each RNA sample, combine the following in a PCR tube: 4 μL 5x reaction mix, 1 μL reverse transcriptase, 25 ng RNA, and RF H2O to a final volume of 20 μL.
- Mix gently by pipetting and spin down briefly.
- Incubate: 25°C, 5 min; 42°C, 30 min; 85°C, 5 min.
- Dilute 1:4 (add 60 μL RNase free water).
- Store at -20°C or keep on ice until ready to use within hours.
- qPCR for TNF-α.
- Prepare a master mix for each primer pair.
- Combine 12.5 μL iQ SYBR green, 1 μL primer mix and 10.5 μL water per sample.
- For both our Rpl13a and TNF-α primers, 200 nM is the optimal concentration.
- Adjust the amount of primers and volume of waster as necessary in a 1.5 mL RF microcentrifuge tube and keep on ice while you set up the plate.
- Set up a qPCR plate with controls/shams/experimentals. Use 1 μL of cDNA per sample in duplicate for each gene.
- Add 24 μL of your master mix to each well and pipette up and down to mix.
- Be very careful to avoid bubbles, as they will interfere with fluorescence readings.
- Run 40 cycles of PCR amplification:
- 95°C, 8.5min;
- 40 cycles: 95°C, 10 sec; 60°C, 30 sec; 72°C, 20 sec/cycle.
- Melt curve: 55°C, 1 min; 80 cycles of 0.5°C increments for 10 sec each starting at 55°C.
- Analyze data (ΔΔCt method; Fig. 4).13
- cDNA synthesis for PCR arrays.
- Calculate volume (μL) of each sample to have 250 ng of RNA.
- Choose an amount of RNA (100 ng to 1 μg) that you can consistently extract from your samples.
- RT2 First Strand Kit - as per manufacturer's instructions. Briefly:
- Thaw and spin down all reagents.
- For each RNA sample, combine the following in a PCR tube:
- 250 ng of RNA, 2 μL of the 5x genomic DNA (gDNA) elimination buffer and water to a final volume of 10 μL.
- Mix gently by pipetting and spin down briefly.
- Incubate at 42°C for 5 min.
- Chill on ice for 1 min.
- In the mean time, prepare the reverse transcription (RT) cocktail (per sample):
- Combine 4 μL of 5x RT buffer 3, 1 μL primer and external control mix, 1 μL RT enzyme mix 3, and 3 μL of water.
- Add 10 μL of RT cocktail to each sample and mix gently.
- Incubate at 42°C for 15 min.
- Stop reaction by incubating at 95°C for 5 min.
- Add 91 μL of RF H2O to each 20 μL cDNA synthesis reaction (dilute to a final volume of 111 μL).
- Mix well by pipetting.
- Store at -20°C or keep on ice until ready to use within hours.
- RT2 Profiler PCR array.
- The following instructions follow those of the manufacturer and are reiterated here for convenience.
- Prepare reagents
- Thaw and spin down all reagents.
- Prepare experimental cocktail:
- Combine 1350 μL of 2x SABiosciences RT2 qPCR SYBR Green Master Mix, 102 μL of diluted cDNA and 1248 μL of RF H2O to a final volume of 2700 μL.
- Carefully remove PCR array from sealed bag.
- Dispense cocktail into a loading reservoir.
- Add 25 μL to each well with a multi-channel pipettor.
- Carefully seal PCR array plate.
- Centrifuge the plate at 10,000 rpm for 1 min at room temperature.
- Place on ice until PCR program is ready.
- Run the appropriate cycling program for your machine.
- iCycler:
- 95°C, 10 min
- 40 cycles: 95°C, 15 sec; 60°C, 1 min.
- Melt curve: 55°C, 1 min; 80 cycles of 0.5°C increments for 10 sec each starting at 55°C
- Analyze data.
- Select "Auto Calculated" for baseline cycles.
- Select "User Defined" for threshold position to manually set threshold value.
- In "Log View", set threshold in the lower one-third of linear phase of the amplification plot.
- Be sure to keep threshold value the same across runs.
- Export Data.
- Input into an Excel file.
- Upload to SABiosciences PCR Array Data Analysis.
- Select reference genes - we used Rpl13a.
- Our practice is to:
- Confirm results with at least a duplicate PCR array.
- 3-fold array changes need not be confirmed by subsequent specific target PCR amplification.6,7
- Less than 3-fold changes should be confirmed by subsequent specific target PCR amplification or use of 2-3 PCR arrays (to confirm 2x changes.
- DNA pre-amplification is used to analyze targets with expected ultra-low expression (Fig. 6).
- SABiosciences's RT2 Nano PreAMP cDNA synthesis kit and primer mixes are intended for pre-amplification of RNA to allow use of small amounts (1-100 ng) of sample RNA to run a full PCR array.
- Each primer mix allows for the amplification of 89 gene-specific cDNA target templates with minimal bias. We have used this system as a means of amplifying low level signals such as IFN-λ to a detectible level.
- RT2 Nano PreAMP cDNA synthesis kit - as per manufacturer's instructions. Briefly:
- Thaw and spin down all reagents.
- For each RNA sample, combine the following in a PCR tube.
- 1-100ng of RNA, 2 μL of the 5x gDNA elimination buffer and water to a final volume of 10 μL.
- Mix gently by pipetting and spin down briefly.
- Incubate at 42°C for 5 min.
- Chill on ice for 1 min.
- In the meantime, prepare the RT cocktail (per sample).
- Combine 4 μL of 5x RT buffer 3, 1 μL primer and external control mix, 1 μL cDNA enzyme synthesis mix, 1 μL RNase inhibitor, and 3 μL of water.
- Add 10 μL of RT cocktail to each sample and mix gently.
- Incubate at 42°C for 15 min.
- Stop reaction by incubating at 95°C for 5 min.
- Store at -20°C or keep on ice until ready to use.
- cDNA amplification with specific primers.
- Mix the following in a PCR tube:
- 12.5 μL PCR master mix, 7.5 μL specific primers, and 5 μL of undiluted cDNA.
- Run 12 cycles of PCR amplification.
- 95°C, 10 min.
- 12 cycles: 95°C, 15 sec; 60°C, 2 min.
- Hold at 4°C.
- Add 2 μL side reaction reducer to each sample.
- Incubate at 37°C for 15 min.
- Stop reaction by incubating at 95°C for 5 min.
- Add 84 μL RF water to each 27 μL cDNA amplification reaction.
- Store at -20°C or keep on ice until ready to use within hours.
- Amplify individual gene targets using SAB primers and SYBR green master mix as described above.
4. Multiplexed Phosphoprotein Assays of Cytokine Signaling
Mutliplexed phosphoprotein assays are used to define cytokine ligand-receptor downstream signaling (Fig. 7).
- Determine amount of total protein in slice cultures.
- Harvest slice cultures by gently lifting slices off of inserts and place in 1 mL cold (4°C) PBS.
- Centrifuge tubes for 30 sec and remove PBS. Place on dry ice until finished harvesting.
- Store at -80°C.
- Homogenize slice cultures for total protein assay.
- Prepare cell lysis buffer according to manufacturer's instructions.
- Add protease inhibitors to a total volume of buffer needed to place at least 200 μL of lysis buffer on each slice culture.
- For example: add 20 μL of Factor 1, 10 μL of Factor 2, and 20 μL of 500 mM PMSF in DMSO to 4.95 mL cell lysis buffer.
- Thaw slice cultures in 200 μL lysis buffer on ice.
- Sonicate slice cultures five times in 1 sec pulses and place immediately back on ice.
- Vortex samples for 10 sec.
- Centrifuge samples at 4°C for 15 min at 13,000 rpm.
- Transfer supernatant to a clean tube and keep on ice.
- Perform total protein assay.
- Prepare protein standards.
- Reconstitute stock bovine serum albumin (BSA) according to manufacturer's instructions.
- Prepare standards ranging from 0-1000 μg/mL diluted in high purity water for BCA protein assay.
- Calculate volume of working reagent needed based on the number of samples and replicates.
- For example: (8 standards + 18 unknown samples) x (2 replicates per sample) x (0.2 mL working reagent needed per well) = 10.4 mL total volume needed for all samples loaded onto the microplate.
- Round up to 12 mL total volume to ensure enough volume.
- When mixing Reagent A with Reagent B for the working reagent, some turbidity may occur, but should disappear shortly.
- Load 10 μL of samples and standards per well.
- Load 0.2 mL of working reagent into wells.
- Cover plate with sealing tape and shake for 30 sec to mix.
- Incubate plate at 37°C for 30 min.
- Cool plate to room temperature (about 10 min) before reading on a plate reader at 595 nm wavelength.
- Construct a standard curve using Microsoft Excel with measured absorbance vs. expected concentration.
- A good correlation coefficient is equal to or greater than 0.98.
- Calculate protein concentrations in samples using the linear equation derived from the standard curve.
- Dilute samples in lysis buffer to equal amounts of protein and store at -80°C until ready for further processing.
- Protein concentration should range from 200-900 μg/mL according to manufacturer's instructions.
- Perform Multiplex Assay.
- Note: multiplexed phosphoprotein assays take 2 days total to complete, with 1 hour of initial set-up and loading of plate followed by an overnight incubation step and additional incubation and wash steps the following day.
- Map out which wells will contain which lysate according to worksheet in manual.
- Thaw homogenized tissue samples and kit lysates at room temperature and then place on ice. Bring all buffers and reagents provided in kit to room temperature (except streptavidin and beads).
- Prepare beads for multiplex assay.
- Cover tubes containing coupled beads with aluminum foil to protect from light. Vortex tubes for 5 sec and keep on ice.
- Dilute coupled beads 1:50 in wash buffer.
- Each well should contain 1 μL of coupled beads in 49 μL wash buffer for a total volume of 50 μL per well.
- Calibrate the vacuum apparatus.
- Wash the plate with 100 μL wash buffer per well.
- Turn on vacuum and suction off any excess liquid within 2-5 sec.
- If complete removal of buffer takes longer or shorter than 2-5 sec, adjust pressure valve accordingly.
- Dry the bottom of the plate thoroughly before adding samples.
- Vortex the diluted coupled beads for 5 sec and add 50 μL to designated wells.
- Immediately vacuum filter and dry the bottom of the plate with a paper towel.
- Wash the plate twice with 100 μL wash buffer per well. Dry the bottom of the plate after each wash.
- Vortex thawed samples for 3 sec and add 50 μL to designated wells. Place sealing tape over plate and incubate for 15-18 hrs shaking at 300 rpm at room temperature, protected from light.
- Vacuum filter excess liquid and wash plate three times with 100 μL wash buffer. Dry the bottom of the plate after each wash.
- Dilute detection antibody 1:25 in wash buffer.
- Each well requires 1 μL detection antibody in a total volume of 25 μL wash buffer.
- Place sealing tape on plate and protect from light with aluminum foil. Shake for 30 sec, gradually increasing speed to 1100 rpm. Continue shaking at 300 rpm for 30 min at room temperature.
- Vacuum filter the plate and wash three times with 100 μL wash buffer. Dry the bottom of the plate after each wash.
- While protecting the plate from light, prepare a 1:100 dilution of streptavidin-PE with enough volume to add 50 μL per well. Protect solution from light.
- Vortex thoroughly and add 50 μL diluted streptavidin-PE per well. Incubate with shaking at 1100 rpm for 30 sec followed by 10 min at 300 rpm.
- Vacuum filter excess buffer off.
- Wash the plate three times with 100 μL resuspension buffer per well.
- Dry the bottom of the plate thoroughly after each wash.
- Add 125 μL resuspension buffer to each well and shake for 30 sec at 1100 rpm.
- Immediately read plate or store at 4°C away from light for up to 24 hours.
5. Mimicking and Modulation of Cytokine Signaling
- Recombinant cytokine proteins (with carrier) are used to mimic changes of SD.
- Lyophilized proteins (e.g., TNF-α) are stored for up to 1 year at -20°C.
- After diluting to a stock solution with 0.1% bovine serum albumin, aliquots are prepared, stored at -20°C, until use within 3 months.
- Any manipulations to slice cultures are refreshed at least every third day.
- Cytokine signaling is silenced by:
- Use of traditional cytokine signaling cascade pharmacological agents;
- Use of soluble ligand antagonist [e.g., soluble TNF receptor 1 (as described above for recombinant ligands)]; or
- Gene silencing [i.e., small interfering RNA (siRNA)].
- Delivery of siRNA to neuronal cells is often problematic.
- Common lipid-based reagents usually result in low transfection efficiency and loss of cell viability.
- Instead, we use Thermo Scientific's Dharmacon Accell siRNAs, which allow for high efficiency knockdown without use of a transfection reagent.
- Here we show knockdown of cyclophilin B, a non-essential protein expressed in all cell types, as confirmation of the use of this method in slice cultures.
- Delivery protocol was adapted for slice cultures from manufacturer's instructions for use in adherent cells.
- Dilute 5x siRNA buffer to 1x with RNase free water.
- Prepare a 100μM siRNA solution in 1x buffer
- Cyclophilin B siRNA is provided at 5 nmol. Resuspend in 50 μL of 1x buffer for a 100 μM stock.
- Mix siRNA with Accell delivery medium to a final concentration of 1μM siRNA.
- Add 11 μL of 100 μM stock siRNA to 1100 μL of Accell delivery medium in a 6-well plate.
- Place in the incubator and allow temperature to equilibrate to normal incubation conditions for at least 20 min.
- Using a BSL-2 hood and sterile technique, transfer inserts into the delivery mix-containing wells.
- Incubate with delivery mix for 96 hrs for protein knockdown.
- Assess protein knockdown by Western blot or intensified immunostaining.
- Western blots, while a potential first choice for knockdown efficiency, may be too insensitive for low-level immune signaling associated with the non-injurious phenomenon of SD.
- Accordingly, we have followed negative Western blot measurements with silver enhanced diaminobenzidine (DAB) immunostaining and computer-based densitometry measurements (see below).
6. Proteomic Confirmations
- Cell specificity of phosphoprotein changes is established using immunostaining.6,7
- Fix slice cultures for 24 hrs with PLP fixative consisting of:
- 10 mL 16% paraformaldehyde, 1.096 g lysine, 0.42 g sodium phosphate, and 0.17 g sodium periodate, filled to a total volume of 80 mL with ultrapure water (fixative is 6.2 pH).
- After 24 hrs, gently remove slice cultures from the inserts with a fine brush and transfer to PBS containing sodium azide (100 mg/L) until ready to section.
- Then, incubate slices at least 24 hrs in PBS-20% sucrose, cut whole slice cultures to 20 μm sections, and mount to gel coated slides.
- Fluorescent immunostaining (e.g., for NFκB) is accomplished as follows with all steps coupled to shaking at ~50 rpm.
- Wash slice cultures in 3 mL PBS three times for 10 min.
- Place slides with mounted 20 μm slice cultures in Coplin jars with ~40 mL PBS for all wash steps.
- Wash slice cultures in PBS three times, 10 min per wash.
- Place a few drops of SFX signal enhancer on slice cultures and incubate in a humid chamber for 30 min.
- Incubate slice cultures for 2 hrs at 37°C with rabbit anti-NFκβ antibody at 1:100 diluted in 0.75% Triton X-100 in PBS.
- Pipette antibody solution directly onto slices.
- Remove slices from antibody solution and wash three times in PBS for 10 min per wash.
- Incubate slices at room temperature for one hour in goat anti-rabbit AlexaFluor 594 antibody at 1:50 in 0.75% Triton X-100 in PBS.
- Centrifuge AlexaFluor 594 antibody for 15 min at 10,000 rpm to remove any precipitate that may have formed in solution.
- Wash three times in PBS, 10 min per wash.
- Dip slides in distilled water to rinse off any salts from PBS.
- Coverslip with ProLong antifade medium and let dry for at least 2 hours, covered away from light.
- Visualize with traditional or confocal (Fig. 7) fluorescence microscopy.
- Protein production changes from siRNA knockdown can be sensitively assessed by enhancing traditional DAB immunostaining7 via silver intensification16 and subsequent densitometric quantification (e.g., as shown here for cyclophilin B) (Video Figure 2).
- Immunostaining for cyclophilin B follows standard procedures for bright field imaging we recently detailed,7 using:
- Anti-mouse cyclophilin B (1:100) for 24 hours at 4°C;
- Followed by horseradish peroxidase secondary antibody labeling (1:100);
- And DAB visualization.
- f the DAB reactions are too faint to allow digital densitometric quantification17, they can be intensified with a differentiated silver procedure according to Quinn and Graybiel.16
- Rehydrate slides and then thoroughly wash x 3 for 10 minutes in high purity water.
- Place slides in high purity water in a Coplin jar and warm to 56°C.
- Prepare ammoniacal silver nitrate solution (0.5%):
- Stock ammonium chloride (25-30%) is freshly diluted (1:1) with high purity water.
- Add ammonium hydroxide (~500-600 μL per 100 mL) drop by drop with stirring to the 0.5% silver nitrate solution which will briefly turn cloudy and then clear.
- Warm the solution to 56°C.
- Incubate sections for 10-12 min.
- Wash slides for 15 sec in high purity water (this and all subsequent steps are done at room temperature using Coplin jars).
- Dip slides for 15 sec in 1% sodium thiosulfate (pentahydrate).
- Dip slides for 15 sec in high purity water.
- Tone the slides for 2 min in 0.2% gold chloride.
- Dip the slides in high purity water for 15 sec.
- Expose the slides to 0.5% oxalic acid for 2 min.
- Dip the slides for 15 sec in high purity water.
- Treat the slides for 5 min in 5% sodium thiosulfate.
- Wash slides thoroughly in high purity water, dehydrate and coverslip.
- Slides are now ready for densitometric quantification.17
- All equipment, including bright field lighting, is turned on for at least 20 min before imaging.
- Whole slice culture sections are imaged at 5-10x on an inverted microscope.
- Light intensity is set to a uniform level (e.g., ~2.6 V) and not changed throughout all image acquisitions.
- Neutral density filters are used as needed to reduce full image transmitted light intensity to ~ 1500 on a 12-bit scale (0-4095) where "0" is completely black and 4095 is completely white.
- Digital images are then acquired and stored electronically for all (i.e., sham control and experimental samples), including a background image derived form an area of the slide without slice culture sections.
- The background image is subtracted from all images (sham and experimental) and an area of interest (AOI) drawn around each slice culture and transmitted light intensity registered.
- Image intensity range is set to a constant range for all experiments.
- For clarity, resultant expressed as a relative density compared to control levels (Fig. 8).
7. Representative Results:
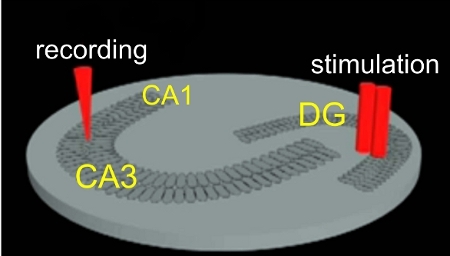 Figure 1. Slice culture electrophysiological recording paradigm. Recording microelectrode is first placed in the CA3 pyramidal neuron layer and then a bipolar stimulating electrode is placed in the dentate gyrus (DG).
Figure 1. Slice culture electrophysiological recording paradigm. Recording microelectrode is first placed in the CA3 pyramidal neuron layer and then a bipolar stimulating electrode is placed in the dentate gyrus (DG).
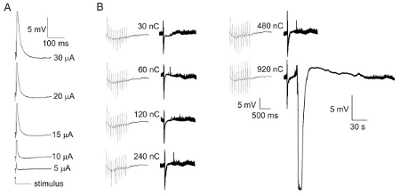 Figure 2. CA3 evoked field potentials and synaptically induced spreading depression. (A) Experiments begin with determination of the current needed to maximize standard CA3 area field potential responses and then half-maximal intensity is used to elicit CA3 area evoked field potentials. This documents the normalcy of evoked synaptic responses between preparations. If a slice's field excitatory post synaptic potentials are not at least 3 mV, slices are discarded. (B) Next, trans-synaptically evoked threshold for triggering spreading depression is determined (right, slow responses) while at the same time field potentials (left, fast responses) are used to document that preparations are healthy enough to follow 10 Hz stimulation (e.g., amplitudes of vertical deviations in fast potential records are similar).
Figure 2. CA3 evoked field potentials and synaptically induced spreading depression. (A) Experiments begin with determination of the current needed to maximize standard CA3 area field potential responses and then half-maximal intensity is used to elicit CA3 area evoked field potentials. This documents the normalcy of evoked synaptic responses between preparations. If a slice's field excitatory post synaptic potentials are not at least 3 mV, slices are discarded. (B) Next, trans-synaptically evoked threshold for triggering spreading depression is determined (right, slow responses) while at the same time field potentials (left, fast responses) are used to document that preparations are healthy enough to follow 10 Hz stimulation (e.g., amplitudes of vertical deviations in fast potential records are similar).
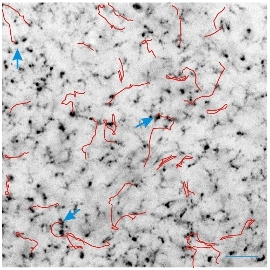 Figure 3. Microglial motion associated with reduced synaptic activity. Evidence shows that microglial branches extend and retract with increased synaptic activity, but long-distance motion of these cells in response to synaptic activity has not been described in uninjured brain. Our results using fluorescent isolectin B4 labeling of microglia show that a fraction of these cells move long distances under normal conditions. Furthermore, the number of these traveling microglia is significantly increased when synaptic activity is abolished by culture exposure to tetrodotoxin as shown in the accompanying image. Red lines mark paths traveled by microglia over a period of 6 hrs with blue arrows marking the origin of a few exemplary microglia. Calibration bar, 50 μm.
Figure 3. Microglial motion associated with reduced synaptic activity. Evidence shows that microglial branches extend and retract with increased synaptic activity, but long-distance motion of these cells in response to synaptic activity has not been described in uninjured brain. Our results using fluorescent isolectin B4 labeling of microglia show that a fraction of these cells move long distances under normal conditions. Furthermore, the number of these traveling microglia is significantly increased when synaptic activity is abolished by culture exposure to tetrodotoxin as shown in the accompanying image. Red lines mark paths traveled by microglia over a period of 6 hrs with blue arrows marking the origin of a few exemplary microglia. Calibration bar, 50 μm.
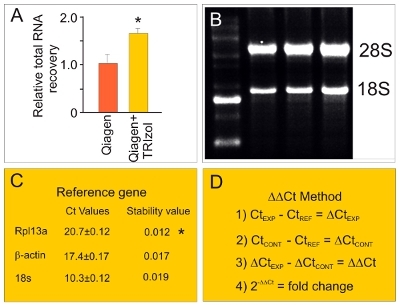 Figure 4. PCR screening activities. (A) We use TRIzol along with Qiagen kits for RNA isolation since, in our hands, this results in significantly greater (P < 0.001; t-test) RNA yield from hippocampal slice cultures than use of Qiagen kits alone. (B) In addition, we periodically confirm that our RNA extraction procedures produce high quality intact RNA as determined via a gel that shows sharp RNA bands. (C) We use NormFinder software to choose a reference gene with a best stability value. For example, slice cultures exposed to lipopolyssacharide (100 ng/mL for 2 hrs) were analyzed for three reference genes (Rpl13a, β-actin, 18s). Ct values are used to calculate a stability value. Our data here [and that for other experiments using spreading depression (data not shown)] show that Rpl13a is an optimum reference gene. (D) We use the "ΔΔCt" method as a sensitive and reproducible means to detect fold-change RNA differences between experimental groups and sham controls.
Figure 4. PCR screening activities. (A) We use TRIzol along with Qiagen kits for RNA isolation since, in our hands, this results in significantly greater (P < 0.001; t-test) RNA yield from hippocampal slice cultures than use of Qiagen kits alone. (B) In addition, we periodically confirm that our RNA extraction procedures produce high quality intact RNA as determined via a gel that shows sharp RNA bands. (C) We use NormFinder software to choose a reference gene with a best stability value. For example, slice cultures exposed to lipopolyssacharide (100 ng/mL for 2 hrs) were analyzed for three reference genes (Rpl13a, β-actin, 18s). Ct values are used to calculate a stability value. Our data here [and that for other experiments using spreading depression (data not shown)] show that Rpl13a is an optimum reference gene. (D) We use the "ΔΔCt" method as a sensitive and reproducible means to detect fold-change RNA differences between experimental groups and sham controls.
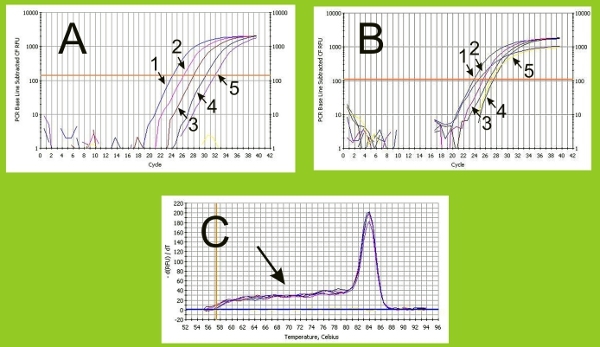 Figure 5. PCR reaction checks. (A) We measure dilution curve amplifications for primers as a means to verify the quality of PCR reactions. For example, optimal amplifications show a uniform (1-5) increase in Ct thresholds. (B) In contrast, amplifications are not uniform with poor primers (1-5). (C) In addition, we perform a melt curve at the conclusion of PCR assays to confirm that the desired amplicons are detected (i.e., a single peak curve), which indicates the absence of any double stranded DNA including primer dimers, contaminating DNA and misannealed PCR product (which would appear as multiple peak curves).
Figure 5. PCR reaction checks. (A) We measure dilution curve amplifications for primers as a means to verify the quality of PCR reactions. For example, optimal amplifications show a uniform (1-5) increase in Ct thresholds. (B) In contrast, amplifications are not uniform with poor primers (1-5). (C) In addition, we perform a melt curve at the conclusion of PCR assays to confirm that the desired amplicons are detected (i.e., a single peak curve), which indicates the absence of any double stranded DNA including primer dimers, contaminating DNA and misannealed PCR product (which would appear as multiple peak curves).
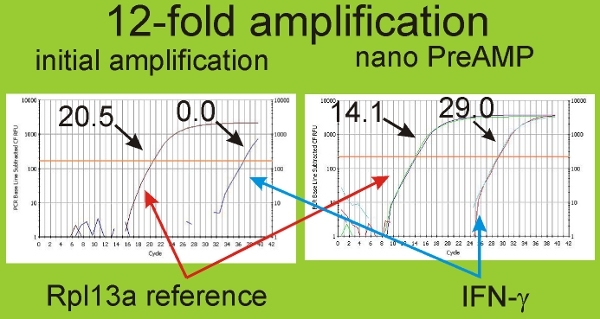 Figure 6. Nano pre-amplification allows detection of IFN-λ in hippocampal slice cultures. Since only ~50 T-cells are found in a slice culture (out of ~55,000-90,000 total cells), we used the RT2 nano PreAMP cDNA Synthesis Kit as a means to detect potential ultra-low level expression of IFN-λ. This means of cDNA preamplification provided a 12-fold increase in sensitivity to RNA levels. The results shown above illustrate the capacity of preamplification to increase detection sensitivity. Ct threshold for reference gene Rpl13a was 20.5 with initial amplification and this was increased to 14.1 with use of the preamplification kit, a 12-fold increase corresponding to 12 cycles of PCR amplification of cDNA. In parallel, IFN-λ was not detectable (0.0) but was well within detection range (29.0) with preamplification.
Figure 6. Nano pre-amplification allows detection of IFN-λ in hippocampal slice cultures. Since only ~50 T-cells are found in a slice culture (out of ~55,000-90,000 total cells), we used the RT2 nano PreAMP cDNA Synthesis Kit as a means to detect potential ultra-low level expression of IFN-λ. This means of cDNA preamplification provided a 12-fold increase in sensitivity to RNA levels. The results shown above illustrate the capacity of preamplification to increase detection sensitivity. Ct threshold for reference gene Rpl13a was 20.5 with initial amplification and this was increased to 14.1 with use of the preamplification kit, a 12-fold increase corresponding to 12 cycles of PCR amplification of cDNA. In parallel, IFN-λ was not detectable (0.0) but was well within detection range (29.0) with preamplification.
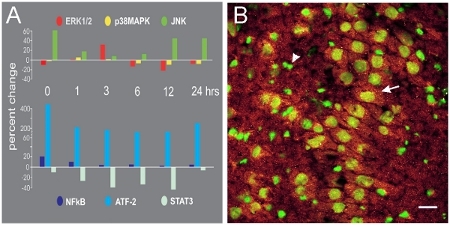 Figure 7. Innate cytokine pathway phosphoprotein phosphorylation changes. (A) Exemplary effect of TNF-α pretreatment (i.e., neuroprotection) on innate cytokine pathway phosphoprotein phosphorylation 24 hrs after excitotoxic injury in hippocampal slice cultures. Results show CA1 area changes between slices pretreated for 3 days with 100 ng/mL TNF-α before exposure to N-methyl-d-aspartate (NMDA) compared to those exposed to NMDA alone (i.e., experimental vs. sham) expressed as a percentage. Notice that TNF-α induced a sustained phosphorylation increase in JNK and ATF-2 as well as a transient increase in NFκB. (B) The spatial distribution of NFκB activation (i.e., presence of phosphorylated NFκB in the nucleus) is revealed by immunostaining (red). YoYo stains nuclei green in a slice culture section [e.g., in astrocytes (arrow head)]. Pyramidal neurons, which otherwise would also appear green, are instead yellow because of concomitant marking with phosphorylated NFκB (red). Calibration bar, 25 μm.
Figure 7. Innate cytokine pathway phosphoprotein phosphorylation changes. (A) Exemplary effect of TNF-α pretreatment (i.e., neuroprotection) on innate cytokine pathway phosphoprotein phosphorylation 24 hrs after excitotoxic injury in hippocampal slice cultures. Results show CA1 area changes between slices pretreated for 3 days with 100 ng/mL TNF-α before exposure to N-methyl-d-aspartate (NMDA) compared to those exposed to NMDA alone (i.e., experimental vs. sham) expressed as a percentage. Notice that TNF-α induced a sustained phosphorylation increase in JNK and ATF-2 as well as a transient increase in NFκB. (B) The spatial distribution of NFκB activation (i.e., presence of phosphorylated NFκB in the nucleus) is revealed by immunostaining (red). YoYo stains nuclei green in a slice culture section [e.g., in astrocytes (arrow head)]. Pyramidal neurons, which otherwise would also appear green, are instead yellow because of concomitant marking with phosphorylated NFκB (red). Calibration bar, 25 μm.
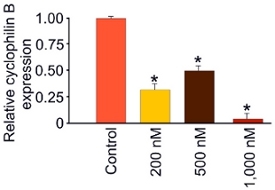 Figure 8. Proteomic confirmation of siRNA efficiency. Differentiated silver intensification of peroxidase-diaminobenzidine based immunostaining for cyclophilin B shows that siRNA can significantly (P < 0.001; ANOVA-Holm-Sidak method) reduce global hippocampal slice cyclophilin B expression 3 days after application of 200, 500, or 1,000 nM siRNA.
Figure 8. Proteomic confirmation of siRNA efficiency. Differentiated silver intensification of peroxidase-diaminobenzidine based immunostaining for cyclophilin B shows that siRNA can significantly (P < 0.001; ANOVA-Holm-Sidak method) reduce global hippocampal slice cyclophilin B expression 3 days after application of 200, 500, or 1,000 nM siRNA.
Discussion
SD is a paroxysmal perturbation of brain whose characteristics are highly conserved in all species and brain regions examined to date. SD is commonly studied in neocortex. Yet, the neural circuit complexities of this structure (compared to hippocampus), as well as the inability to keep neocortex alive in vitro with quiescent immune functions for protracted periods, make it less useful as an in vitro preparation for defining the long-term consequences of SD susceptibility. In contrast, hippocampus is not only the brain structure that is most sensitive to SD, it is also an archicortical structure with a more simplified neural circuit structure and function, which is well suited for defining the neuroimmune means by which neural activity can modulate SD susceptibility.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NS-19108), the National Institute of Child Health and Disease (PO1-HD09402), the Migraine Research Foundation and the White Foundation. Ms. Marcia P. Kraig assisted in the preparation and maintenance of culture systems.
References
- Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci. 1998;18(9):3416–3425. doi: 10.1523/JNEUROSCI.18-09-03416.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, Hulse RE, Schmitt MW, Nicholson C, Kraig RP. Optical current source density analysis in hippocampal organotypic culture shows that spreading depression occurs with uniquely reversing currents. J Neurosci. 2005;25(15):3952–3961. doi: 10.1523/JNEUROSCI.0491-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse RE, Swenson WG, Kunkler PE, White DM, Kraig RP. Monomeric IgG is neuroprotective via enhancing microglial recycling endocytosis and TNF-αlpha. J Neurosci. 2008;28(47):12199–12211. doi: 10.1523/JNEUROSCI.3856-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer NP, Jarhult JD, Nitsch R. Resting microglial cells in vitro: analysis of morphology and adhesion molecule expression in organotypic hippocampal slice cultures. Glia. 1996;18(4):319–331. doi: 10.1002/(sici)1098-1136(199612)18:4<319::aid-glia6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Mitchell HM, White DM, Domowicz MS, Kraig RP. Cold pre-conditioning neuroprotection depends on TNF-αlpha and is enhanced by blockade of interleukin-11. J Neurochem DOI. 2010.
- Mitchell HM, White DM, Kraig RP. Strategies for study of neuroprotection from cold-preconditioning. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Kunkler PE, Kraig RP. Reactive astrocytosis from excitotoxic injury in hippocampal organ culture parallels that seen in vivo. J Cereb Blood Flow Metab. 1997;17(1):26–43. doi: 10.1097/00004647-199701000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg YY, Milton JG, Kraig RP. Spreading depression sends microglia on Lévy flights. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koroleva VI, Oitzl MS, Bures J. Threshold of synaptically elicited cortical spreading depression: drug-induced changes in rats. Electroencephalogr Clin Neurophysiol. 1985;60(1):55–64. doi: 10.1016/0013-4694(85)90951-4. [DOI] [PubMed] [Google Scholar]
- Gubern C. Validation of housekeeping genes for quantitative real-time PCR in in-vivo and in-vitro models of cerebral ischaemia. BMC Mol Biol. 2009;10:57–57. doi: 10.1186/1471-2199-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian YF. The quantification of ADAMTS expression in an animal model of cerebral ischemia using real-time PCR. Acta Anaesthesiol Scand. 2007;51(2):158–164. doi: 10.1111/j.1399-6576.2006.01161.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45–e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Pusic AD, Kraig RP. Inflammatory gene micro array profiling demonstrates "T-cell-like" activation after recurrent spreading depression - implications for migraine pathogenesis. Soc Neurosci abst. 2010.
- Quinn B, Graybiel AM. A differentiated silver intensification procedure for the peroxidase-diaminobenzidine reaction. J Histochem Cytochem. 1996;44(1):71–74. doi: 10.1177/44.1.8543785. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence induction of reactive gliosis from spreading depression in microglia but not astrocytes. J Comp Neurol. 1996;369(1):93–108. doi: 10.1002/(SICI)1096-9861(19960520)369:1<93::AID-CNE7>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]