

Abstract
Cancer susceptibility is due to interactions between inherited genetic factors and exposure to environmental carcinogens. The genetic component is constituted mainly by weakly acting low-penetrance genetic variants that interact among themselves, as well as with the environment. These low susceptibility genes can be categorized into two main groups: one includes those that control intrinsic tumor cell activities (i.e. apoptosis, proliferation or DNA repair), and the other contains those that modulate the function of extrinsic tumor cell compartments (i.e. stroma, angiogenesis, or endocrine and immune systems). Genome-Wide Association Studies (GWAS) of human populations have identified numerous genetic loci linked with cancer risk and behavior, but nevertheless the major component of cancer heritability remains to be explained. One reason may be that GWAS cannot readily capture gene–gene or gene–environment interactions. Mouse model approaches offer an alternative or complementary strategy, because of our ability to control both the genetic and environmental components of risk. Recently developed genetic tools, including high-throughput technologies such as SNP, CGH and gene expression microarrays, have led to more powerful strategies for refining quantitative trait loci (QTL) and identifying the critical genes. In particular, the cross-species approaches will help to refine locations of QTLs, and reveal their genetic and environmental interactions. The identification of human tumor susceptibility genes and discovery of their roles in carcinogenesis will ultimately be important for the development of methods for prediction of risk, diagnosis, prevention and therapy for human cancers.
Introduction
Cancer is a highly heterogeneous disease, both in terms of the time of individual cancer development, and the biological properties of each tumor. Only a subset of human individuals exposed to the same carcinogen, as for example after radiation exposure,1 actually developed a tumor, and even among those who were susceptible, tumors did not appear at the same time. Individual tumors also vary enormously in terms of tumor evolution and behavior (i.e. local growth, distant dissemination, treatment response, and relapse or tumor dormancy).2–5 Thus, individuals that share the same apparent histopathological type of tumor and TNM (Tumor, Node, Metastasis) stage, and also receive the same treatment, could have tumors with completely different evolutionary histories.
One of the most important aspects that remains to be clarified in this field is the identification of the genetic and molecular components that determine the diverse tumor behaviors among different patients who seemingly have the same histopathological disease. It is assumed that most parts of the genetic component that contribute to this variability is mainly constituted by the sum of actions of weakly acting low-penetrance genes, whose allelic forms interact among themselves and with the environment to determine the clinical variability among individuals. These genes (also called modifier genes), mostly follow a trend of quantitative inheritance.6 Additionally, a major part of cancer growth is due to non-cell autonomous processes that consist in an aberrant tissue, growing in an uncontrolled manner within the context of the physiology of a complex organism.7 Therefore, it is a disease that not only depends on the properties of the tumor cells themselves, but also on other compartments like the immune and endocrine systems, stroma, vasculature, and others, all of which play key roles in the development and evolution of cancer. Consequently, differences in tumor behavior are not only determined by intrinsic factors to the tumor cells (such as proliferation, apoptosis, etc.), but also by extrinsic factors outside the tumor compartment per se. Modifier genes could regulate both the molecular and cellular functions of these different compartments, and this fact could explain the differences in tumor behavior among patients who seemingly suffer the same disease. The identification of those modifier genes is one of the major challenges of the future in cancer research. In this review, we mainly use breast cancer as paradigm to illustrate this issue.
Genetics analysis strategies are the only tools that allow us to consider the global scenario, because the susceptibility loci can contain risk genes controlling either intrinsic or extrinsic factors.6,8 Genome Wide Association Studies (GWAS) have permitted the identification of different susceptibility regions, genes and pathways, but the reproducibility between studies is difficult, probably due to the high heterogeneity of human genetics and its complex interaction with environmental factors.6,8 Crosses among inbred mouse strains of defined genetic backgrounds that exhibit strong differences in cancer susceptibility, have simplified the identification of Quantitative Trait Loci (QTL) and their interactions responsible for variable tumor behavior. Although the refinement of the QTLs to the gene level using mouse models was a very difficult task, the new high-throughput technologies recently developed in genetics, genomics and bioinformatics help to tackle this complicated duty with success. All these new technologies are improving our understanding of the genetic component/networks that control the variability in tumor risk, development and clinical evolution. The final goal is to obtain a better understanding of the molecular factors that determine the variability of the disease, which will finally result in the development of more personalized clinical applications for the benefit of the patient.
Cancer heritability
Cancer heritability is still very poorly understood. No more than a modest portion of cancers present an obvious trend of heredity (the considered “real” hereditary cancers). This is actually the situation of breast cancer where just a small portion of tumors could be identified by the inheritance of mutated variants of high penetrance genes like BRCA1 and BRCA2. However, mutations of these genes only account for a small percentage of the human tumor predisposition, resulting in quite rare hereditary cancer syndromes. These kinds of uncommon, but severe alleles have been additionally implicated in most forms of hereditary cancer syndromes. Hereditary susceptibility to breast cancer has become connected with germline mutations in at least eighteen genes.9 A huge number of distinct loss-of-function mutations have been discovered within BRCA1 as well as BRCA2 genes; most of these variations are usually individually rare, and each one confers quite high susceptibility for breast and ovarian malignancies. Uncommon germline mutations of other genes are also connected with an elevated risk of breast cancer, ranging from two-fold for CHEK2 to ten-fold for P53. Interestingly, all of these genes function in networks that are crucial to DNA repair and preservation of genomic integrity. In most cases, the inherited mutation is followed by somatic loss of the corresponding wild type allele, resulting in mistakes in DNA repair that ultimately lead to cancer development.10 However, it is important to emphasize that only a low percentage of global cancer risk can be attributed to hereditary mutations in the high-penetrance care-taker genes, and present obvious patterns of Mendelian inheritance.
The environmental component may play a more essential role in sporadic tumors than in hereditary cancer, and in some cases may over-ride the genetics.11–13 Nevertheless, there is significant evidence from large scale epidemiological studies indicating that the chance of developing sporadic cancer also has a significant hereditary component. One of these studies, which was carried out on several thousand pairs of twins, demonstrated that when one twin developed cancer, the other had a significantly increased risk of generating the same type of disease, but without any obvious Mendelian inheritance pattern. In fact, many scientific studies have concluded that common cancers are polygenic diseases with a quantitative genetic pattern.11–14
Even in families carrying specific mutant alleles of high-penetrance risk genes with potent effects such as BRCA1 and BRCA2, phenotypes tend to be influenced by the hereditary background, becoming much more comparable between affected twins, but varying among more distant family members with the same gene alteration;6,14,15 this would indicate that low-penetrance genes could also modify the behavior of hereditary cancer. A deeper knowledge of the genetic component would be essential to estimate the individual genetic susceptibility to develop cancer, to improve early detection and diagnosis of the disease, and to understand the fundamental biochemical and physiological pathways governed by those low-susceptibility genes as a critical step for the development of new cancer treatments.16
Cancer has a non-cell autonomous disease component
Cancer is, in part, a non-cell autonomous process; it is an aberrant tissue that grows in an uncontrolled manner in the context of the physiology and pathophysiology of a complex organism. Tumor growth, as that of any other tissue, depends not only on the intrinsic properties of the parenchymal component (tumor cells), but also on other organism compartments such as the immune and endocrine systems, stroma, vascular system, etc. (Fig. 1). All of them play key roles in the development and evolution of cancer. Consequently, tumor behaviour (i.e. susceptibility, development and clinical evolution) is not only going to be determined by factors intrinsic to the tumor cell, involved in processes such as proliferation, apoptosis, DNA repair etc., but it will also be influenced by those extrinsic factors from other compartments. Furthermore, these two main compartments are not independent, but rather they continuously crosstalk and interact with each other, so that the intrinsic factors are capable of recruiting the extrinsic ones, and the availability of the extrinsic factors determines the intrinsic cellular activity.
Fig. 1.

Cancer is not purely a cell-autonomous disease. There are connections between cancer cells and immune and endocrine systems, vasculature and stroma surrounding them that modify the tumor behaviour and susceptibility.
The fact is, that connections among cancer cells, the stroma and the rest of the organism have their roots in the physiological responses that take part in regular tissue homeostasis.17–19 The equilibrium between cell-renewal and cell-reduction is tightly governed through connections between parenchyma stem cells and the microenvironment to carry out the tissue remodelling or respond to the stress caused by tissue injury. Cancer cells virtually do not react in response to normal physiological regulators of cell growth, and are constantly sending remodelling signals for the stroma to be reorganized in an activated form to permit tumour growth; to some degree tumors behave like a wound that does not heal.20 Tumor modifier genes could play a role in controlling molecular and cellular factors of these two main compartments that would explain not only discrete physiological differences among individuals, but also differences in the susceptibility, development and the different clinical evolution among patients who seemingly suffer the same cancer disease.
The relevance of the microenvironment is highlighted by new studies that demonstrate how the apparently normal stromal cells can manipulate epithelial cancer cell activity in reconstitution experiments, and by recognition of particular somatic genetic alterations in the stromal element of the tumor.21,22 It has been proposed that the global microenvironment mostly functions as an epigenetic tumor modifier.23 In fact, the genetic inactivation of Pten in stromal fibroblasts associated with mouse mammary glands speeds up the initiation, progression and malignant transformation of mammary epithelial tumors.24 Furthermore, malignant cells can be reverted to a quiescent state simply by incorporation into an embryonic microenvironment.25 This suggests that the microenvironment is dominating over malignancy. Thus, for tumors to advance into a more malignant stage, they must change their own microenvironment to a promoting one. The change in microenvironment probably originates from oncogenic mutations in proliferating tumor cells that send signals to the stroma, but possibly also mutations in the stroma itself.21–24
The resulting tumors are complicated structures of malignant cancer cells surrounded by vasculature and associated with an active tumor stroma composed of several non-malignant cell types, such as fibroblasts and myeloid cells with an important role in global tumor behaviour. For example, evidence suggests that tumor initiation, progression and metastasis are influenced by particular subpopulations of macrophages,26,27 and also other inflammatory cells, such as B and T-lymphocytes and mastocytes, have been shown to play a role in tumor promotion.28 In fact, the milieu of the tumor microenvironment is similar to the one found in the inflammatory reactions within a restorative healing injury, which stimulates angiogenesis, turnover of the extracellular matrix (ECM), as well as tumor cell motility.7 And, as occurs in inflammation, a growing body of data support the perspective that extracellular proteinases, like the matrix metalloproteinases (MMPs), mediate numerous modifications within the microenvironment in the course of tumor development.29 Additionally, one of the most critical pathways controlling both inflammation and tumor microenvironment, is the TGF-beta signalling pathway, together with important cell-autonomous effects. GWAS have identified many SNPs close to genes that belong to the TGF-beta superfamily, such as CREM1 and SMAD7. In addition, constitutively reduced TGFBR1 expression is a powerful modifier of colorectal cancer susceptibility. All these data indicate that germline variations of the TGF-beta superfamily might account for a very important percentage of colorectal cancer susceptibility.30
The modulation of stroma function by tumor susceptibility modifier genes is well-known. The first tumor-modifier gene identified was a modifier of the Apc (Adenomatous Polyposis Coli) gene function, located in the QTL named Mom1 (“Modifier of Min1”, which in turn means “Multiple Intestinal Neoplasia-1”). The gene responsible encodes a secretory phosphatase type II phospholipase A (Pla2s). Pla2s was proposed to modify the polyp number by altering the cellular microenvironment within the intestinal crypt.31 Interestingly, this gene has been widely implicated in the inflammatory process,32 angiogenesis, and has pro-atherogenic activity.33 More importantly, later studies demonstrated that the PLA2S gene has a role in human cancer pathogenesis of the digestive tract,34 supporting the importance of mouse tools to identify cancer modifier genes in the human population. Thus, it is feasible for a number of these genetic determinants to be involved in the pathogenesis of different physiological and/or pathophysiological events at the same time; this effect is named “pleiotropy”. This concept refers to those genes that concurrently have effects on different phenotypes. This has been demonstrated not only for the diverse subtypes of cancer, such as 8q12 abnormalities that are related with various types of tumors,35 but also for autoimmune diseases36 or very different pathologic conditions. Many parallels exist between different diseases and pathologic situations; for example, hypertension, hypercholesterolemia and obesity are included in known metabolic syndromes; or the existence of an association between those processes and particular types of cancer; or the relationship between certain autoimmune diseases and cancer.37–39 All of these data indicate that complex interactions take place among genes that simultaneously control different processes. It is also possible to relate all the disorders that share common conditions and the gene interactions that control them. This fact has recently generated the interesting concept of diseasome,40 which can be represented by two networks: first, by the human disease network, in which nodes represent disorders, and two disorders are connected to each other if they share at least one gene; and second, by a disease gene network where nodes represent disease genes, and two genes are connected if they are associated with the same disorder.40
One of the most important challenges in cancer research is to understand the underlying basis of heterogeneity of tumor susceptibility, development, and evolution in the context of the physiology and pathophysiology of the organism. It would be desirable to tackle the cancer problem with tools that permit visualization of this global picture, integrating both intrinsic and extrinsic factors with the behaviour of the tumor cell. Genetic analysis offers a unique tool to embrace the global scenario, because each QTL region could contain both intrinsic and/or extrinsic modifier genes, and can help to explain cancer as a complex disease.
Identification of cancer susceptibility genes in human populations
Even though rare alleles with strong effects could be substantial contributors to sporadic cancer risk, the searching for tumor susceptibility has focused mainly on the common disease-common variant model that presumes that cancer susceptibility originates from the additive effects of combinations of common low-penetrance variants.41 With this model, every susceptibility variant is assumed to contribute a small amount of risk, without any variant conferring enough by itself to result in tumor development. Cancer origin and evolution have been proposed to be the consequence of the merged effects of a number of such alleles, which may control intrinsic and/or extrinsic functions. The search for tumor susceptibility genes has mainly been carried out by GWAS, in which allele frequencies at thousands of polymorphic sites (i.e. SNPs) are compared in a large number of cases versus a similar number of controls. As discussed in later sections, in spite of their limitations, these studies have successfully identified some of the common susceptibility variants for different common diseases and traits, including cancer.10
A. Identification of susceptibility genes in breast cancer
We will use breast cancer as a model for this discussion, as major efforts have been made to identify genetic components of both hereditary and sporadic versions of the disease. Studies of susceptibility genes in breast cancer initially focused on the detection of high-penetrance susceptibility genes through the analysis of linkage in family pedigrees comprising of several affected members. These familial studies involve fewer patients, and need significantly reduced marker density, in comparison with current GWAS, but the two approaches can be complementary. The results of pedigree evaluation can offer important and persuasive signs of genetic effects, because they are primarily based on genetic transmission of disease-causing alleles between affected family members. Inherited variations in the two main susceptibility genes already known for breast cancer, BRCA1 and BRCA2, together account for only around 20% of hereditary breast cancer.35 A few additional genes have been identified,42,43 but all these known mutations can only elucidate a small portion of familial breast cancers, and around less than 5% of the total breast cancer susceptibility. A number of linkage studies have described candidate loci that contain breast cancer susceptibility genes. However, these loci were not clearly statistically significant presumably due to the fact that the number of families affected by each locus was low. In a recent linkage study in Spanish breast cancer families, three more regions of interest came out, located on 3q25, 6q24 and 21q22;43 it will be very important to further confirm these results in new populations.
The majority of studies to identify susceptibility genes in breast cancer have been carried out by GWAS. These studies have identified several common variants that have an influence on breast cancer susceptibility, but only four were replicated in two or more GWAS (Table 1).44–51 Meta-analysis of suggestive loci utilizing three published GWAS resulted in the detection of an extra locus on 5p1252 that appeared to be linked particularly with estrogen-receptor positive cancers of the breast. It must be taken into account that statements for associations with particular categories require much more cautious replication studies. For instance, the 2q35 locus was initially associated particularly with estrogen-receptor positive breast cancer, but a later study reported comparable results irrespective of the estrogen receptor status.53
Table 1.
Main regions found by human GWAS for breast cancer susceptibility
| Susceptibility region | Reference |
|---|---|
| 5q11.2; 8q24; 10q26; 11p15.5; 16q12.1 | Easton et al., 200744 |
| 2q35; 16q12 | Stacey et al., 200745 |
| Three ERBB4 SNPs | Murabito et al., 200746 |
| 10q26 (intron 2 of FGFR2) | Hunter et al., 200747 |
| 6q22.33 | Gold et al., 200848 |
| 6q25.1 | Zheng et al., 200949 |
| 1p11.2; 14q24.1 | Thomas et al., 200950 |
| 3p24; 17q23 | Ahmed et al., 200951 |
| 5p12 | Stacey et al., 200852 |
| 2q35 | Milne et al., 200953 |
Almost all of the individual low-penetrance variations discovered to date have weak effects (odds ratios per-allele are less than 1.41) and explain much less of the heritability of breast cancer, compared to the known BRCA1 and BRCA2 mutations; and maybe a few others such as a common variant within the Fibroblast Growth Factor Receptor 2 gene (FGFR2). Nonetheless, a very significant portion of the breast cancer susceptibility presently continues to be uncharacterized and may be due to the sum of combinations or interactions of low-penetrance genes. These allele variants, together with the environmental exposure, may contribute to breast cancer susceptibility. The causative environmental exposures continue to be evasive, because many of the formerly suggestive environmental and life style risk factors (e.g. nutrition) for breast cancer have recently been refuted by large studies in the last decade.54,55
In conclusion, around twenty different presumed breast cancer susceptibility loci have already been identified using GWAS studies, but few loci were replicated in different studies.44 In addition, almost all of these variants identified in breast cancer and other studies, have no demonstrated biological or mechanistic relevance to the disease, or medical utility for diagnosis or therapy. This could mean that causality within this framework can hardly ever be solved by large-scale association or case-control studies exclusively.10 A reason for this could be the genetic heterogeneity. A number of genes are presumed to play an important role in the susceptibility to breast cancer, but those genes could be important exclusively within a limited number of families, and could be absolutely lost as soon as they are diluted in the general population.16
B. The challenges of genome-wide association studies
Within the last few years, over fifty GWAS have been performed to search for cancer susceptibility genes. As discussed by other authors, a few repetitive conclusions can be obtained from them: first, only a few variants were found in each GWAS; second, each locus has a tiny effect; and third, there is a relative deficit of replication of allele variants identified by diverse GWAS.16 One explanation for this last problem could be that, despite having large sample numbers, there is a restricted potential to identify modest genetic effects due to the strict levels of significance demanded in these studies. Therefore, variants that attain significant p-values, for instance P > 10−8, are usually real,56,57 while those associated with significantly more modest P-values (e.g. 10−5 or 10−6) might indicate false positives. Furthermore, a large number of those variants will not be replicated when screened in other samples. For instance, in GWAS the chance that a variant with a P-value of 10−5 shows a genuine association is actually lower than one percent.58,59
A different situation that could play a role in clarifying the current incongruence of GWAS is the fact that cancer susceptibility is an extremely complicated phenotype and, together with the incomplete penetrance of the inherited tumor risk alleles, the interaction with environmental risk factors could substantially alter hereditary susceptibility. Based on environmental exposures, a person with high genetic susceptibility to develop malignancy may never be affected, while a person at low cancer risk, but high exposure, might suffer the affliction (Fig. 2).60 This question is still widely debated for breast cancer, which has been linked to nutritional61 as well as reproductive factors,62 and alcohol,63 along with other exposures. Inability to take into account this kind of variable in GWAS may well decrease the strength of analysis or even reduce our ability to discover genuine causative susceptibility loci.64 This may also clarify the fact that, even though the hereditary element of developing prostate cancer was estimated to be around 40%,11 early studies including quite large families with high risk, have not confirmed this data.65 This might be due to genetic heterogeneity (i.e. the causal polymorphisms that are the responsible for the phenotype vary among families).
Fig. 2.

Tumor risk is the consequence of the interaction between constitutional genetics and environmental exposures. The combination between the genetic background (modifier genes, mainly low susceptibility cancer genes) and the environmental factors varies among individuals and might explain the different tumor susceptibility and behaviour observed in patients.
Additionally, even though quite a few studies have discovered genes with important phenotypic effects,44,47,66–69 as we indicated above, the majority of cancer risk is most likely due to a selection of genes with additive effects. However, it is also possible that many genes regulate cancer susceptibility mainly via non-additive interactions. These interactions could be multiplicative or conditional, in such a way that the principal effect of one gene would depend on the existence of a specific allele within a second locus, and so on, forming a network of gene interactions where the next interaction is possible only if a particular allelic form is already present. It might be also possible that those numerous weak-interacting loci would simply achieve suitable levels of significance within particular series of patient samples, depending on the hereditary background or environmental factors (Fig. 2). Thus, on top of this currently complicated situation of the GWAS scenario would be the spectrum of hereditary interactions that depend on the genetic background, a fact that has been clearly demonstrated in animal models like the mouse, among others.6,70–72 Moreover, genome-wide studies in mouse models of cancer have discovered loci that arise as a result of genetic interactions that are not viewed as individual QTL with major effects, utilizing common methods of analysis.70–73 These studies demonstrated, first, the power of mouse models to simplify the problem, and second, that more advanced statistical methods used to discover interactions among loci in linkage analyses might be required to discover the locations of multiple weak susceptibility alleles.6,74,75
In summary, extrapolation of the final results obtained from GWAS to other human populations raises the uncomfortable possibility that a specific SNP discovered as a tumor modifier in a particular population lacks any effect (or even might work in the opposite direction) within another ethnic background. For that reason, even though hereditary background in individual patients is consequently capable of controlling illness development, as it has been evidently demonstrated in animal models,76,77 very few of these human low susceptibility genes have been convincingly identified. Therefore, even though the present flood of GWAS show the strength of this strategy, there are natural restrictions of this whole-genome association analysis that circumvent the capture of most pertinent scientific data. Thus, GWAS are afflicted by implicit limitations and cannot provide us with an entire understanding of the intricate genetic and environmental interactions connected with common disease phenotypes. In fact, today no individual method is good enough to permit an extensive knowledge of cancer etiology and pathogenesis, in particular within the extremely complicated area of human genetics. But there have been great improvements in QTL research within the last ten years, mainly by utilizing mouse cancer models. Mouse QTL analysis, as well as GWAS, will become complementary strategies that will improve the knowledge of the actual genetic basis of the human disease.8
The mouse as a complementary approach
The use of mouse models can complement human GWAS by allowing a higher level of control over hereditary variance and environmental exposure. An intercross or a backcross carried out between two inbred mouse strains with divergent tumor phenotypes generates offspring where each mouse is genetically and phenotypically unique for different quantitative sub-phenotypes, controlled by different interacting QTL. These strategies reproduce a simplified model of cancer as a polygenic complex disease.8 Furthermore, these approaches are facilitated by the production of a large number of inbred and outbred strains of various Mus species which have different evolutionary genealogies, together with recombinant inbred strains, congenics,69 consomics,78 and genetically engineered mice (GEM), all of them constituting a unique genetic resource among animal models that can greatly simplify the identification of susceptibility genes. Certainly, the enormous number of GEMs available, in particular through programs like the Knock-Out Mouse Project (KOMP) whose goal is to mutate all protein-encoding genes and make all these mice available to the scientific community,79 provide important tools for narrowing down QTL candidate genes. Knockouts are used in this context to test the candidature of a driver gene at a QTL by what has been named the QTL-knockout interaction test, by which the interaction between the null allele and the QTL is tested, and compared with the interaction with the wild type allele.80 The use of a GEM strain carrying a knock-out or a knock-in allele located in a QTL, can help to validate the participation of that gene in the QTL effect by linkage analysis.81
Additionally, there is increasing evidence showing that hereditary risk factors have a comparable role in complex disease pathogenesis within human and mouse models regardless of interspecies dissimilarities. Rodents develop cancer that appears to be amazingly similar in most cases to human tumors, and they accumulate mutations in a comparable spectrum involving the same genes and pathways.82 These facts suggest that, at least some of the numerous QTLs containing tumor risk genes that have been mapped in the mouse may be highly relevant to the human scenario and serve as an effective method of complementing observations within human populations.6 This has been demonstrated for example, for plasma levels of cholesterol.83 Mouse QTLs have already been proven to be equivalent to human disease susceptibility loci in a number of cases including cancer,84–87 and although mouse QTL analysis is not without drawbacks, there have been important technical advances in the last few years (see below) (Fig. 3). Certainly, the introduction of novel techniques and resources can help to unravel the exceptionally complicated interwoven factors influencing cancer etiology. These data suggest that mouse studies, carried out in parallel with human sample analysis, may accelerate development of a deeper understanding of the hereditary risk component of complex diseases (Fig. 4).8
Fig. 3.
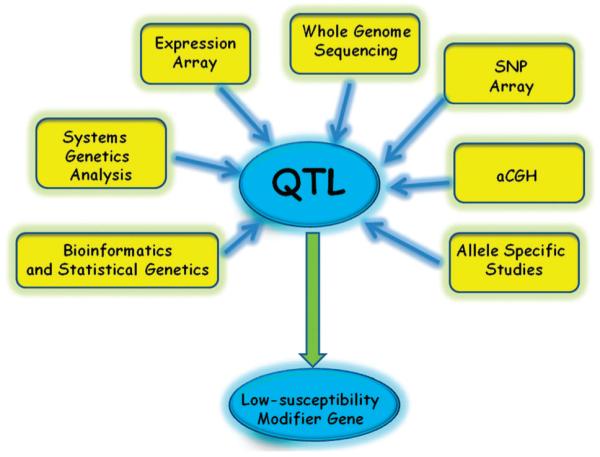
Recent technical advances have improved QTL research: The use of genetically more complex mice (outbred and artificial strains) in combination with high resolution SNPs arrays and new techniques of statistical genetics greatly improve the definition of new QTLs. In tumor cells, the analysis of changes in copy number and expression by whole genome comparative hybridization and expression arrays allow the further refinement of QTL.
Fig. 4.

Mouse models are a good tool to identify QTLs in an environmentally controlled way that could be extrapolated to the human population. At the same time, they are very useful to verify and refine candidate loci found in humans by GWAS. Technical advances such as whole genome sequencing are readily making the recognition of orthologous chromosomal regions between species straight forward, simplifying the refinement of QTLs found in both kinds of studies.
Technological advances for identification of QTLs in mouse models
Although mouse quantitative trait locus mapping has been demonstrated to be an effective tool for identifying trait risk, this strategy is not without drawbacks.8 These have prompted numerous researchers to examine substitute approaches for candidate gene detection.77 Among the drawbacks are low mapping resolution; the difficulty of identifying specific genes and nucleotides associated with complex genetic traits; and thirdly, modelling multiple QTL, which although easier than in GWAS carried out in human populations, requires complex statistical strategies. Technological improvements in the meantime have partially resolved several of these issues (Fig. 3), including:
1. Analysis of haplotype structure and in silico mapping
Understanding the parts of the genome that are the same by ancestry (i.e., have the same haplotype organization) among mouse strains is a useful approach for refining loci of interest. The strategy utilizes genetically more complex mice from natural origin such as partially inbred and outbred strains like Mus spretus,86 or heterogeneous stocks generated by combinations of various inbred mouse strains. In both cases, this strategy may quickly minimize the number of candidate genes that have to be tested.86,88,89 The main idea is to identify frequent haplotype sections that segregate inside the genetically characterized candidate locus to restrict the quest for genes presumed to be of interest.88–91
Heterogeneous stock mice like MF192 have been developed through the arbitrary reproduction of progeny from normally four to eight inbred strains.93,94 QTL should be found in a region in which sequence divergence matches genetic origin. Therefore, whenever QTLs have been mapped in heterogeneous stock populations, the markers of strain distribution structure within the initial QTL can be joined with mapping data to refine the area which contains the functional variant.
This strategy has been utilized to carry out mapping at an exceptionally high resolution and to identify candidate genes.88–90,95 In summary, due to the fact that heterogeneous stocks are produced from known ancestral inbred strains, it is possible, by some statistical genetics and in silico analysis, to obtain the origin of every allele and to map QTL at sub-centimorgan resolution.
Similar to the use of heterogeneous stocks of mice artificially generated, is the utilization of natural outbred stocks which accumulate recombinants with time, so they provide a substantial increase in mapping resolution, possibly sufficient enough to identify candidate genes. Specifically, outbred Mus spretus have been utilized with successful results to discover Aurora Kinase A (Aurka/Stk6)96,97 as a skin tumor susceptibility gene,86 and an outbred population of CD1 mice has been utilized to chart a predisposing region for lung cancer.98 These kinds of natural outbred stocks may well provide greater resolution than artificial versions, however they miss the benefits from parental information within theheterogeneous stocks. In addition, these approaches require many animals as well as high density genotyping. In fact, genome-wide mapping in heterogeneous stocks demands a minimum of 6000 genetic markers (i.e. SNPs). It is very important to consider that to reduce false-positive results to appropriate levels with such amount of markers, it is necessary to utilize strict significance thresholds for the p-value.99,100
Recently the Collaborative Cross project was launched to generate the largest panel of recombinant inbred (RI) strains with more than one thousand RI lines of mice. These strains originated from the crossing between five inbred and three wild-derived strains. This strategy will allow high resolution mapping equivalent to the heterogeneous stocks, together with the reproducibility of the inbred strains. The main aim is to reach a mapping resolution of about a megabase. Additionally, the genetic variation will be homogeneously distributed along all the genome without regions where there is no variation, so every single gene can potentially be tested for involvement in a particular phenotype.101,102
2. Analysis of tumors using whole genome array comparative genomic hybridization (aCGH) and loss of heterozygosity (LOH) analysis by SNP arrays
High penetrance germline susceptibility genes are often linked to somatic loss of the wild type allele in tumors (the “two hit” Knudson hypothesis). The same could happen with at least some of the low-penetrance susceptibility genes that control intrinsic cellular activities. Cancer low-susceptibility genes could drive copy number gains in tumors in an allele-specific manner, while cancer resistance alleles may possibly be lost as a result of deletion or mitotic recombination leading to loss of heterozigosity (LOH). These types of allele-specific somatic losses and gains can be used to identify cancer risk genes;87,103 this technique facilitated the recognition of Stk6 as a low-penetrance tumor susceptibility gene.86
3. Genome-wide expression arrays
The observation that most part of SNPs are located outside coding regions has led numerous investigators to hypothesize that many QTLs are probably attributed to delicate alterations in gene expression instead of to missense or nonsense mutations, as is the case for Kras2 in cancer induced by urethane.81 This idea has consequently resulted in the screening for genes in QTL regions that exhibit differential expression regarding the strains of interest.104 This particular strategy, initially specified as genetical-genomics,105 offered a good impartial method for quickly screening hundreds of possible candidate genes at the same time to reduce the list for additional evaluation to a workable quantity.90,106 Within this technology, researchers could include co-regulated networks of expression and QTL evaluation. This allows identification of a group of genes that are operating collectively to impact a susceptibility phenotype based on the network of genes that are significantly correlated with each other, and their expression levels controlled by common genetic loci. In some informative circumstances, it would be possible to find the susceptibility locus, the candidate gene is affected in cis by that locus, and downstream genes which are influenced in trans. Thus, by adding automatic finding and manual curation, it is possible to define networks of genes with a common function and that are controlled by a common mechanism.72,74,75
4. The next generation of sequencing techniques together with the culmination of the human107,108 as well as mouse109 genome sequencing projects
Thanks to the completion of the human genome project, it is possible to identify most genes within a specified location. Next-generation sequencing will make it possible to investigate particular candidate genes without prior genomic screening. The power to discover and define candidate loci has continued to grow considerably since the whole genomes of many species have been sequenced.110,111 This has allowed recognition of evolutionary conserved sequence domains, and much more recently has allowed direct visualizations of SNPs among some of the widely used inbred mouse strains through the use of chip-based sequencing, as well as large-scale polymorphism screening.112 Interestingly, sequence accessibility throughout species has allowed additional speeding of candidate gene recognition for all those traits which have already been mapped in several species. Recognition of orthologous chromosomal sections and their breakpoints inside genetically identified loci might help to refine QTL localization and candidate gene databases by restricting searches to those regions shared between the two species.8,106
Future perspectives for human cancer risk
As we have discussed, the majority of studies in human families with higher cancer susceptibility continue to be centred on the chance that a single or even a small number of powerful genes could be the cause of the “missing” hereditary element of tumor risk. Even though there may still be a number of high penetrance genes remaining to be discovered, mostly we will be confronted with the difficulties of the existence of numerous low-penetrance modifier genes and their interconnections. As it has already been suggested using mouse models of cancer, combinations of these low-penetrance modifier genes may be responsible for the variable risk of both hereditary and sporadic cancer.6 In the last few years, the efforts to identify this cancer genetic component in the human population have mainly been focused on the use of GWAS. Although these studies have proven to be a very useful tool for the identification of some common genetic variants, how much this technique has contributed to clarification of the “missing heritability” of different complex diseases is a matter of controversy.10,113,114 There have been substantial attempts to recognize low penetrance cancer susceptibility genes by GWAS.44,115–117 Even though this research has found a few allelic variants which influence cancer risk, the majority of them will probably be challenged by this method, because of the tiny impact that any single one confers on the total tumor risk. These low penetrance-genes are subject to strong interactions among themselves as well as with the natural environment, and the results can be quite inconsistent within different populations under the influence of diverse environmental elements. Therefore, the actual identification of low penetrance cancer risk genes within the human population is a challenging endeavour because of the huge heterogeneity within human genetics and the environment. This could explain why the majority of the heritable portion of tumor and complex traits has not yet been identified by GWAS. For the same reasons, even though some of the genetic loci discovered through GWAS initially possess robust statistical significance regarding association with specific tumors, the informative potential of these loci to predict individual tumor susceptibility is restricted by their small impact on global cancer risk, so the clinical importance of this kind of variant will be very limited. Therefore, with our current information, we can say that single SNPs will have limited utility in predicting if someone will suffer from cancer. But, although the diagnostic benefit of any genetic polymorphism alone is limited, we can anticipate that understanding of the combined interactions among those allelic variants that collectively possess considerably more potent consequences on risk would likely exert a significantly larger effect on the prognosis as well as on cancer therapy and its general clinical management.
Concluding remarks
Even though mouse models have become an invaluable tool for QTL mapping, a refining of QTL locations remains problematic. This task is beginning to be tackled successfully with the help of newly developed technologies such as high throughput gene expression arrays, together with systems genetics approaches,74 whole genome SNP arrays and aCGH with allele specific analysis,86 and high-coverage whole genome sequencing that will probably become the technique of reference as soon as it becomes cost-effective. In the following years, as a result of the application of these technical innovations, we ought to start to see the refinement of several loci containing mouse cancer risk alleles and also the identification of clusters of them, jointly with their interactions, that may help selecting presumed genes and pathways to become analyzed in human populations.6 Moreover, considering the current speed of technical advancement, it is quite possible that in the near future, with the advent of new technologies such as whole genome sequencing, positional cloning may be unnecessary and fine mapping of significant loci may lead straight to their identification.
Mouse models not only are a good tool for identifying QTL regions that can be extrapolated to human populations, but also offer a parallel system for immediate testing and verification of the results obtained from human epidemiology and GWAS. Also, moving back and forth between mouse and human systems will be a good strategy to recognize the causal genetic variant of a presumed candidate gene (Fig. 4). Moreover, it is known that the environmental influences and way of life options have an important effect on tumor susceptibility in humans. Gene–environment interactions could also be investigated using mouse models, and will allow us to recognize how genes work together with particular environmental influences recognized by epidemiological studies. Enrolling together systems genetics and epidemiology ought to enable us to clarify the connections involving hereditary background and environmental factors that are the reason for part of the “obscure” cancer heritability.75
The knowledge acquired by means of these genetics studies will have a significant effect on medical sciences, and should certainly lead to improved prognosis prediction and therapy of human cancer, leading to a more individualized clinical management of the disease.
Insight, Innovation, Integration.
Mouse models are an excellent strategy to identify QTLs in a genetically and environmentally controlled manner that could be extrapolated to human populations. Additionally, they are very useful to validate and refine candidate loci found in humans by GWAS. The integration of new technical innovations has improved QTL research: the employment of high resolution SNP, CGH and gene expression arrays speeds up the refinement of QTLs. Other technical advances such as whole genome sequencing are readily making the recognition of orthologous regions between both species straight forward, simplifying the refinement of QTLs found in human and mouse, and facilitating a cross-species strategy to identify QTL-driver genes.
Acknowledgements
We apologize for these contributions that could not be cited in the manuscript due to space restrictions. We thank Drs Allan Balmain, Isidro Sánchez Garcia and Cesar Cobaleda for useful comments. J. H. Mao is supported by Office of Biological & Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, by Laboratory Directed Research & Development Program (LDRD), and by the National Institutes of Health, National Cancer Institute grant R01 CA116481. J. Pérez-Losada is partially supported by FEDER and MICINN (PLE2009-119), FIS (PI070057; PI10/00328), CSIC (200920I137), Junta de Castilla y León (SAN126/SA66/09; SA079A09). A. Castellanos-Martín is supported by FEDER and MICINN (PLE2009-119).
Footnotes
Published as part of an Integrative Biology themed issue in honour of Mina J. Bissell: Guest Editor Mary Helen Barcellos-Hoff.
References
- 1.Carmichael A, Sami AS, Dixon JM. Breast cancer risk among the survivors of atomic bomb and patients exposed to therapeutic ionising radiation. Eur. J. Surg. Oncol. 2003;29:475–9. doi: 10.1016/s0748-7983(03)00010-6. [DOI] [PubMed] [Google Scholar]
- 2.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer. 2007;7:834–46. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 4.Smigal C, Jemal A, Ward E, Cokkinides V, Smith R, Howe HL, Thun M. Trends in breast cancer by race and ethnicity: update 2006. Ca-Cancer J. Clin. 2006;56:168–83. doi: 10.3322/canjclin.56.3.168. [DOI] [PubMed] [Google Scholar]
- 5.Winter SF, Hunter KW. Mouse modifier genes in mammary tumorigenesis and metastasis. J. Mammary Gland Biol. Neoplasia. 2008;13:337–42. doi: 10.1007/s10911-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 6.Balmain A. Cancer as a complex genetic trait: tumor susceptibility in humans and mouse models. Cell. 2002;108:145–52. doi: 10.1016/s0092-8674(02)00622-0. [DOI] [PubMed] [Google Scholar]
- 7.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunter KW, Crawford NP. The future of mouse QTL mapping to diagnose disease in mice in the age of whole-genome association studies. Annu. Rev. Genet. 2008;42:131–41. doi: 10.1146/annurev.genet.42.110807.091659. [DOI] [PubMed] [Google Scholar]
- 9.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 10.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 11.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 12.Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int. J. Cancer. 2002;99:260–6. doi: 10.1002/ijc.10332. [DOI] [PubMed] [Google Scholar]
- 13.Borecki IB, Province MA. Linkage and association: basic concepts. Adv. Genet. 2008;60:51–74. doi: 10.1016/S0065-2660(07)00403-8. [DOI] [PubMed] [Google Scholar]
- 14.Fodor FH, Weston A, Bleiweiss IJ, McCurdy LD, Walsh MM, Tartter PI, Brower ST, Eng CM. Frequency and carrier risk associated with common BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer patients. Am. J. Hum. Genet. 1998;63:45–51. doi: 10.1086/301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nathanson KL, Wooster R, Weber BL. Breast cancer genetics: what we know and what we need. Nat. Med. 2001;7:552–6. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- 16.Galvan A, Ioannidis JP, Dragani TA. Beyond genome-wide association studies: genetic heterogeneity and individual predisposition to cancer. Trends Genet. 2010;26:132–41. doi: 10.1016/j.tig.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donjacour AA, Cunha GR. Stromal regulation of epithelial function. Cancer Treat Res. 1991;53:335–64. doi: 10.1007/978-1-4615-3940-7_16. [DOI] [PubMed] [Google Scholar]
- 18.Paus R, Peters EM, Eichmuller S, Botchkarev VA. Neural mechanisms of hair growth control. J. Investig. Dermatol. Symp. Proc. 1997;2:61–8. doi: 10.1038/jidsymp.1997.13. [DOI] [PubMed] [Google Scholar]
- 19.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat. Rev. Cancer. 2003;3:434–43. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 21.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002;32:355–7. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 23.Weaver VM, Gilbert P. Watch thy neighbor: cancer is a communal affair. J. Cell Sci. 2004;117:1287–90. doi: 10.1242/jcs.01137. [DOI] [PubMed] [Google Scholar]
- 24.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, Naidu S, Wei G, Sharma SM, Stephens JA, Fernandez SA, Gurcan MN, Weinstein MB, Barsky SH, Yee L, Rosol TJ, Stromberg PC, Robinson ML, Pepin F, Hallett M, Park M, Ostrowski MC, Leone G. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–91. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29:309–16. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellam N, Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- 31.MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell. 1995;81:957–66. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 32.Pruzanski W, Vadas P. Phospholipase A2–a mediator between proximal and distal effectors of inflammation. Immunol. Today. 1991;12:143–6. doi: 10.1016/S0167-5699(05)80042-8. [DOI] [PubMed] [Google Scholar]
- 33.Jaross W, Eckey R, Menschikowski M. Biological effects of secretory phospholipase A(2) group IIA on lipoproteins and in atherogenesis. Eur. J. Clin. Invest. 2002;32:383–93. doi: 10.1046/j.1365-2362.2002.01000.x. [DOI] [PubMed] [Google Scholar]
- 34.Leung SY, Chen X, Chu KM, Yuen ST, Mathy J, Ji J, Chan AS, Li R, Law S, Troyanskaya OG, Tu IP, Wong J, So S, Botstein D, Brown PO. Phospholipase A2 group IIA expression in gastric adenocarcinoma is associated with prolonged survival and less frequent metastasis. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16203–8. doi: 10.1073/pnas.212646299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 36.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wysocki PJ, Wierusz-Wysocka B. Obesity, hyperinsulinemia and breast cancer: novel targets and a novel role for metformin. Expert Rev. Mol. Diagn. 2010;10:509–19. doi: 10.1586/erm.10.22. [DOI] [PubMed] [Google Scholar]
- 38.Tai P, Yu E, Joseph K, Miale T. A review of autoimmune diseases associated with cancer. Frontiers in Bioscience. 2010;2:122–6. doi: 10.2741/e73. [DOI] [PubMed] [Google Scholar]
- 39.Siddiqui AA. Metabolic Syndrome and Its Association With Colorectal Cancer: A Review. Am. J. Med. Sci. 2010 doi: 10.1097/MAJ.0b013e3181df9055. [DOI] [PubMed] [Google Scholar]
- 40.Goh I, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8685–90. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 42.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002;31:55–9. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 43.Rosa-Rosa JM, Pita G, Urioste M, Llort G, Brunet J, Lazaro C, Blanco I, Ramon y Cajal T, Diez O, de la Hoya M, Caldes T, Tejada MI, Gonzalez-Neira A, Benitez J. Genome-wide linkage scan reveals three putative breast-cancer-susceptibility loci. Am. J. Hum. Genet. 2009;84:115–22. doi: 10.1016/j.ajhg.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2007;39:865–9. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- 46.Murabito JM, Rosenberg CL, Finger D, Kreger BE, Levy D, Splansky GL, Antman K, Hwang SJ. A genome-wide association study of breast and prostate cancer in the NHLBI’s Framingham Heart Study. BMC Medical Genetics. 2007;8(Suppl 1):S6. doi: 10.1186/1471-2350-8-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr., Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007;39:870–4. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P, Kosarin K, Olsh A, Bergeron J, Ellis NA, Klein RJ, Clark AG, Norton L, Dean M, Boyd J, Offit K. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4340–5. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, Wen W, Levy S, Deming SL, Haines JL, Gu K, Fair AM, Cai Q, Lu W, Shu XO. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009;41:324–8. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, Chatterjee N, Garcia-Closas M, Gonzalez-Bosquet J, Prokunina-Olsson L, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver R, Prentice R, Jackson R, Kooperberg C, Chlebowski R, Lissowska J, Peplonska B, Brinton LA, Sigurdson A, Doody M, Bhatti P, Alexander BH, Buring J, Lee IM, Vatten LJ, Hveem K, Kumle M, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr., Hoover RN, Chanock SJ, Hunter DJ. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat. Genet. 2009;41:579–84. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, Morrison J, Maranian M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat. Genet. 2009;41:585–90. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, Jakobsdottir M, Bergthorsson JT, Gudmundsson J, Aben KK, Strobbe LJ, Swinkels DW, van Engelenburg KC, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Saez B, Lambea J, Godino J, Polo E, Tres A, Picelli S, Rantala J, Margolin S, Jonsson T, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Sveinsdottir SG, Alexiusdottir K, Saemundsdottir J, Sigurdsson A, Kostic J, Gudmundsson L, Kristjansson K, Masson G, Fackenthal JD, Adebamowo C, Ogundiran T, Olopade OI, Haiman CA, Lindblom A, Mayordomo JI, Kiemeney LA, Gulcher JR, Rafnar T, Thorsteinsdottir U, Johannsson OT, Kong A, Stefansson K. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2008;40:703–6. doi: 10.1038/ng.131. [DOI] [PubMed] [Google Scholar]
- 53.Milne RL, Benitez J, Nevanlinna H, Heikkinen T, Aittomaki K, Blomqvist C, Arias JI, Zamora MP, Burwinkel B, Bartram CR, Meindl A, Schmutzler RK, Cox A, Brock I, Elliott G, Reed MW, Southey MC, Smith L, Spurdle AB, Hopper JL, Couch FJ, Olson JE, Wang X, Fredericksen Z, Schurmann P, Bremer M, Hillemanns P, Dork T, Devilee P, van Asperen CJ, Tollenaar RA, Seynaeve C, Hall P, Czene K, Liu J, Li Y, Ahmed S, Dunning AM, Maranian M, Pharoah PD, Chenevix-Trench G, Beesley J, Bogdanova NV, Antonenkova NN, Zalutsky IV, Anton-Culver H, Ziogas A, Brauch H, Justenhoven C, Ko YD, Haas S, Fasching PA, Strick R, Ekici AB, Beckmann MW, Giles GG, Severi G, Baglietto L, English DR, Fletcher O, Johnson N, dos Santos Silva I, Peto J, Turnbull C, Hines S, Renwick A, Rahman N, Nordestgaard BG, Bojesen SE, Flyger H, Kang D, Yoo KY, Noh DY, Mannermaa A, Kataja V, Kosma VM, Garcia-Closas M, Chanock S, Lissowska J, Brinton LA, Chang-Claude J, Wang-Gohrke S, Shen CY, Wang HC, Yu JC, Chen ST, Bermisheva M, Nikolaeva T, Khusnutdinova E, Humphreys MK, Morrison J, Platte R, Easton DF. Risk of estrogen receptor-positive and -negative breast cancer and single-nucleotide polymorphism 2q35-rs13387042. J. Natl. Cancer Inst. 2009;101:1012–8. doi: 10.1093/jnci/djp167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyle P, Boffetta P, Autier P. Diet, nutrition and cancer: public, media and scientific confusion. Ann. Oncol. 2008;19:1665–7. doi: 10.1093/annonc/mdn561. [DOI] [PubMed] [Google Scholar]
- 55.Travis RC, Reeves GK, Green J, Bull D, Tipper SJ, Baker K, Beral V, Peto R, Bell J, Zelenika D, Lathrop M. Gene-environment interactions in 7610 women with breast cancer: prospective evidence from the Million Women Study. Lancet. 2010;375:2143–51. doi: 10.1016/S0140-6736(10)60636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Mol. Genet. Epidemiol. 2008;32:179–85. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 57.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat. Genet. 2001;29:306–9. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 58.Khoury MJ, Bertram L, Boffetta P, Butterworth AS, Chanock SJ, Dolan SM, Fortier I, Garcia-Closas M, Gwinn M, Higgins JP, Janssens AC, Ostell J, Owen RP, Pagon RA, Rebbeck TR, Rothman N, Bernstein JL, Burton PR, Campbell H, Chockalingam A, Furberg H, Little J, O’Brien TR, Seminara D, Vineis P, Winn DM, Yu W, Ioannidis JP. Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. American Journal of Epidemiology. 2009;170:269–79. doi: 10.1093/aje/kwp119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ioannidis JP. Calibration of credibility of agnostic genome-wide associations. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2008;147B:964–72. doi: 10.1002/ajmg.b.30721. [DOI] [PubMed] [Google Scholar]
- 60.Ioannidis JP, Loy EY, Poulton R, Chia KS. Researching genetic versus nongenetic determinants of disease: a comparison and proposed unification. Sci. Transl.Med. 2009;1:7ps8. doi: 10.1126/scitranslmed.3000247. [DOI] [PubMed] [Google Scholar]
- 61.Linos E, Willett WC. Diet and breast cancer risk reduction. J. Natl. Compr. Canc. Netw. 2007;5:711–718. doi: 10.6004/jnccn.2007.0072. [DOI] [PubMed] [Google Scholar]
- 62.Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr. Relat. Cancer. 2007;14:907–33. doi: 10.1677/ERC-07-0137. [DOI] [PubMed] [Google Scholar]
- 63.Schatzkin A, Longnecker MP. Alcohol and breast cancer. Where are we now and where do we go from here? Cancer. 1994;74:1101–10. doi: 10.1002/1097-0142(19940801)74:3+<1101::aid-cncr2820741519>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 64.Ambrosone CB. The promise and limitations of genome-wide association studies to elucidate the causes of breast cancer. Breast Cancer Res. 2007;9:114. doi: 10.1186/bcr1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ostrander EA, Stanford JL. Genetics of prostate cancer: too many loci, too few genes. Am. J. Hum. Genet. 2000;67:1367–75. doi: 10.1086/316916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–2. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 67.Oldenburg RA, Kroeze-Jansema K, Kraan J, Morreau H, Klijn JG, Hoogerbrugge N, Ligtenberg MJ, van Asperen CJ, Vasen HF, Meijers C, Meijers-Heijboer H, de Bock TH, Cornelisse CJ, Devilee P. The CHEK2*1100delC variant acts as a breast cancer risk modifier in non-BRCA1/BRCA2 multiple-case families. Cancer Res. 2003;63:8153–7. [PubMed] [Google Scholar]
- 68.Wang-Gohrke S, Rebbeck TR, Besenfelder W, Kreienberg R, Runnebaum IB. p53 germline polymorphisms are associated with an increased risk for breast cancer in German women. Anticancer Res. 1998;18:2095–9. [PubMed] [Google Scholar]
- 69.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 70.van Wezel T, Stassen AP, Moen CJ, Hart AA, van der Valk MA, Demant P. Gene interaction and single gene effects in colon tumour susceptibility in mice. Nat. Genet. 1996;14:468–70. doi: 10.1038/ng1296-468. [DOI] [PubMed] [Google Scholar]
- 71.Fijneman RJ, Jansen RC, van der Valk MA, Demant P. High frequency of interactions between lung cancer susceptibility genes in the mouse: mapping of Sluc5 to Sluc14. Cancer Res. 1998;58:4794–8. [PubMed] [Google Scholar]
- 72.Mackay TF. Quantitative trait loci in Drosophila. Nature Reviews Genetics. 2001;2:11–20. doi: 10.1038/35047544. [DOI] [PubMed] [Google Scholar]
- 73.Nagase H, Mao JH, de Koning JP, Minami T, Balmain A. Epistatic interactions between skin tumor modifier loci in interspecific (spretus/musculus) backcross mice. Cancer Res. 2001;61:1305–8. [PubMed] [Google Scholar]
- 74.Quigley DA, To MD, Perez-Losada J, Pelorosso FG, Mao JH, Nagase H, Ginzinger DG, Balmain A. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature. 2009;458:505–8. doi: 10.1038/nature07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quigley D, Balmain A. Systems genetics analysis of cancer susceptibility: from mouse models to humans. Nat. Rev. Genet. 2009;10:651–7. doi: 10.1038/nrg2617. [DOI] [PubMed] [Google Scholar]
- 76.Balmain A, Nagase H. Cancer resistance genes in mice: models for the study of tumour modifiers. Trends Genet. 1998;14:139–44. doi: 10.1016/s0168-9525(98)01422-x. [DOI] [PubMed] [Google Scholar]
- 77.Nadeau JH, Frankel WN. The roads from phenotypic variation to gene discovery: mutagenesis versus QTLs. Nat. Genet. 2000;25:381–4. doi: 10.1038/78051. [DOI] [PubMed] [Google Scholar]
- 78.Matin A, Collin GB, Asada Y, Varnum D, Nadeau JH. Susceptibility to testicular germ-cell tumours in a 129.MOLF-Chr 19 chromosome substitution strain. Nat. Genet. 1999;23:237–40. doi: 10.1038/13874. [DOI] [PubMed] [Google Scholar]
- 79.Collins FS, Finnell RH, Rossant J, Wurst W. A new partner for the international knockout mouse consortium. Cell. 2007;129:235. doi: 10.1016/j.cell.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 80.Darvasi A. Dissecting complex traits: the geneticists’ “Around the world in 80 days”. Trends Genet. 2005;21:373–6. doi: 10.1016/j.tig.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 81.To MD, Perez-Losada J, Mao JH, Hsu J, Jacks T, Balmain A. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nat. Genet. 2006;38:926–30. doi: 10.1038/ng1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Balmain A, Harris CC. Carcinogenesis in mouse and human cells: parallels and paradoxes. Carcinogenesis. 2000;21:371–7. doi: 10.1093/carcin/21.3.371. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Paigen B. Genetics of variation in HDL cholesterol in humans and mice. Circ. Res. 2005;96:27–42. doi: 10.1161/01.RES.0000151332.39871.13. [DOI] [PubMed] [Google Scholar]
- 84.Crawford NP, Qian X, Ziogas A, Papageorge AG, Boersma BJ, Walker RC, Lukes L, Rowe WL, Zhang J, Ambs S, Lowy DR, Anton-Culver H, Hunter KW. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet. 2007;3:e214. doi: 10.1371/journal.pgen.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crawford NP, Ziogas A, Peel DJ, Hess J, Anton-Culver H, Hunter KW. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 2006;8:R16. doi: 10.1186/bcr1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ewart-Toland A, Briassouli P, de Koning JP, Mao JH, Yuan J, Chan F, MacCarthy-Morrogh L, Ponder BA, Nagase H, Burn J, Ball S, Almeida M, Linardopoulos S, Balmain A. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat. Genet. 2003;34:403–12. doi: 10.1038/ng1220. [DOI] [PubMed] [Google Scholar]
- 87.Ewart-Toland A, Dai Q, Gao YT, Nagase H, Dunlop MG, Farrington SM, Barnetson RA, Anton-Culver H, Peel D, Ziogas A, Lin D, Miao X, Sun T, Ostrander EA, Stanford JL, Langlois M, Chan JM, Yuan J, Harris CC, Bowman ED, Clayman GL, Lippman SM, Lee JJ, Zheng W, Balmain A. Aurora-A/STK15 T+91A is a general low penetrance cancer susceptibility gene: a meta-analysis of multiple cancer types. Carcinogenesis. 2005;26:1368–73. doi: 10.1093/carcin/bgi085. [DOI] [PubMed] [Google Scholar]
- 88.Park YG, Clifford R, Buetow KH, Hunter KW. Multiple cross and inbred strain haplotype mapping of complex-trait candidate genes. Genome Res. 2003;13:118–21. doi: 10.1101/gr.786403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, Korstanje R, Higgins D, Paigen B. Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res. 2004;14:1767–72. doi: 10.1101/gr.2668204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hitzemann R, Malmanger B, Cooper S, Coulombe S, Reed C, Demarest K, Koyner J, Cipp L, Flint J, Talbot C, Rademacher B, Buck K, McCaughran J., Jr. Multiple cross mapping (MCM) markedly improves the localization of a QTL for ethanol-induced activation. Genes Brain Behav. 2002;1:214–22. doi: 10.1034/j.1601-183x.2002.10403.x. [DOI] [PubMed] [Google Scholar]
- 91.Hitzemann R, Malmanger B, Reed C, Lawler M, Hitzemann B, Coulombe S, Buck K, Rademacher B, Walter N, Polyakov Y, Sikela J, Gensler B, Burgers S, Williams RW, Manly K, Flint J, Talbot C. A strategy for the integration of QTL, gene expression, and sequence analyses. Mamm. Genome. 2003;14:733–47. doi: 10.1007/s00335-003-2277-9. [DOI] [PubMed] [Google Scholar]
- 92.Yalcin B, Willis-Owen SA, Fullerton J, Meesaq A, Deacon RM, Rawlins JN, Copley RR, Morris AP, Flint J, Mott R. Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat. Genet. 2004;36:1197–202. doi: 10.1038/ng1450. [DOI] [PubMed] [Google Scholar]
- 93.Mott R, Flint J. Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics. 2002;160:1609–18. doi: 10.1093/genetics/160.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chia R, Achilli F, Festing MF, Fisher EM. The origins and uses of mouse outbred stocks. Nat. Genet. 2005;37:1181–6. doi: 10.1038/ng1665. [DOI] [PubMed] [Google Scholar]
- 95.Manenti G, Galbiati F, Gianni-Barrera R, Pettinicchio A, Acevedo A, Dragani TA. Haplotype sharing suggests that a genomic segment containing six genes accounts for the pulmonary adenoma susceptibility 1 (Pas1) locus activity in mice. Oncogene. 2004;23:4495–504. doi: 10.1038/sj.onc.1207584. [DOI] [PubMed] [Google Scholar]
- 96.Nagase H, Bryson S, Cordell H, Kemp CJ, Fee F, Balmain A. Distinct genetic loci control development of benign and malignant skin tumours in mice. Nat. Genet. 1995;10:424–9. doi: 10.1038/ng0895-424. [DOI] [PubMed] [Google Scholar]
- 97.Nagase H, Mao JH, Balmain A. A subset of skin tumor modifier loci determines survival time of tumor-bearing mice. Proc. Natl. Acad. Sci. U. S. A. 1999;96:15032–7. doi: 10.1073/pnas.96.26.15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Manenti G, Galbiati F, Noci S, Dragani TA. Outbred CD-1 mice carry the susceptibility allele at the pulmonary adenoma susceptibility 1 (Pas1) locus. Carcinogenesis. 2003;24:1143–8. doi: 10.1093/carcin/bgg065. [DOI] [PubMed] [Google Scholar]
- 99.Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 2005;6:271–86. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- 100.Flint J. Mapping quantitative traits and strategies to find quantitative trait genes. Methods. 2010 doi: 10.1016/j.ymeth.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 102.Iraqi FA, Churchill G, Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm. Genome. 2008;19:379–81. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 103.Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat. Genet. 2003;33(Suppl):238–44. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- 104.Arbilly M, Pisante A, Devor M, Darvasi A. An integrative approach for the identification of quantitative trait loci. Animal Genetics. 2006;37(Suppl 1):7–9. doi: 10.1111/j.1365-2052.2006.01472.x. [DOI] [PubMed] [Google Scholar]
- 105.Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. 2001;17:388–91. doi: 10.1016/s0168-9525(01)02310-1. [DOI] [PubMed] [Google Scholar]
- 106.DiPetrillo K, Wang X, Stylianou IM, Paigen B. Bioinformatics toolbox for narrowing rodent quantitative trait loci. Trends Genet. 2005;21:683–92. doi: 10.1016/j.tig.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 107.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 108.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 109.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 110.The Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 111.Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–19. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 112.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–3. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 113.Klein RJ, Xu X, Mukherjee S, Willis J, Hayes J. Successes of genome-wide association studies. Cell. 2010;142:350–1. doi: 10.1016/j.cell.2010.07.026. author reply 353–5. [DOI] [PubMed] [Google Scholar]
- 114.Wang K, Bucan M, Grant SF, Schellenberg G, Hakonarson H. Strategies for genetic studies of complex diseases. Cell. 2010;142:351–3. doi: 10.1016/j.cell.2010.07.025. author reply 353-5. [DOI] [PubMed] [Google Scholar]
- 115.Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O’Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellie C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 2007;39:989–94. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- 116.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet. 2007;39:984–8. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 117.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009;10:241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]