

Abstract
Background
The breadth of the clinical spectrum underlying Pelizaeus-Merzbacher disease and spastic paraplegia type 2 is due to the extensive allelic heterogeneity in the X-linked PLP1 gene encoding myelin proteolipid protein (PLP). PLP1 mutations range from gene duplications of variable size found in 60-70% of patients to intragenic lesions present in 15-20% of patients.
Methods
Forty-eight male patients from 38 unrelated families with a PLP1-related disorder were studied. All DNA samples were screened for PLP1 gene duplications using real-time PCR. PLP1 gene sequencing analysis was performed on patients negative for the duplication. The mutational status of all 14 potential carrier mothers of the familial PLP1 gene mutation was determined as well as 15/24 potential carrier mothers of the PLP1 duplication.
Results and Conclusions
PLP1 gene duplications were identified in 24 of the unrelated patients whereas a variety of intragenic PLP1 mutations were found in the remaining 14 patients. Of the 14 different intragenic lesions, 11 were novel; these included one nonsense and 7 missense mutations, a 657-bp deletion, a microdeletion and a microduplication. The functional significance of the novel PLP1 missense mutations, all occurring at evolutionarily conserved residues, was analysed by the MutPred tool whereas their potential effect on splicing was ascertained using the Skippy algorithm and a neural network. Although MutPred predicted that all 7 novel missense mutations would be likely to be deleterious, in silico analysis indicated that four of them (p.Leu146Val, p.Leu159Pro, p.Thr230Ile, p.Ala247Asp) might cause exon skipping by altering exonic splicing elements. These predictions were then investigated in vitro for both p.Leu146Val and p.Thr230Ile by means of RNA or minigene studies and were subsequently confirmed in the case of p.Leu146Val. Peripheral neuropathy was noted in four patients harbouring intragenic mutations that altered RNA processing, but was absent from all PLP1-duplication patients. Unprecedentedly, family studies revealed the de novo occurrence of the PLP1 duplication at a frequency of 20%.
Background
Mutations in the PLP1 gene are responsible for a wide range of X-linked white matter disorders, collectively termed PLP1-related disorders, which together represent a continuum of neurological features, that characterize conditions ranging from Pelizaeus-Merzbacher disease (PMD, MIM# 312080) with severe CNS involvement to the milder spastic paraplegia type 2 (SPG2, MIM# 312920) [1-3]. The PMD phenotype, which includes both the severe 'connatal' and 'classic' forms of the disease, typically manifests with nystagmus, hypotonia, cognitive impairment, severe spasticity and ataxia. In the 'connatal' form, nystagmus is present at birth and may be associated with stridor and hypotonia. Ambulation and speech are usually not achieved. The classical form of PMD is usually characterized by the onset in early infancy of nystagmus, initial hypotonia leading to spasticity, ataxia, variable extrapyramidal involvement and cognitive impairment. Supported ambulation may be achieved, although it may eventually be lost during follow-up. The so called "PLP1 null syndrome" is characterized by the occurrence of peripheral neuropathy that is typically absent in the other PLP1-related disorders. SPG2 may be either 'complicated' or 'pure' according to the presence or absence of symptoms other than spastic gait and autonomic dysfunction [3]. Brain magnetic resonance imaging (MRI) reveals a diffuse hypomyelinating leukoencephalopathy in children with the PMD phenotype, whereas in SPG2, either patchy abnormalities on T2-weighted images or a more diffuse leukoencephalopathy may occur [3].
The proteolipid protein gene (PLP1; MIM# 300401) contains seven exons and spans ~17 kb of chromosome Xq22.2 [4,5]. PLP1 encodes both the 277 amino acid proteolipid protein 1 (PLP) and its 242 amino acid isoform, DM20, which is derived from the use of a developmentally regulated internal splice donor site within PLP1 exon 3 [6,7]. The in vivo functional significance of the two distinct isoforms has not yet been fully elucidated [8,9]. PLP and DM20 are however known to be differentially expressed both during development and within different regions of the nervous system [10]. DM20, ubiquitously present both in the myelinating Schwann cells and in non-myelinating cells, is the predominant product during embryonic stages of development [11,12], but is overtaken postnatally by PLP which is abundantly expressed in oligodendrocytes and accounts for 17% of the total myelin protein [13]. The amino acid sequence of PLP has been highly conserved during mammalian evolution, with human, mouse and rat PLP sequences being completely homologous to each other [14].
The remarkable variety of mutations in the PLP1 gene that lead to the arrest of myelination can be grouped into two main categories: PLP1 gene duplications of variable size (60-70%) and intragenic sequence variants (15-20%) [2,3,15,16]. The deletion of the entire PLP1 gene has been reported in barely 2% of patients [17,18] whilst position effect rearrangements have been reported in a very small number of individuals with both PMD and SPG2 [19,20]. Finally, triplication and quintuplication of the PLP1 gene also occur, albeit very rarely [2,3]. To date, more than 160 different disease-causing PLP1 mutations have been reported [see the Human Gene Mutation Database [http://www.hgmd.org] [21] and the Leiden Locus-Specific Database (http://grenada.lumc.nl/LOVD2/MR/home.php)].
To date, our clinical and diagnostic center has provided a definitive diagnosis for 47 unrelated patients with PLP1-related disorders. We have previously reported the study of 9 unrelated patients, 8 of whom were males carrying the PLP1 gene duplication [22,23] whilst the remaining female harboured a very large duplication of Xq with random X inactivation that led to brain hypomyelination together with multiple congenital anomalies [24]. Here we report the molecular characterization of the PLP1 defect in a further 38 families with PLP1-related disorders, comprising a total of 43 patients. Molecular findings from this patient series have revealed that 24 of the unrelated patients possessed PLP1 gene duplications whereas the remaining 14 patients harboured mostly novel PLP1 coding sequence or splicing-relevant mutations.
Methods
Patients
The present patient series comprised a total of 43 male patients from 38 unrelated families with a diagnosis of a PLP1-related disorder. The diagnosis was predicated upon clinical evaluation and was supported by MRI studies performed in all patients except one. The main clinical and neuroradiological findings of these patients are summarized in Table 1.
Table 1.
Clinical, neuroradiological and molecular findings of the patients
| Fam/Pt ID |
Age at onset | Age at last evaluation | Clinical symptoms at onset |
Developmental delay/ Mental retardation |
Neurological findings | Peripheral neuropathy | Clinical severity score# | Brain MRI findings | Molecular findings | |
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Nature of mutation ♣ | |||||||||
| 1 | Birth | 8 y | Nystagmus Muscular hypotonia |
Severe MR | Axial hypotonia Limb spasticity Pyramidal signs |
+ | 0 | Hypo (Supra/Infra) | c.453G>T (p.Lys151Asn)♠ | Inherited |
| 2 | Birth | 5 m | Nystagmus Stridor |
+ | Spastic tetraparesis | Absent | 0 | Hypo | c.552C>G (p.Cys184Trp) |
Inherited |
| 3 | Birth | 4 y | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia | NT | 0 | Hypo (Supra/Infra) | c.689C>T (p.Thr230Ile) |
Inherited |
| 4 | Birth | 12 y | Nystagmus Muscular hypotonia |
Severe MR | Muscular hypotonia Pyramidal signs | Absent | 1 | Hypo (Supra/Infra) | c.89C>A (p.Ala30Glu) |
Inherited |
| 5 | 2 m | 2 y | Nystagmus | + | Spastic tetraparesis | Absent | 1 | Hypo (Supra/Infra) | c.505T>C (p.Cys169Arg) |
Inherited |
| 6 | Birth | 3 y | Nystagmus Muscular hypotonia |
Mild MR | Muscular hypotonia Pyramidal signs |
Absent* | 1 | Hypo | c.554_564del11 ♠ (p.Gln185LeufsX15) | Inherited |
| 7 | Birth | 18 y | Nystagmus Muscular hypotonia |
Severe MR | Muscular hypotonia Pyramidal signs |
Absent | 1 | Hypo | c.634T>C | Inherited |
| Birth | 2 y | Nystagmus Muscular hypotonia |
Mild MR | Muscular hypotonia Pyramidal signs |
Absent | 1 | Hypo | (p.Trp212Arg) | ||
| 8 | 3 m | 4 y | Nystagmus Muscular hypotonia |
Mild MR | Spastic tetraparesis | + | 2 | Hypo (Supra/Infra) | c.205C>T (p.Gln69X)♠ |
Inherited |
| 9 | 8 m | 9 y | Nystagmus Developmental delay |
Mild MR | Spastic tetraparesis | + | 3 | Hypo | c.1-329_c.1_c.4 +324del657♠ | Inherited |
| 10 | 4 m | 14 y | Nystagmus Muscular hypotonia |
+ | Spastic tetraparesis | + | 3 | Hypo (Supra/Infra) |
c.134_140dup7 (p.Ile47IlefsX4)♠ | Inherited |
| 11 | 6 m | 30 y | Nystagmus | - | Limb spasticity Poor tendon reflexes |
NT | 4 | NT | c.436C>G (p.Leu146Val)♠ | Inherited |
| 12 | Birth | 11 m | Nystagmus Stridor |
+ | Muscular hypotonia | NT | ♦ | Hypo | c.83G>T (p.Gly28Val) |
Inherited |
| 13 | Birth | 11 m | Nystagmus | Mild MR | Muscular hypotonia | Absent | ♦ | Hypo (Supra/Infra) | c.476T>C (p.Leu159Pro) |
De novo |
| 14 | 1 m | 3 m | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia | NT | ♦ | Hypo | c.740C>A (p.Ala247Asp) |
Inherited |
| 15 | Birth | 5 y | Nystagmus Muscular hypotonia |
Severe MR | Muscular hypotonia Pyramidal signs |
Absent | 0 | Hypo (Supra/Infra) | PLP1dup | NA |
| 16 | Birth | 18 m | Nystagmus Muscular hypotonia Stridor |
Mild MR | Muscular hypotonia | Absent | 0 | Hypo (Supra/Infra) | PLP1dup | De novo |
| 17 | 1 m | 17 y | Nystagmus | Mild MR | Muscular hypotonia | Absent | 1 | Hypo (Supra/Infra) | PLP1dup | NA |
| 18 | 3 m | 10 y | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia Pyramidal signs Dystonia Ataxia |
Absent | 1 | Hypo | PLP1dup | Inherited |
| 2 m | 11 y | Nystagmus Muscular hypotonia |
Moderate MR | Spastic tetraparesis Dystonia |
Absent | 1 | Hypo | |||
| 19 | 1 m | 2 y | Nystagmus | Moderate MR | Muscular hypotonia Pyramidal signs Dystonia |
Absent | 1 | Hypo (Supra/Infra) | PLP1dup | Inherited |
| 20 | 1 m | 4 y | Nystagmus | Mild MR | Muscular hypotonia Pyramidal signs |
Absent | 2 | Hypo | PLP1dup | Inherited |
| 21 | 2 m | 4 y | Nystagmus | + | Muscular hypotonia | Absent | 2 | Hypo | PLP1dup | NA |
| 22 | 2 m | 4 m | Nystagmus Muscular hypotonia |
Mild MR | Muscular hypotonia | Absent | 2 | Hypo | PLP1dup | NA |
| 23 | 4 m | 2 y | Nystagmus | Mild MR | Muscular hypotonia | Absent | 2 | Hypo | PLP1dup | Inherited |
| 24 | Birth | 22 y | Nystagmus | Severe MR | Spastic tetraparesis Dystonia |
Absent | 2 | Hypo | PLP1dup | Inherited |
| 25 | 8 m | 2 y | Nystagmus Developmental delay | + | Spastic tetraparesis | Absent | 2 | Hypo | PLP1dup | Inherited |
| 26 | 1 m | 4 y | Nystagmus | Severe MR | Pyramidal signs Dystonia |
Absent | 2 | Hypo | PLP1dup | Inherited |
| 27 | 5 m | 2 y | Nystagmus | Severe MR | Muscular hypotonia | Absent | 2 | Hypo | PLP1dup | De novo |
| 15 d | 5 y | Nystagmus Seizures |
Moderate MR | Spastic tetraparesis Dystonia |
Absent | 2 | Hypo | |||
| 28 | 1 m | 8 y | Nystagmus | + | Pyramidal signs Dystonia |
Absent | 2 | Hypo | PLP1dup | Inherited |
| 4 m | 29 y | Nystagmus Seizures |
+ | Pyramidal signs Dystonia |
Absent | 3 | Hypo | |||
| 29 | 1 y | 25 y | Nystagmus Developmental delay |
Severe MR | Spastic tetraparesis | Absent | 3 | Hypo | PLP1dup | Inherited |
| 1 y | 23 y | Nystagmus Developmental delay |
Severe MR | Spastic tetraparesis | Absent | 3 | Hypo | |||
| 30 | 3 m | 5 y | Nystagmus | Mild MR | Muscular hypotonia Pyramidal signs |
Absent | 3 | Hypo (Supra/Infra) | PLP1dup | Inherited |
| 31 | Birth | 3 m | Nystagmus Muscular hypotonia |
Mild MR | Muscular hypotonia | Absent | 3 | Hypo | PLP1dup | NA |
| 32 | 2 m | 6 y | Nystagmus | Moderate MR | Pyramidal signs Ataxia |
Absent | 3 | Hypo | PLP1dup | Inherited |
| 33 | 6 y | 30 y | Nystagmus Learning difficulties Behavioural problems |
Moderate MR | Limb spasticity Ataxia |
Absent | 4 | Hypo | PLP1dup | NA |
| 34 | 3 m | 11 m | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia Pyramidal signs |
Absent | ♦ | Hypo (Supra/Infra) | PLP1dup | Inherited |
| 35 | 1 m | 7 m | Nystagmus | + | Muscular hypotonia | Absent | ♦ | Hypo | PLP1dup | NA |
| 36 | 3 m | 11 m | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia Dystonia |
Absent | ♦ | Hypo | PLP1dup | NA |
| 37 | 1 m | 11m | Nystagmus | + | Muscular hypotonia | Absent | ♦ | Hypo | PLP1dup | De novo |
| 38 | 3 m | 8 m | Nystagmus Muscular hypotonia |
+ | Muscular hypotonia | Absent | ♦ | Hypo (Supra/Infra) | PLP1dup | NA |
Legend: Fam = Family; Pt = Patient; d = day(s) m = month(s); y = year(s); NT = not tested; * indicates peripheral neuropathy present in the symptomatic mother; MR = mental retardation; MRI = magnetic resonance imaging; Hypo = hypomyelination; Supra = supratentorial; Infra = infratentorial; # according to Cailloux et al. [35]; ♦indicates score not assessable in patients below 12 months of age; NA = not available; ♠flags mutations predicted to alter RNA processing; ♣indicates data based on molecular findings in the mother's DNA samples
Ethical aspects
Following ethical guidelines, all cell and nucleic acid samples were obtained for analysis and storage with the patients' (and/or a family member's) written informed consent. The consent was sought using a form approved by the local Ethics Committee.
Cell culture
Fibroblast and lymphoblast cells were cultured according to standard procedures. The cell lines were cultured and maintained in RPMI medium (EuroClone, Gibco, Paisley, UK) containing 15% FCS and penicillin/streptomycin, in a humidified atmosphere containing 5% CO2 at 37°C.
Oli-neu (murine oligodendrocyte precursor) cells (kindly provided by Dr. J. Trotter, University of Mainz, Germany) were cultured in Sato medium containing 1% horse serum [25].
Molecular analysis
All DNA samples were initially screened for the presence of the PLP1 gene duplication using real-time PCR. In a subsequent step, sequencing analysis was performed on every patient who was found to be negative with respect to the gene duplication. Where necessary, further investigations on RNA samples were carried out whenever possible. Genomic DNA was extracted from peripheral blood leukocytes and/or cultured cell lines using standard methods or suitable kits [QIAmp DNA blood mini kit (Qiagen Inc., Valencia, CA, USA) or the Nucleon BACC3 kit for blood and cell cultures (Amersham Biosciences, Bucks, UK)].
Total RNA was extracted from patient fibroblasts using an RNeasy mini plus kit (Qiagen, Valencia, CA, USA) and reverse transcribed by means of an Advantage RT-for-PCR kit (BD Biosciences Clontech, Mountain View, CA, USA).
PLP1 gene dosage determination by real-time PCR
DNA samples from the patients were assessed for PLP1 gene dosage by real-time PCR using amplicon PLP1gen, located in exon 3 of the PLP1 gene, and amplicon GAPgen, located in exon 7 of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene which was used as a reference sequence [26].
PLP1 gene dosage was determined by real-time PCR amplification from genomic DNA. Amplicons defined by two primers and a TaqMan probe were designed for the real-time PCR runs using the Primer Express 1.5 software (Applied Biosystems, Foster City, CA, USA) according to previously reported requirements [26]. The real-time PCR experiments were performed using the Applied Biosystems 7500 Real-Time PCR System with TaqMan chemistry as previously described [26]. The standard curve method, with amplification of the target and reference sequences in separate tubes, was employed (User Bulletin #2. Relative quantitation of gene expression; http://www.appliedbiosystems.com/). Samples were run in quadruplicate whereas standards were run in duplicate. All TaqMan probes used were labelled with FAM as the reporter fluorophore at their 5'-end and with BHQ1 as the quencher at their 3'-end. Primers and probes were purchased by TIB Molbiol (Genoa, Italy).
PLP1 Mutation Analysis
All PLP1 gene exons and exon-intron boundaries were PCR amplified to yield 5 amplicons using 5 sets of primers designed by reference to the PLP1 genomic sequence (GenBank-EMBL Accession No. NC_000023.10). Reverse transcript-PCR (RT-PCR) was performed using sets of primers designed by reference to the PLP1 mRNA sequence (GenBank accession No. NM_000533.3) Primers and PCR reaction conditions are given in Additional file 1, Table S1a. All amplicons were purified and directly sequenced using an ABI 377 DNA automated sequencer with dye terminator cycle sequencing kits (Applied Biosystems, Foster City, CA, USA).
Putative mutations were confirmed by sequencing duplicate PCR products or by digesting PCR products with a specific restriction endonuclease whose recognition site was consequently altered. If the mutation neither created nor destroyed a restriction site, amplification was carried out using PCR-mediated site-directed mutagenesis that served to introduce a new restriction enzyme cleavage site [27]. The possibility that the novel mutations were polymorphisms was excluded by determining that none of 50 female healthy control subjects (100 alleles) carried any of these alterations. Further, by definition, none of the novel variants had ever been reported before (as either a disease-causing mutation or a polymorphism) in any previous study.
Bioinformatic analysis of PLP1 variants
Missense variants in the PLP1 gene were analysed by means of a computational model, MutPred [28,29]. MutPred was designed to model the effect of observed changes in structural and functional sites within a protein between wild-type and mutant protein sequences. MutPred can also be used to generate hypotheses as to the underlying molecular mechanism(s) responsible for disease pathogenesis for any given mutation. The effects of coding region variants upon splicing [splice site disruption, cryptic splice site activation and exon skipping, as a consequence either of the loss of an exonic splicing enhancer (ESE) and/or the gain of an exonic splicing silencer (ESS)] were ascertained using Skippy [30] and a neural network for splice site prediction [31]. The NI-ESR hexamers [32] (979 ESEs and 496 ESSs) formed the basis of the ESE and ESS motifs used in this analysis, as this set had previously been identified as providing the strongest signal for identifying exon skipping variants [30].
Mutation c.436C>G (p.Leu146Val): minigene splicing construct and quantitative real-time RT-PCR-based evaluation of PLP1 and DM20 transcript content
A PCR product containing portions of exon/intron 2, exon/intron 3 and exon 4 was obtained from a healthy control using primers PP2F and PP4R that introduced ClaI and EcoRI sites (Additional file 1, Table S1b). The product was cleaved with ClaI and EcoRI and cloned into a similarly cleaved pcDNA3.1/V5-His-TOPO/LacZ vector (Invitrogen, San Diego, CA). A recombinant in-frame LacZ-PLP1-LacZ minigene, containing the genomic region between exons 2 and 4 of the PLP1 gene, was then obtained. The cloned fragment was sequenced, thereby confirming its identity to the PLP1 gene reference sequence. Subsequently, the c.436C>G (p.Leu146Val) mutation was introduced into the vector using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene Agilent, Santa Clara, California, USA) according to the manufacturer's instructions. Wild-type and mutant plasmid constructs were transfected into murine oligodendroglial Oli-neu cells using the lipofectamine 2000 Transfection Reagent (Invitrogen, San Diego, CA) according to the manufacturer's instructions. Transfected cells were harvested 72 hrs after transfection, and RNA was extracted using an RNeasy Plus Mini kit (Qiagen, Valencia, CA, USA) and reverse transcribed using the Advantage RT-for-PCR kit (BD Biosciences Clontech, Mountain View, CA, USA). Reverse transcription was performed using a LacZ reverse primer (LACT2R) in order to avoid retrotranscription of endogenous Plp1 gene transcripts from the Oli-neu cells. RT-PCR was performed using the primers reported in Additional file 1, Table S1b.
To evaluate PLP and DM20 transcript content quantitatively in cells transfected with wild-type and mutant constructs, a real-time PCR analysis was performed, as previously described, using a DM20 transcript-specific amplicon (D2), a PLP transcript-specific amplicon (P2), both encompassing exons 3 and 4, and a (DM20+PLP) transcript-specific amplicon (P23B) encompassing exons 2 and 3 (Additional file 1, Table S1c). While amplicons D2 and P2 were used as targets, amplicon P23B was used as a reference. Using this experimental approach, we were able to derive the DM20/(DM20+PLP) and PLP/(DM20+PLP) ratios for cells transfected with wild-type and mutant constructs. Wild-type and mutant cDNAs were run in quadruplicate, each well containing the cDNA obtained from 10 ng RNA. A plasmid containing the DM20 cDNA was used to generate the standard curve for the D2 amplicon whilst a plasmid containing the PLP cDNA was used to generate the standard curves for the P2 and the P23B amplicons, respectively. Standards were run in duplicate. Standard wells contained 10-2, 10-3, 10-4, 10-5, 10-6, 10-7 ng/μl plasmid DNA.
Mutation nomenclature
All mutations are described according to the recommended nomenclature [33,34]. Nucleotide numbering was derived from the human PLP1 cDNA sequence (GenBank-EMBL Accession No. NM_000533.3) ascribing the A of the first ATG translational initiation codon as nucleotide +1. Amino acid residue numbering was as derived for the human PLP protein (GenBank-EMBL Accession No. NP_000524.3).
Results and Discussion
In this study we report the molecular genetic analysis of 43 patients with PLP1-related disorders from 38 unrelated families. The clinical, neuroradiological and molecular findings obtained in relation to the 43 patients are summarized in Table 1.
Clinical aspects
As reported in Table 1, 15 patients (pts) (from 14 families) were found to possess PLP1 mutations whereas the remaining 28 patients (from 24 families) harboured a PLP1 duplication. Age at onset of clinical symptoms ranged from birth to 1 year old, with the exception of one patient (pt #33) who was reported as having nystagmus, learning difficulties and behavioural problems in childhood. The age at last evaluation ranged from 3 months to 30 years (Table 1). Nystagmus, either isolated or associated with other symptoms, was the presenting symptom in all patients. The other presenting symptoms in the 43 patients were: seizures (2/43), stridor (3/43), developmental delay (4/43) and muscular hypotonia (18/43). Developmental delay and mental retardation (ranging from mild to severe) were reported during follow-up in all patients except for pt #11. Neurological findings revealed abnormal muscular tone with prevailingly axial hypotonia or diffuse muscular hypotonia in the youngest subjects associated with pyramidal signs in 11/43. Increased muscle tone, either limb spasticity (3 pts) or tetraparesis (11 pts), was evident in older patients. Dystonia was present in 9 patients (in association with pyramidal signs in 8/9) and ataxia in three patients. Peripheral neuropathy (confirmed by nerve conduction velocity studies) was present in four subjects with PLP1 mutations but was absent in all patients harbouring a PLP1 duplication. A reduced amplitude of the compound muscle action potential, consistent with an axonal neuropathy, was identified in two subjects (pts #1 and #9), whereas a mixed axonal and demyelinating process was diagnosed in two further patients (pts #8 and #10).
Clinical severity, assessed according to the score proposed by Cailloux et al. [35], ranged from 0 to 4 on the basis of the maximal level of motor acquisition. Three out of five patients with the lowest score (i.e. no postural achievement) belonged to the PLP1 mutation group, whereas the remaining two patients harboured PLP1 duplications. Nine patients, having achieved head control, had a score of 1: of these, five were in the PLP1 mutation group whereas four were in the PLP1 duplication group. Eleven patients, 10 of whom carried a PLP1 duplication, were able to sit without assistance, thereby obtaining a score of 2. Of the 8 patients who acquired the ability to walk with support (viz. score 3), two were in the PLP1 mutation group whereas six were in the PLP1 duplication group. Finally, only two patients, one with a PLP1 mutation and the other with a PLP1 duplication, acquired the ability to walk without support, thereby obtaining a score of 4. Brain MRI indicated hypomyelination in all 42 patients who had undergone brain MRI. In 14/17 patients, from whom more detailed neuroradiological data were available, infratentorial myelin deficiency was present in addition to the supratentorial hypomyelination that was evident in all patients.
Molecular studies
The laboratory diagnostic protocol included an initial screening, by real-time PCR, of all DNA samples for the presence of the PLP1 gene duplication. In the next step, patients negative for the gene duplication underwent PLP1 sequencing analysis. Analysis of the present patient series revealed a PLP1 gene duplication in 24 of the unrelated patients whereas a variety of intragenic PLP1 mutations was found to underlie disease pathogenesis in the remaining 14 unrelated patients (Table 1). Table 2 reports the characteristics of the 14 different genomic lesions identified in these patients as a result of sequencing the exons and exon-intron boundaries of the PLP1 gene. Figure 1 depicts the distribution of the coding sequence mutations in relation to the proposed model of the tetra-span proteolipid proteins, PLP and DM20 [36,37].
Table 2.
Characteristics of PLP1 gene mutations identified in 14 unrelated patients and MutPred analysis of the missense mutations
| Location | cDNA mutation* | Mutation Effect | MutPred analysis of missense mutations | Refer ence | |||||
|---|---|---|---|---|---|---|---|---|---|
| Probability of deleterious mutation |
No. of ESE binding sites loses | No. of ESS binding sites gained | Skippy Log Odds Ratio (LOR) Total | Splice site disruption prediction | |||||
| 5'UTR-Ex 1-Intr 1 | c.1-329_c.1_c.4 +324del657 | r.? | . | . | . | . | Present study | ||
| c.134_140dup7 | p.Ile47IlefsX4 | . | . | . | . | Present study | |||
| Exon 2 | c.83G>T | p.Gly28Val | 0.80 | SS Loss of loop (P = 0.0252) |
0 | 1 | 0.052 | Present study | |
| c.89C>A | p.Ala30Glu | 0.84 | 0 | 0 | -5.884 | Present study | |||
| c.205C>T | p.Gln69X | . | 0 | 2 | 1.290 | Present study | |||
| c.436C>G | p.Leu146Val | 0.67 | 0 | 5 | 0.356 | Present study | |||
| Exon 3 | c.453G>T | p.Lys151Asn | 0.63 | 0 | 0 | -1.284 | [predicted to abolish 5'SS with Neural Network] already reported in the cited ref | Hobson et al. [38] |
|
| c.476T>C | p.Leu159Pro | 0.87 | SS Helix > Sheet (P = 0.0266), Gain of glycosylation at T160 (P = 0.0342) |
6 | 0 | 1.360 | Present study | ||
| Exon 4 | c.505T>C | p.Cys169Arg | 0.91 | 4 | 0 | 1.360 | Mimault et al. [48] |
||
| c.552C>G | p.Cys184Trp | 0.88 | 2 | 0 | -0.929 | Present study | |||
| c.554_564del11 | p.Gln185LeufsX15 | . | Present study | ||||||
| Exon 5 | c.634T>C | p.Trp212Arg | 0.79 | Loss of catalytic residue at L210 (P = 0.0114), Gain of methylation at W212 (P = 0.0245) |
0 | 0 | -1.284 | Cailloux et al. [35] |
|
| c.689C>T | p.Thr230Ile | 0.68 | 5 | 1 | 2.696 | Present study | |||
| Exon 6 | c.740C>A | p.Ala247Asp | 0.88 | 1 | 2 | 2.006 | Present study | ||
* Nucleotide numbers are derived from cDNA PLP1 sequence (GenBank-EMBL accession no. NM_000533.3) taking as nucleotide +1 the A of the first ATG translation initiation codon; Bold type denotes novel mutation; Ex = exon; Intr = Intron; ESE = exonic splicing enhancers; ESS = exonic splicing silencers; Skippy Log Odds Ratio (LOR) score is the output from the Skippy tool [30], the Skippy LOR score represents the likelihood that the combination of ESR changes consequent to a substitution are associated with exon skipping. The higher the Skippy LOR score, the more likely the combination of ESR changes will be associated with an exon skipping event. The prediction of splice site disruption was evaluated using a neural network. The column 'Location' refers to the exon within which the mutation occurs and also the exon that could potentially be skipped due to ESE loss and/or ESS gain.
Figure 1.
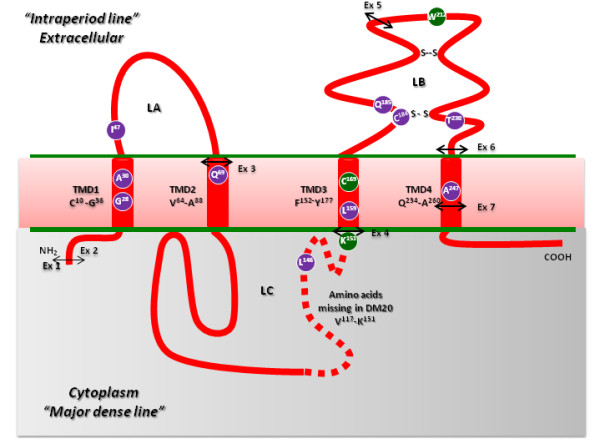
Distribution of PLP1 gene mutations identified in our series of patients with PLP1-related disorders in relation to the proposed model* of the tetra-span proteolipid proteins, PLP and DM20. With respect to the missense mutations reported here, the amino acid residues affected by novel mutations are shown in purple, whereas residues harbouring previously reported mutations are given in green. The four predicted transmembrane domains (TMD1-4) are depicted, the first and last amino acids being indicated for each TMD. The PLP1-specific region (which is absent from DM20) is denoted by a dotted line. The locations of the two disulfide bridges (Cys184-Cys228 and Cys201-Cys220) within loop B (LB) are marked "S--S". The double arrow indicates the relative placement of the exon-exon (Ex) junctions superimposed upon the PLP1 protein. *model according to Popot et al, [36] and Weimbs and Stoffel [37]
Eleven of the mutations were novel and included (i) 7 missense mutations (p.Gly28Val, p.Ala30Glu, p.Leu146Val, p.Leu159Pro, p.Cys184Trp, p.Thr230Ile, p.Ala247Asp) and one nonsense mutation (p.Gln69X); (ii) a large deletion (c.1-329_c.1_c.4+324del657) involving the removal of 657-bp from the untranslated, exonic and intronic regions surrounding exon 1; (iii) a microdeletion (c.554_564del11) which results in a frameshift (p.Gln185LeufsX15) that is predicted to introduce a premature stop codon 15 residues downstream; and (iv) a microduplication in exon 2 (c.134_140dup7) which would also be predicted to lead to a frameshift (p.Ile47IlefsX4) with premature truncation of the protein four amino acids downstream.
The question of the pathological authenticity of the novel PLP1 sequence alterations detected was addressed by (i) searching dbSNP (http://www.ncbi.nlm.nih.gov/SNP) for their annotation and excluding their presence, (ii) screening 50 healthy female control subjects (100 alleles) for each alteration and determining that none carried any of these alterations, (iii) analysing the evolutionary conservation of the amino acid residues affected, (iv) employing the MutPred program [28,29] to assess each missense mutation, and (v) using Skippy [30] and a neural network for splice site prediction [31] to explore the potential for mutational consequences for splicing.
Analysis of the evolutionary conservation of amino acid residues affected by missense mutations
Support for the pathological relevance of the missense mutations identified in the present study, came from the analysis of the extent of evolutionary conservation of the mutated residues in 9 orthologous (vertebrate) PLP proteins. Computational analysis (http://www.ensembl.org/) revealed that all missense mutations occurred at evolutionarily conserved amino acid residues. Moreover, seven of the novel mutations affected amino acid residues (namely Gly28, Ala30, Leu159, Cys184, Trp212, Thr230 and Ala247) that were invariant even when zebrafish PLP was considered (Additional file 2, Table S2).
MutPred analysis
In an attempt to assess the functional relevance or otherwise of the novel PLP1 missense mutations identified, we employed the in silico analysis tool, MutPred [28,29]. MutPred predicted that all 7 novel missense mutations listed in Table 2 would be deleterious (MutPred general score > 65) whilst confident in silico hypotheses for the underlying mechanism of pathogenesis were generated for two of them. Thus, protein secondary structure was predicted to be altered by the missense mutations p.Gly28Val (P = 0.025) and p.Leu159Pro (Helix to Sheet; P = 0.027); the latter replacement (p.Leu159Pro) was also predicted to give rise to a gain of glycosylation at p.Thr160 (P = 0.034).
The effect of the coding region variants upon splicing [including splice site disruption, cryptic splice site activation and exon skipping via loss of exonic splicing enhancers (ESE), and/or gain of exonic splicing silencers (ESS)] was ascertained using Skippy [30] and a neural network for splice site prediction [31]. Whereas this latter algorithm predicted that only p.Lys151Asn would abolish the 5' splice site, as already reported by Hobson et al. [38], the results of the Skippy analysis (Table 2) indicated that four other missense mutations (p.Leu146Val, p.Leu159Pro, p.Thr230Ile, p.Ala247Asp) could potentially cause exon skipping by altering exonic splicing elements (ESR). In all four instances, the exon predicted to be skipped would be the same as that harbouring the mutation (Table 2, Location). The dramatic copy number changes of ESE or ESS motifs for p.Leu146Val and p.Thr230Ile further identified these novel missense mutations as being high confidence 'exon skipping' candidates suitable for further in vitro analysis. Indeed, c.436C>G (p.Leu146Val) appears to lead to a net gain of 5 exon splicing silencer (ESS) motifs, whilst c.689C>T (p.Thr230Ile) results in the net loss of 5 exon splicing enhancers (ESE) as well as the gain of an ESS motif. The potential impact of both p.Leu146Val and p.Thr230Ile on PLP1 mRNA processing was therefore investigated by means of RNA or minigene studies.
Deduced consequences of PLP1 mutations for mRNA processing
The postulated effect of mutations c.436C>G (p.Leu146Val) and c.689C>T (p.Thr230Ile) on PLP1 mRNA processing was explored in vitro as follows:
Mutation c.436C>G (p.Leu146Val)
Since no RNA sample was available from pt #11, the functional consequences of the mutation c.436C>G (p.Leu146Val) were assessed using a recombinant minigene construct containing a PLP1 exon 2-exon 4 fragment, cloned into an expression vector. Oli-neu cells were then transfected with the wild-type (c.436C) and mutant (c.436G) versions of the construct, and RNA samples extracted. To avoid the synthesis of endogenous murine Oli-neu Plp1 transcripts, first strand cDNA was reverse transcribed using a minigene-specific primer. RT-PCR products from the mutant construct (c.436G) revealed only the presence of the DM20-specific transcript (the PLP-specific transcript being totally absent) when compared with the wild-type construct (c.436C) (Figure 2a). To further corroborate these data, we evaluated the [DM20]/[DM20+PLP] and [PLP]/[DM20+PLP] ratios in cells transfected with the wild-type and mutant constructs, by real-time PCR quantification using [DM20]- [PLP]- and [DM20+PLP]-specific amplicons. As expected, whereas the [DM20]/[DM20+PLP] ratio displayed by the mutant construct was 0.998 (compared to 0.707 for the wild-type control), the [PLP]/[DM20+PLP] ratio associated with the mutant construct was 0.019 vs 0.192 for the wild-type construct (Figure 2b). These findings indicate that the mutation c.436C>G (p.Leu146Val) abolished PLP isoform-specific splicing, thereby confirming our in silico predictions. Thus, only the transcript corresponding to the DM20 isoform was found in association with the p.Leu146Val PLP1 mutation.
Figure 2.
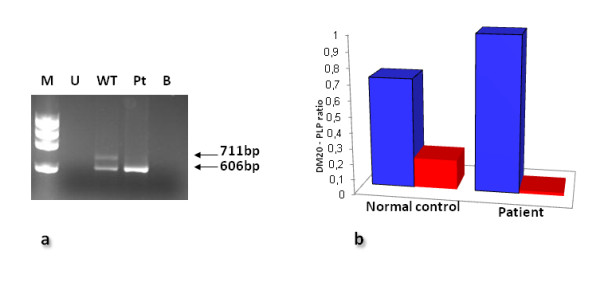
Mutation c.436C>G (p.Leu146Val). a) RT-PCR performed with the minigene-specific 31GF/LACT2R primers (Additional file 1, Table S1b) on cDNA transfected into Oli-neu cells. Whereas the expected PLP1 and DM20 products were present in cells transfected with the wild-type construct (WT), only the DM20 product was present in cells transfected with the patient mutant construct (Pt). No product was present in untransfected cells (U) or in the negative control lacking DNA (B). ΦX174 DNA HaeIII digest used as a marker (M). b) Real-time PCR quantification using [DM20]- [PLP]- and [DM20+PLP]-specific amplicons confirmed the altered ratio of DM20/(DM20+PLP) (blue bars) and PLP/(DM20+PLP) (red bars) in the patient carrying the mutation Leu146Val as compared to the healthy control.
Mutation c.689C>T (p.Thr230Ile)
Based on the in silico prediction (loss of 5 ESE motifs and gain of 1 ESS motif), an alteration of the splicing phenotype was expected for this missense mutation. RT-PCR analysis of PLP1 RNA isolated from the fibroblasts of pt #3 (using the PLPF-PLP1R primer set reported in Additional file 1, Table S1c), revealed instead the presence only of normally-sized 1040-bp and 915-bp products of the PLP and DM20 isoforms, respectively, both of which harboured the missense mutation.
Characterization of the gross deletion (c.1-329_c.1_c.4 +324del657)
The genomic breakpoints of the large genomic deletion were found to overlap 4 nucleotides of exon 1 (including the start codon ATG), the flanking upstream 329-bp untranslated region (5'UTR) and the downstream 324-bp intronic regions, respectively. We then evaluated its consequences for RNA processing by means of quantitative real-time PCR. These experiments, performed on RNA extracted from the fibroblasts of pt #9, indicated that the PLP transcript isoform was completely absent, whereas the DM20 transcript isoform was present at a reduced level, ~50% as compared to healthy controls (data not shown).
The recombinational mechanism underlying the gross deletion was then explored. Since no significant homologies were detected at the deletion breakpoints, we infer that non-homologous end-joining (NHEJ) may have been responsible for this deletion [39,40]. The underlying mechanism may also have involved sequence elements present at the 5' breakpoint [i.e. an A-rich sequence and a polypurine tract (both 17 bp long)] and 3' breakpoint [a tract highly homologous to the translin site consensus sequence], previously reported as being over-represented at translocation breakpoints [41].
Genotype-phenotype correlations
A remarkable number of different PLP1 mutations have been reported as being responsible for disease pathogenesis in PLP1-related disorders. It has generally been observed that patients with PLP1 gene duplications display a range of clinical severity, independent of the extent of the duplication [22,42]. This variation in clinical severity is also evident in our patient series; indeed, the clinical severity scores varied between 0 and 4 in different patients.
Peripheral neuropathy, never reported in patients with PLP1 gene duplications, has been frequently found to be associated with intragenic PLP1 mutations that either abrogate PLP1 expression (null alleles), affect the PLP-specific region or alter certain PLP1 splice sites [43,44]. In accordance with these general observations, peripheral neuropathy was absent in all 24 families with a PLP1 duplication (Table 1). Interestingly, only those patients (#1, #8, #9, #10) who harboured PLP1 mutations that were predicted to result in the absence of the PLP isoform, exhibited peripheral neuropathy [as confirmed by nerve conduction velocity studies (NCVs)]. Although NCVs in another patient (#6) who harboured a null allele were within the normal range for his age (2 years), his carrier mother exhibited spastic paraplegia and peripheral neuropathy with severe slowing of nerve conduction velocity.
Analysing the clinical severity score of each patient in the PLP1 mutation group in relation to the type of mutation, the most severe phenotype (score = 0) (Table 1 pts #1, #2 and #3) was found to be associated with (i) the p.Lys151Asn mutation, which occurred at the alternative 5' splice donor site of the PLP and DM20 isoforms, and which altered levels of both the PLP- and DM20-encoding RNAs [38]; (ii) the novel substitution (p.Cys184Trp), that replaced a cysteine known to be involved in disulfide bridge formation (Cys184-Cys228) (Figure 1) with a tryptophan (Trp), and which is likely to destabilize the folded protein and induce the UPR (unfolded protein response) pathway leading to apoptosis [45]. Recently, the Cys184Arg substitution has been reported as a cause of the 'classic' form of PMD [46] thereby supporting our contention that any defect involving the disulfide bridge (Cys184-Cys228) would strongly impair correct PLP folding; (iii) the substitution of an amino acid residue (p.Thr203Ile) that is common to both the PLP and DM20 isoforms and is evolutionarily invariant even when zebrafish PLP is considered (Additional file 2, Table S2).
The least severe phenotype (clinical severity score = 4) was noted in patient #11 who harboured the lesion c.436C>G (p.Leu146Val); in vitro studies confirmed that this mutation results in altered PLP1 RNA processing, leading to the production of the DM20 isoform in the absence of the PLP isoform (Figure 2). The findings outlined above are therefore in accord with previous observations of patients with PLP1-related disorders in whom a relatively mild syndrome is associated with mutations that affect the PLP isoform but not the DM20 isoform [2,3,16].
Family studies
The mutation analysis was extended wherever possible to all female family members. So far, although de novo mutations have been reported in several instances of PLP1 point mutations, de novo mutation appears at the very least to be much rarer (and is perhaps unprecedented) in the case of PLP1 duplications (see Hodes et al. [47], and Mimault et al. [48], for discussion of this still contentious issue). Mimault et al. [48] analysed the maternal mutation status of 56 families; whereas in 22 families with PLP1 gene mutations, 32% of mothers were found not to be carriers of the mutation concerned, these authors found that only three (9%) of 34 mothers of PLP1 duplication patients (analyzed by a multiplex endpoint PCR-based approach) were not carriers. On the basis of a statistical analysis which revealed a significant male duplication imbalance, these authors suggested that the PLP1 duplications could have arisen in the grandpaternal germline.
In our own patient series, we analyzed all 14 obligate carrier mothers of the familial PLP1 gene mutation and 15/24 obligate carrier mothers of the PLP1 duplication. In contrast to the previously reported results, the PLP1 mutations were all found to be maternally inherited with the sole exception of pt #13, in whom the missense mutation appears to have occurred de novo (7%). However, the PLP1 duplication was found to have occurred de novo in 3 of the 15 mothers analyzed (20%) (Table 1). The proportion of PLP1 duplications occurring de novo in our series is significantly higher than the 9% reported by Mimault et al. [48] The limited number of cases analyzed here (15 mothers) notwithstanding, the de novo occurrence of the PLP1 gene duplication at a frequency of 20% is quite unprecedented. If these novel and unexpected findings are confirmed on a larger number of patients, the previously postulated high prevalence of a grandpaternal origin for the PLP1 duplication will have to be re-evaluated. The relatively high frequency of de novo PLP1 duplication reported here would certainly concur with recent findings indicating a rather higher de novo mutation rate for CNVs than for point mutations [49,50]. It may well be that the discrepancy between our own data and those previously reported is due to the greatly improved analytical methods currently in use for the identification of the PLP1 duplication.
Although a high rate of de novo duplication was documented in our study, gonadal mosaicism could not be formally excluded in the patient's mothers who were found to be negative with respect to a specific mutation. Therefore, in clinical practice, prenatal testing is recommended for those couples potentially at risk. Female carriers of PLP1-related disorders are generally normal neurologically, but may manifest mild to moderate signs of late onset disease [3]. It is however known that those mutant PLP1 alleles that are responsible for hemizygous males being affected with comparatively mild neurological disease symptoms, may also give rise to neurological manifestations in female heterozygotes [51]. In addition, owing to favourably skewed X inactivation in heterozygous females with a PLP1 duplication, the risk of a carrier female being clinically affected is lowest in the case of a PLP1 duplication and highest in the presence of a PLP1 null allele [52]. None of the 15 mothers studied here who were heterozygous for the PLP1 duplication were clinically affected. On the other hand, of the 14 mothers who were carriers of an intragenic PLP1 mutation, neurological impairment (spastic paraplegia and peripheral neuropathy) was reported only in the mother (Table 1, Fam #6) who harboured a putative null allele (p.Gln185LeufsX15).
Competing interests
The authors are not aware of any financial or non-financial competing interests related to this manuscript.
Authors' contributions
SR, SG, and CVB performed the molecular genetic studies and the sequence alignment, and helped to draft the manuscript. SL assisted with the molecular studies. FC performed and managed cell line cultures and biobanking. MM carried out the bioinformatics analysis. RB, EB, GU, OBT, AS, ED, VM, AP, FS contributed to the acquisition, organization and analysis of the clinical data. AR contributed to the evaluation of neuroradiological data. RB coordinated the clinical data and helped to draft the manuscript. MF coordinated the study and drafted the manuscript with the assistance of DNC who also made a substantial contribution to placing the findings in their broader scientific context. All authors read and approved the final manuscript.
Contributor Information
Serena Grossi, Email: seregro@libero.it.
Stefano Regis, Email: stefanoregis@ospedale-gaslini.ge.it.
Roberta Biancheri, Email: roberta@biancheri.com.
Matthew Mort, Email: mortm@cardiff.ac.uk.
Susanna Lualdi, Email: susannalualdi@iol.it.
Enrico Bertini, Email: ebertini@tin.it.
Graziella Uziel, Email: uziel@istituto-besta.it.
Odile Boespflug-Tanguy, Email: odile.boespflug-tanguy@rdb.aphp.fr.
Alessandro Simonati, Email: alessandro.simonati@univr.it.
Fabio Corsolini, Email: fabiocorsolini@yahoo.it.
Ercan Demir, Email: ercdemir28@yahoo.com.
Valentina Marchiani, Email: valentina.marchiani@aosp.bo.it.
Antonio Percesepe, Email: percesepe.antonio@unimo.it.
Franco Stanzial, Email: franco.stanzial@asbz.it.
Andrea Rossi, Email: andrearossi@ospedale-gaslini.ge.it.
Catherine Vaurs-Barrière, Email: Catherine.BARRIERE@u-clermont1.fr.
David N Cooper, Email: cooperdn@cardiff.ac.uk.
Mirella Filocamo, Email: mirellafilocamo@ospedale-gaslini.ge.it.
Acknowledgements
The patient samples were obtained from the "Cell Line and DNA Biobank from Patients Affected by Genetic Diseases" (G. Gaslini Institute) - Telethon Genetic Biobank Network (Project No. GTB07001A). This work was partially supported by grants from FP7-HEALTH - LeukoTreat no.241622
References
- Hudson LD. In: The metabolic and molecular basis of inherited diseases. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, editor. McGraw-Hill, New York; 2001. Pelizaeus-Merzbacher disease and the allelic disorder X-linked spastic paraplegia type 2; pp. 5789–5798. [Google Scholar]
- Woodward KJ. The molecular and cellular defects underlying Pelizaeus-Merzbacher disease. Expert Rev Mol Med. 2008;10:e14. doi: 10.1017/S1462399408000677. [DOI] [PubMed] [Google Scholar]
- Garbern JY, Hobson GH. PLP1-Related Disorders. GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993. http://www.genetests.org (Updated [March 16, 2010])
- Mattei MG, Alliel PM, Dautigny A, Passage E, Pham-Dinh D, Mattei JF, Jollès P. The gene encoding for the major brain proteolipid (PLP) maps on the q-22 band of the human X chromosome. Hum Genet. 1986;72:352–353. doi: 10.1007/BF00290964. [DOI] [PubMed] [Google Scholar]
- Diehl HJ, Schaich M, Budzinski RM, Stoffel W. Individual exons encode the integral membrane domains of human myelin proteolipid protein. Proc Natl Acad Sci USA. 1986;83:9807–9811. doi: 10.1073/pnas.83.24.9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Lai C, Bloom FE, Milner RJ. Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc Natl Acad Sci USA. 1987;84:5665–5669. doi: 10.1073/pnas.84.16.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouyiouklis DA, Barrie JA, Griffiths IR, Thomson CE. A proteolipid protein-specific pre-mRNA (Ppm-1) contains intron 3 and is up-regulated during myelination in the CNS. J Neurochem. 2000;74:940–948. doi: 10.1046/j.1471-4159.2000.0740940.x. [DOI] [PubMed] [Google Scholar]
- Boison D, Bussow H, D'Urso D, Muller HW, Stoffel W. Adhesive properties of proteolipid protein are responsible for the compaction of CNS myelin sheaths. J Neurosci. 1995;15:5502–5513. doi: 10.1523/JNEUROSCI.15-08-05502.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugmann M, Schwab MH, Pühlhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave KA. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59–70. doi: 10.1016/S0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- Campagnoni AT, Skoff RP. The pathobiology of myelin mutants reveal novel biological functions of the MBP and PLP genes. Brain Pathol. 2001;11:74–91. doi: 10.1111/j.1750-3639.2001.tb00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spörkel O, Uschkureit T, Büssow H, Stoffel W. Oligodendrocytes expressing exclusively the DM20 isoform of the proteolipid protein gene: myelination and development. Glia. 2002;37:19–30. doi: 10.1002/glia.10014. [DOI] [PubMed] [Google Scholar]
- Jacobs EC, Bongarzone ER, Campagnoni CW, Campagnoni AT. Embryonic expression of the soma-restricted products of the myelin proteolipid gene in motor neurons and muscle. Neurochem Res. 2004;29:997–1002. doi: 10.1023/b:nere.0000021244.38279.c4. [DOI] [PubMed] [Google Scholar]
- Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer J, Becker T, Schachner M, Nave KA, Werner H. Evolution of myelin proteolipid proteins: gene duplication in teleosts and expression pattern divergence. Mol Cell Neurosci. 2006;31:161–177. doi: 10.1016/j.mcn.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: genetic and cellular pathogenesis. Cell Mol Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskind WH, Williams CA, Hudson LD, Bird TD. Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus-Merzbacher disease. Am J Hum Genet. 1991;49:1355–1360. [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet. 2002;71:838–853. doi: 10.1086/342728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncke N, Wogatzky BS, Breuning M, Sistermans EA, Endris V, Ross M, Vetrie D, Catsman-Berrevoets CE, Rappold G. Position effect on PLP1 may cause a subset of Pelizaeus-Merzbacher disease symptoms. J Med Genet. 2004;41:e121. doi: 10.1136/jmg.2004.019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball E, Howells K, Phillips A, Thomas NST, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regis S, Biancheri R, Bertini E, Burlina A, Lualdi S, Bianco MG, Devescovi R, Rossi A, Uziel G, Filocamo M. Genotype-phenotype correlation in five Pelizaeus-Merzbacher disease patients with PLP1 gene duplications. Clin Genet. 2008;73:279–287. doi: 10.1111/j.1399-0004.2007.00961.x. [DOI] [PubMed] [Google Scholar]
- Regis S, Grossi S, Corsolini F, Biancheri R, Filocamo M. PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus-Merzbacher disease patients. Biochim Biophys Acta. 2009;1792:548–554. doi: 10.1016/j.bbadis.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Carrozzo R, Arrigo G, Rossi E, Bardoni B, Cammarata M, Gandullia P, Gatti R, Zuffardi O. Multiple congenital anomalies, brain hypomyelination, and ocular albinism in a female with dup(X) (pter→q24::q21.32→qter) and random X inactivation. Am J Med Genet. 1997;72:329–334. doi: 10.1002/(SICI)1096-8628(19971031)72:3<329::AID-AJMG15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Regis S, Grossi S, Lualdi S, Biancheri R, Filocamo M. Diagnosis of Pelizaeus-Merzbacher disease: detection of proteolipid protein gene copy number by real-time PCR. Neurogenetics. 2005;6:73–78. doi: 10.1007/s10048-005-0214-7. [DOI] [PubMed] [Google Scholar]
- Schwartz EI, Shevtsov SP, Kuchinski AP, Kovalev YP, Plutalov OV, Berlin YA. Approach to identification of a point mutation in apo B100 gene by means of a PCR-mediated site-directed mutagenesis. Nucleic Acids Res. 1991;19:3752. doi: 10.1093/nar/19.13.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort M, Evani US, Krishnan VG, Kamati KK, Baenziger PH, Bagchi A, Peters BJ, Sathyesh R, Li B, Sun Y, Xue B, Shah NH, Kann MG, Cooper DN, Radivojac P, Mooney SD. In silico functional profiling of human disease-associated and polymorphic amino acid substitutions. Hum Mutat. 2010;31:335–346. doi: 10.1002/humu.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A, Mullikin JC, Elnitski L. Genomic features defining exonic variants that modulate splicing. Genome Biol. 2010;11:R20. doi: 10.1186/gb-2010-11-2-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Shomron N, Yeo GW, Schneider A, Xiao X, Burge CB. Inference of splicing regulatory activities by sequence neighborhood analysis. PLoS Genet. 2006;2:e191. doi: 10.1371/journal.pgen.0020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Paalman MH. Standardizing mutation nomenclature: why bother? Hum Mutat. 2003;2:181–182. doi: 10.1002/humu.10262. [DOI] [PubMed] [Google Scholar]
- Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G, Dastugue B, Boespflug-Tanguy O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- Popot JL, Pham Dinh D, Dautigny A. Major myelin proteolipid: the 4-alpha-helix topology. J Membr Biol. 1991;123:278. doi: 10.1007/BF01870411. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Stoffel W. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry. 1992;31:12289–12296. doi: 10.1021/bi00164a002. [DOI] [PubMed] [Google Scholar]
- Hobson GM, Huang Z, Sperle K, Sistermans E, Rogan PK, Garbern JY, Kolodny E, Naidu S, Cambi F. Splice-site contribution in alternative splicing of PLP1 and DM20: molecular studies in oligodendrocytes. Hum Mutat. 2006;27:69–77. doi: 10.1002/humu.20276. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Férec C, Kehrer-Sawatzki H, Patrinos GP. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20:222–233. doi: 10.1016/j.semcancer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- Woodward KJ, Cundall M, Sperle K Sistermans EA, Ross M, Howell G, Gribble SM, Burford DC, Carter NP, Hobson DL, Garbern JY, Kamholz J, Heng H, Hodes ME, Malcolm S, Hobson GM. Heterogeneous duplications in patients with Pelizaeus-Merzbacher disease suggest a mechanism of coupled homologous and nonhomologous recombination. Am J Hum Genet. 2005;77:966–987. doi: 10.1086/498048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy ME, Hobson G, Jain M, Boespflug-Tanguy O, Garbern J, Sperle K, Li W, Gow A, Rodriguez D, Bertini E, Mancias P, Krajewski K, Lewis R, Kamholz J. Schwann cell expression of PLP1 but not DM20 is necessary to prevent neuropathy. Ann Neurol. 2003;53:354–365. doi: 10.1002/ana.10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaurs-Barrière C, Wong K, Weibel TD, Abu-Asab M, Weiss MD, Kaneski CR, Mixon TH, Bonavita S, Creveaux I, Heiss JD, Tsokos M, Goldin E, Quarles RH, Boespflug-Tanguy O, Schiffmann R. Insertion of mutant proteolipid protein results in missorting of myelin proteins. Ann Neurol. 2003;54:769–780. doi: 10.1002/ana.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Fukumura S, Adachi N, Nagao M, Tsutsumi H. A novel proteolipid protein 1 gene mutation causing classical type Pelizaeus-Merzbacher disease. Brain Dev. 2010. [DOI] [PubMed]
- Hodes ME, Aydanian A, Dlouhy SR, Whelan DT, Heshka T, Ronen G. A de novo mutation (C755T, Ser252Phe) in exon 6 of the proteolipid protein gene responsible for Pelizaeus-Merzbacher disease. Clin Genet. 1998;54:248–249. doi: 10.1111/j.1399-0004.1998.tb04295.x. [DOI] [PubMed] [Google Scholar]
- Mimault C, Giraud G, Courtois V, Cailloux F, Boire JY, Dastugue B, Boespflug-Tanguy O. Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher disease: duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. Am J Hum Genet. 1999;65:360–369. doi: 10.1086/302483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst S, Garbern J, Trepanier A, Gow A. Quantifying the carrier female phenotype in Pelizaeus-Merzbacher disease. Genet Med. 2006;8:371–378. doi: 10.1097/01.gim.0000223551.95862.c3. [DOI] [PubMed] [Google Scholar]
- Woodward K Kirtland K, Dlouhy S, Raskind W, Bird T, Malcolm S, Abeliovich D. X inactivation phenotype in carriers of Pelizaeus-Merzbacher disease: skewed in carriers of a duplication and random in carriers of point mutations. Eur J Hum Genet. 2000;8:449–454. doi: 10.1038/sj.ejhg.5200480. [DOI] [PubMed] [Google Scholar]