

Abstract
Here we provide the first genome-wide, high-resolution map of the phylogenetic origin of the genome of most extant laboratory mouse inbred strains. Our analysis is based on the genotypes of wild caught mice from three subspecies of Mus musculus. We demonstrate that classical laboratory strains are derived from a few fancy mice with limited haplotype diversity. Their genomes are overwhelmingly M. m. domesticus in origin and the remainder is mostly of Japanese origin. We generated genome-wide haplotype maps based on identity by descent from fancy mice and demonstrate that classical inbred strains have limited and non-randomly distributed genetic diversity. In contrast, wild-derived laboratory strains represent a broad sampling of diversity within M. musculus. Intersubspecific introgression is pervasive in these strains and contamination by laboratory stocks has played role in this process. The subspecific origin, haplotype diversity and identity by descent maps can be visualized and searched online.
Introduction
Most mouse laboratory strains are derived from Mus musculus, a species with multiple lineages including three major subspecies, M. m. domesticus, M. m. musculus and M. m. castaneus, with distinct geographical ranges1. In historical times mice followed human migratory patterns and colonized new regions. In regions of secondary contact between subspecies there is evidence of gene flow1-3. Hybridization between M. m. musculus and M. m. castaneus in Japan resulted in the M. m. molossinus subspecies4.
Laboratory strains can be classified into two groups based on their origin. Classical inbred strains were derived during the 20th century from “fancy” mice. These strains have been the preferred tools in biomedical research. Historical sources and genetic studies suggest that fancy mice had significant inbreeding5. These sources indicate that three subspecies of Mus musculus were represented in the genome of fancy mice making classical strains artificial hybrids between multiple subspecies found in the wild. However, there is wide disagreement about the relative contribution of each subspecies to classical inbred strains6,7. Classical strains have substantial population structure because of the limited genetic diversity present in fancy mice and the complex schema used in their derivation.
Wild-derived laboratory strains are derived directly from wild caught mice8. Each strain has been assigned to a subspecies or represents a natural hybrid between subspecies. The population structure of wild-derived strains can be accounted for by their taxonomical classification.
The genome sequence and annotation of the C57BL/6J classical inbred strain was reported in 2002 (9), followed by an extensive SNP discovery effort in 15 laboratory strains6 and the ongoing whole genome sequencing of 17 inbred strains10. These data will inform hundreds of projects that use the mouse as a model for biomedical research including the International Knockout projects and the Collaborative Cross11,12.
Despite this wealth of sequence data, our understanding of genetic diversity in mice is shallow and biased. SNP discovery has involved only a limited number of strains resulting in SNP panels with substantial ascertainment bias13. Pedigree records continue to serve as the primary source of information about the origin and relationships among laboratory strains5. Although such records are valuable, genetic studies and the experience of mouse breeders indicate that contamination is common7. We have previously reported the presence of intersubspecific introgression in three commonly used wild-derived strains7. However, this conclusion has been controversial and the lack of data from wild caught mice has prevented consensus. Finally, the M. musculus subspecies are undergoing the early stages of speciation. There is shared variation among subspecies mostly due to polymorphisms that have persisted from a common ancestor and introgression between subspecies in the wild. Thus selection of a single reference genome for each subspecies cannot accurately reflect the population structure of these recently diverged taxa. Furthermore, the choice of a single inbred strain to represent an entire taxon is particularly problematic because laboratory strains were subject to many generations of selective mating in an artificial setting where there is high potential for contamination7.
Given the contradictory conclusions reached regarding the origin of the genome of classical and wild-derived laboratory mouse strains6,7,14-16 it is crucial to select representative reference samples along with a platform that can address the limitations of previous studies. We have collected a geographically diverse sample of mice from natural populations of the three major M. musculus subspecies to use as references and a large and diverse set of laboratory strains that can be effectively used to infer the genome of most remaining strains through imputation13. Our platform is a custom high-density genotyping array for the mouse17.
Results
Sample and genotypes
We selected 198 samples for genotyping including 36 wild caught mice, 62 wild-derived laboratory strains and 100 classical strains (Supplementary Table 1). Wild caught mice, including representatives from M. m. domesticus, M. m. musculus and M. m. castaneus, were used as references to infer the phylogenetic origin of laboratory strains (Supplementary Figure 1). Our laboratory samples include strains derived from different stocks and by different laboratories5 as well as 13 sets of classical substrains that are thought to be closely related to each other.
Every sample was genotyped with the Mouse Diversity array17. We performed additional steps to improve the quality of the genotype calls and to detect residual heterozygosity and deletions larger than 100kb. Our genotype dataset include SNPs and VINOs (Variable INtensity Oligonucleotides). The latter represent previously unknown genetic variants that substantially alter the performance of SNP detection probes (see Methods). We used 549,599 SNPs and 117,203 VINOs with six possible calls: homozygous for either allele, heterozygous, VINO, deletion and no call. In analyses based on SNPs we treated VINOs as no calls. In analyses based on VINOs we treated data as binary for presence and absence of VINOs. SNPs and VINOs have complementary characteristics that can be used to strengthen phylogenetic analyses (see Discussion).
Heterozygosity and deletions in laboratory strains
The local frequency of heterozygous calls was used to identify regions with two distinct haplotypes in a sample. Such regions were deemed heterozygous. Wild caught mice are predominantly heterozygous and the variation in the heterozygosity rate (Supplementary Table 1) among subspecies is as expected from sequencing studies2. Wild-derived strains have wide variation in heterozygosity and most classical strains are fully inbred. There are, however, some blocks of residual heterozygosity of variable size and distribution among lab strains (Supplementary Table 2). We detected the presence of deletions in 102 samples and determined their boundaries by visual inspection of probe intensity plots (Supplementary Table 3). These large deletions were excluded from our phylogenetic analysis. The analysis of structural variation in laboratory strains will be reported elsewhere.
Diagnostic alleles
We used the genotypes of the 36 wild caught mice to determine the ability of each SNP or VINO to discriminate between subspecies allowing for some misclassification due to genotyping error, homoplasy or gene flow in the wild. Alleles found in only one subspecies were considered diagnostic. These include fully informative alleles, in which subspecies are fixed for different alleles and partially informative alleles, in which an allele is restricted to one subspecies but not fixed. We identified 251,676 SNPs and 96,188 VINOs with diagnostic alleles distributed across every chromosome (Supplementary Figure 2). SNPs and VINOs with nondiagnostic alleles are also distributed evenly across the genome but were not used to infer ancestry.
We found significant differences between the number of SNPs and VINOs with diagnostic alleles for each the three subspecies detected. For example, 55% of all informative SNPs carry diagnostic alleles for M. m. domesticus, while only 27% and 18% carry diagnostic alleles for M. m. musculus and M. m. castaneus, respectively. This situation is reversed among VINOs where 17%, 24% and 59% of diagnostic alleles identify M. m. domesticus, M. m. musculus and M. m. castaneus, respectively. These differences reflect two types of biases with compensatory effects. On one hand, the selection criteria for inclusion of SNPs in the array led to the overrepresentation of SNPs with M. m. domesticus diagnostic alleles and underrepresentation of M. m. castaneus SNPs17. On the other hand, our deeper knowledge of the genetic variation present in the M. m. domesticus subspecies allowed screening of candidate SNP probes with internal polymorphisms that could create VINOs. Whereas our limited knowledge of the genetic variation present in the M. m. castaneus subspecies in particular results in an excess of M. m. castaneus diagnostic VINOs2,7.
We confirmed the taxonomic classification of the 36 wild caught samples by generating phylogenetic trees for the autosomes, sex chromosomes and mitochondria. All trees are consistent with the expected subspecific origin (Supplementary Figure 3).
Subspecific origin of classical strains
We used informative SNPs and VINOs to impute the subspecific origin of every region of the genome of each sample. Figure 1 shows the overall contribution of each subspecies to the autosomes while Figure 2a provides a map of the subspecific origin for chromosomes 6 and X (complete data is available at http://msub.csbio.unc.edu/PhylogenyTool.html). The genome of classical inbred strains is predominantly derived from M. m. domesticus (94.3 ± 2.0%) with variable contribution of M. m. musculus (5.4 ± 1.9%) and with a small contribution from M. m. castaneus (0.3 ± 0.1%). The contribution from subspecies other than M. m. domesticus is not distributed randomly across the genome or among strains (Figure 2). In the combined 100 classical inbred strains M. m. musculus haplotypes can be found in only 46.9% of the genome and M. m. castaneus in 2.8%. Importantly, there is a strong bias towards multiple strains sharing the same M. m. musculus haplotype in some regions.
Figure 1.

Overall contribution of each subspecies to the genome of wild and laboratory mice. For each sample the figure depicts the cumulative contribution of M. m. domesticus (D, blue), M. m. musculus (M, red) and M. m. castaneus (C, green) subspecies for the autosomes. H, hybrid strains.
Figure 2.

Subspecific origin and haplotype diversity of chromosomes 6 (left) and X (right). A) Subspecific origin. Colors follow the same conventions as in Figure 1. B-E) Phylogenetic trees for classical and wild-derived strains for two compatible intervals, one spanning positions 143,009,892-143,140,072 on chromosome 6 (C and D) and the other spanning positions 37,770,186-42,329,981 on chromosome X (E and F).
Strikingly, the M. m. castaneus and M. m. musculus contributions are not independent from each other, with the former frequently nested within or contiguous with the latter (Figure 2). This association suggests a M. m. molossinus origin of the M. m. musculus contribution to the classical inbred strains18,19. We tested this hypothesis by comparing the M. m. musculus regions found in classical inbred strains to wild caught M. m. musculus mice from Europe or Asia (Supplementary Figure 3). Over 90% of the M. m. musculus haplotypes found in classical inbred strains cluster with Asian wild caught mice.
Haplotype diversity and identity by descent in classical strains
The subspecific origin of classical inbred strains support the hypothesis that these strains are derived from a small population of fancy mice that was itself subject to significant inbreeding. To estimate the size of the fancy mice population from which classical inbred strains are derived, we divided their genome in overlapping intervals that have no evidence for historical recombination (see Methods). We identified 43,285 intervals (median size = 71kb, median number SNPs = 12). The distribution of the number of haplotypes in each interval (median and mode = 5) indicates that the original population harbored a limited number of distinct chromosomes (Supplementary Figure 4a). Over 97% of the genome can be explained by fewer than ten haplotypes. In conclusion, classical strains can be partitioned locally into a small number of classes within which all strains are identical by descent (IBD) with respect to their common origin. Intervals with larger numbers of haplotypes often reflect accumulation of new mutations in the past century as demonstrated by resequencing projects6,7,10 and our analysis of substrains (Supplementary Figure 5).
Recombination intervals provide a natural scaffold upon which to build genome-wide maps of haplotype diversity and IBD among classical strains. For each interval we estimated the genotype identity among all pairs of strains and defined the minimum number and composition of cliques required to represent the haplotype variation. A critical step in this process was to determine a threshold of genotype identity that corresponds to IBD. This lower bound on genotype identity should be consistent with the accumulation of new mutations over several hundreds of generations and genotyping error. For this purpose we carried out an analysis of local similarity among sister substrains. These closely related sets of strains, such as BALB/cJ and BALBcByJ, do not show evidence of substantial genetic divergence or contamination (Supplementary Figure 5). We established that 99.0% genotype identity is a suitable threshold for provisional assignment of local IBD status among strains. To further refine this assignment and to address the shortcoming of hard thresholding, we used clique completion to define sets of strains that are mutually IBD to each other and we calculated the mean genotype identity within and between cliques. The distribution of number of cliques is similar to the distribution of number of haplotypes per interval (Supplementary Figure 4). Using this approach we generated a map of haplotype diversity in 100 classical inbred strains (http://msub.csbio.unc.edu/PhylogenyTool.html).
Haplotypes can differ from each other just slightly more than our threshold to declare IBD (99%) or by as much as is typically observed between different subspecies (50%, see Supplementary Figure 6). To estimate the local level of haplotype variation and to guide interpretation of the maps, we determined the quantitative similarity between haplotypes at each interval based on phylogenetic distance trees. Figure 2 (c-e) shows two recombination intervals with obvious differences in the number haplotypes and level of similarity among them. This illustrates the complex relationship between haplotype number and haplotype diversity among classical inbred strains.
Intersubspecific introgression in wild-derived laboratory strains
The recombination intervals computed for classical inbred strains cannot be easily extended to the wild-derived strains. Instead, we computed the frequency of diagnostic alleles in non-overlapping 1Mb intervals and for each wild-derived strain. The majority of the genome of the 62 wild-derived laboratory strains originates from the expected subspecies or combination of subspecies (Figure 1). However, only nine strains have a genome derived entirely from a single subspecies, 18 have contributions from two subspecies and 35 have contribution from all three subspecies. The prevalence and extent of multi-subspecific origin is a defining characteristic of wild-derived laboratory strains as a group. Our set of wild-derived strains includes 10 strains derived from natural intersubspecific hybrids (Supplementary Table 1) all of which have, unexpectedly, contributions from all three subspecies. The remarkable discordance in subspecific origin in several strains based on phylogenetic trees (Supplementary Table 1 and Supplementary Figure 7) provides further evidence for intersubspecific introgression. The sharing of patterns of subspecific origin between classical inbred strains and some wild-derived strains (Figure 2) suggests that some of the intersubspecific introgressions in the later group involved cross breeding to classical strains.
Relationship between classical and wild-derived laboratory strains
To characterize the relationship between the classical and wild-derived laboratory strains we determined the maximum local level of genotype identity between each wild-derived strain and all classical inbred strains in non-overlapping 1Mb windows and generated genome-wide similarity distributions (Supplementary Figure 6a). The distributions of local similarity reveal the presence of distinct patterns for wild-derived strains of each of the three major subspecies. M. m. domesticus and M. m. castaneus wild-derived strains have typically unimodal distributions with distinct means (Figure 3). In contrast, M. m. musculus and M. m. molossinus strains have a bimodal distribution of local genotype identity when compared to classical inbred strains.
Figure 3.
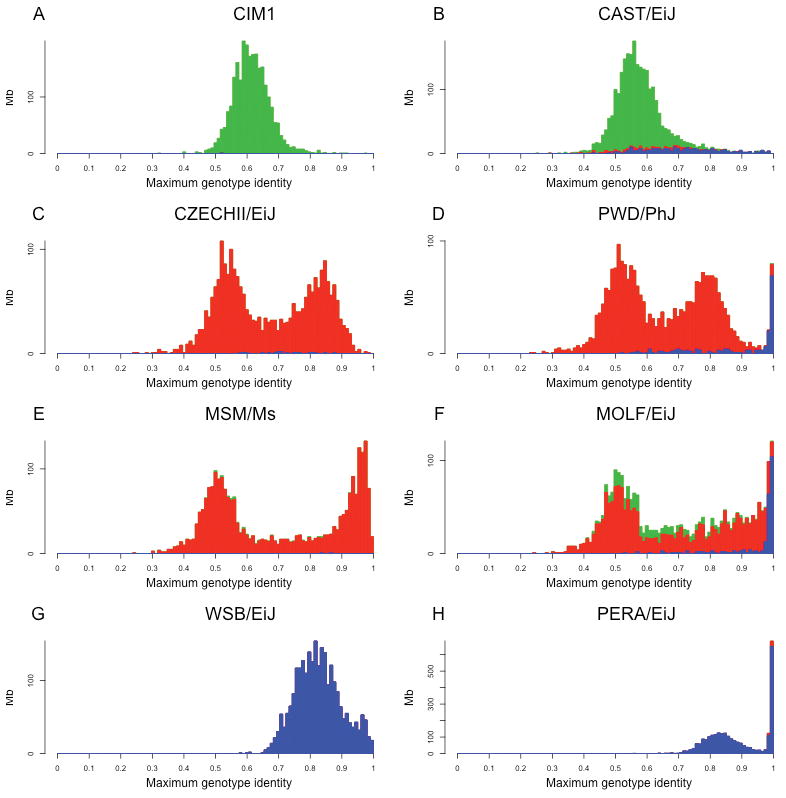
Intersubspecific introgression and contamination by classical strains in the wild-derived inbred strains. For each 1Mb interval we identified the classical inbred strain with maximum genotype similarity to a given wild derived strains. Panels A-H show the frequency distribution of similarity for eight strains. Colors follow the same conventions as in previous figures.
This analysis provides insight into the origins of intersubspecific introgressions that occur in many of the wild-derived strains. Regions of near identity (> 98%) with classical inbred strains indicate cross-breeding to extant classical strains or stocks descended from fancy mice. For example, 15 wild-derived strains (Supplementary Table 1) show a distinct peak at levels of genotype identity (>98%) that are only consistent with recent IBD. The fraction of the genome involved ranges from 3.9 to 64.6%. Three wild-derived strains from three different subspecies, PWD/PhJ, MOLF/EiJ and PERA/EiJ, exemplify this pattern. In all three cases regions of IBD to classical inbred strains are predominantly of M. m. domesticus origin, but also include regions of M. m. musculus introgression (Figure 3). This is particularly striking in the PERA/EiJ strain providing further evidence of the role classical laboratory strains in intersubspecific introgression in wild-derived laboratory stocks.
For each of the 15 wild-derived strains we tested whether a single donor classical strain can explain the overall pattern of IBD with all classical strains. Using this approach we identified the donor of introgressed regions in six wild-derived strains (Supplementary Table 1) including PERA/EiJ. Contamination by CBA/CaJ explains all IBD regions in PERA/EiJ whereas comparison with any of the other 99 classical inbred strains explains only a fraction of intervals of high local similarity (Figure 4). Another six wild-derived strains appear to have been contaminated by classical laboratory mice that are not among our set of classical strains. The remaining 21 wild-derived strains that show evidence of intersubspecific introgression are not contaminated by classical laboratory strains.
Figure 4.
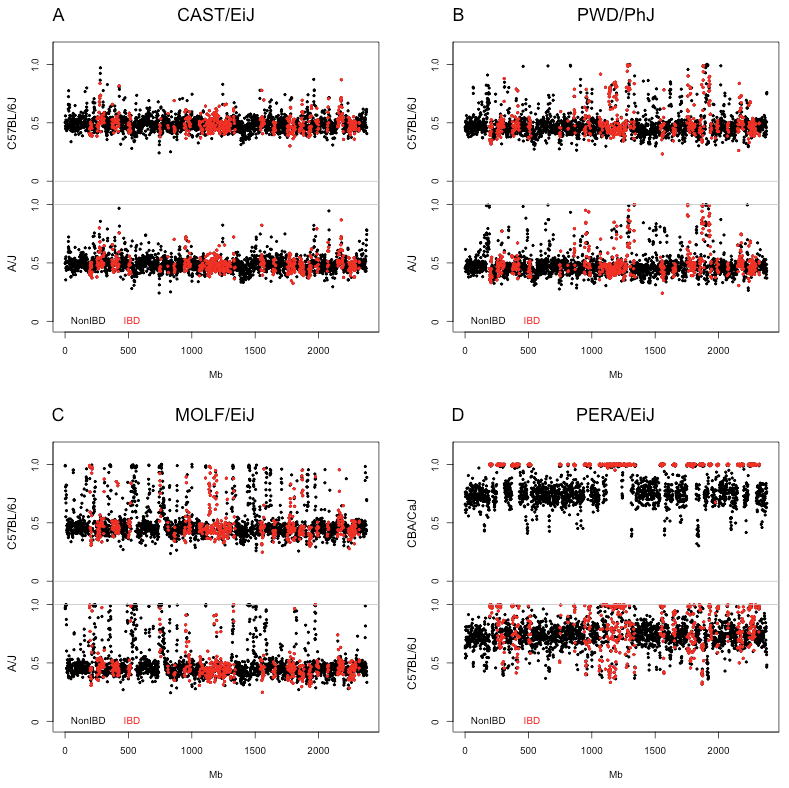
Identification of donor strain. Panels A-D provide examples of the approach used in the identification of the donor classical strain that contaminated a wild-derived strain. Red circles represent 1Mb intervals in which a wild-derived strain is IBD to an haplotype present in classical inbred strains and black circles represent 1Mb intervals that are not IBD.
The distribution of local similarity between wild-derived and classical inbred strains provides further insights into the origins of the non- M. m. domesticus regions in the genomes of classical inbred strains. When wild-derived M. m. musculus strains are compared to classical inbred strains (Figure 3e,f, Supplementary Figure 6), the peak with lower genotype similarity corresponds to genomic regions in which classical inbred strains completely lack M. m. musculus haplotypes. The peak with higher genotype similarity corresponds to regions in which at least one classical inbred strain carries a M. m. musculus haplotype and has an average SNP identity of 83%. When we make the same comparisons with M. m. molossinus wild-derived inbred strains, the high peak is shifted towards near complete identity (∼98%). We conclude that the vast majority of M. m. musculus regions in classical strains are of M. m. molossinus origin.
Discussion
There are two competing views on the origin and composition of the genome of classical inbred strains6,7. The first view claims that the genome of these strains is 68% M. m. domesticus, 10% M. m. molossinus, 6% musculus, 3% M. m. castaneus and 13% of unknown origin6. On the other hand, we concluded that 92% is of M. m. domesticus, 6% of M. m. musculus and 1% of M. m. castaneus origin7. Both studies were based on NIEHS data6 but took different approaches to the use of wild-derived inbred strains as reference genomes to infer subspecific origin. Frazer and coworkers assumed that the four wild-derived strains, WSB/EiJ, PWD/PhJ, CAST/EiJ and MOLF/EiJ, were faithful representative of four subspecies, M. m. domesticus, M. m. musculus, M. m. castaneus and M. m. molossinus, respectively. We concluded that three of these wild-derived strains, PWD/PhJ, CAST/EiJ and MOLF/EiJ, had introgressed haplotypes from other subspecies. Obviously, in regions where a given wild-derived strain has undergone such intersubspecific introgression the genotypes are not suitable as a reference for that subspecies. The results presented here conclusively demonstrate that classical inbred strains are overwhelmingly derived from M. m. domesticus, that the non M. m. domesticus contribution to their genomes is largely of M. m. molosinus origin, and that intersubspecific introgression is common in wild-derived laboratory strains.
The wild caught mice used here represent a wide geographically diverse sample. The genomes of these mice are overwhelmingly derived from a single subspecies (mean: 99.84%; range: 100 – 98.42%). Half of wild caught mice carry small regions with haplotypes from a second subspecies, mostly in heterozygous combinations. We acknowledge that a larger and more geographically diverse set of mice would be of great interest but it would have little impact on our conclusions regarding the origin of the genome of the laboratory mouse. We also acknowledge that our definition of diagnostic alleles in SNPs and VINOs may change with the inclusion of more samples. However, this definition provides a simple and robust method to assign phylogenetic origin while preserving enough flexibility to account for genotyping error, homoplasy and gene flow among subspecies in the wild. Although our method works very well at Mb genomic scale it has limitations in providing subspecific assignments at finer scale (Supplementary Figure 8).
Excluding hybrid strains, 28 wild-derived strains have intersubspecific introgressions covering between 1% and 27% of their genome (Figure 1; Supplementary Table 1). In CAST/EiJ and PWD/PhJ, the two strains that were used as references in previous studies, introgression covers 12% and 7% of their genome, respectively confirming 96% of regions that were declared introgressed in our previous study (Supplementary Figure 9). We have been able to identify additional regions of introgression in CAST/EiJ and PWD/PhJ due to the better reference genotypes for each subspecies and the combined use of SNPs and VINOs. Subspecies, time since derivation, and laboratory history appear to have a strong effect on the prevalence and extent of intersubspecific introgression, which could have occurred in the wild or in the laboratory. The limited extent of introgression in wild caught samples suggests that breeding in the laboratory played a major role in shaping the genomes of wild-derived strains. Independent confirmation was obtained by comparing the genome of wild-derived and classical inbred strains. Fifteen wild-derived strains have inherited haplotypes from classical inbred strains. Contamination by classical strains was expected, and likely intentional, in some cases (i.e., SOD1/EiJ and RBB/DnJ) but not in others (i.e., CASA/EiJ and CALB/RkJ). Introgression in the remaining wild-derived strains probably arose though a combination of gene flow in the wild (in samples captured close to hybrid zones and recently colonized regions) and breeding in the laboratory to non-classical mouse stocks (most likely other wild-derived mice). Wild-derived inbred strains have been used frequently as models in evolutionary studies 20. Our results suggest that new information about the subspecific origin of the strains should be incorporated in the analyses.
A complementary strength of our study was the ability to account and correct for ascertainment biases in the SNPs included in the array. Most of these SNPs were selected on the basis of the local phylogeny among the NIEHS strains. This approach ensured that all major local branches were represented while ignoring minor branches. However, the approach also had limitations because locally all branches represented in the array were allocated the same number of SNPs and, therefore, long and short local branches would appear to be equal in length17. Furthermore, there are subspecies-specific false negative rates in SNP identification in the NIEHS study and prior identification of a SNP is a necessary condition for its presence in the array7. Subspecies-specific false negative rates in SNP discovery should also impact negatively the rate at which selected SNPs are converted into successful genotyping assays17. For example, M. m. castaneus SNPs should be underrepresented compared to the true level of diversity due the combined effects of our selection criteria and the higher assay failure rate. However, we were able to overcome the high failure rate by using VINOs. For the purpose of this study, VINOs have the critical advantage of being less subject to ascertainment biases within a given phylogenetic group. However, VINOs can only be reliably detected in homozygosity resulting in a significant undercounting of VINOs in some samples (Supplementary Table 1). We conclude that the combination of SNP and VINO genotype data in wild caught mice has enormous value for population studies.
Among the most useful results of the present study are the maps of subspecific origin and haplotype diversity of the genome of classical inbred strains (Figure 2). These maps should allow researchers to combine information from multiple crosses to refine candidate intervals. It should also extend the advantages of the very high-density genotype data in the 15 NIEHS strains (and eventually whole genome sequence) to many additional classical strains5,10. Our maps will enable researchers to determine not only which strains share the same haplotype on a given region but the sequence divergence among those strains that do not share them. We have also calculated the number of variants used to infer IBD and a score to guide interpretation of these trees by potential users. In particular we have flagged haplotypes with weak support. Our data and tools should allow researchers to rapidly determine the number of haplotypes in a given region and the level of sequence divergence among them. Both are important considerations for association mapping. These data will also allow researchers to identify discrete regions of genetic divergence between substrains. Finally, they may be used to select strains with the desired level and type of genetic variation in any given region of the genome.
The spatial distribution of mean genetic variation observed in the 100 classical strains analyzed here is very similar to the one reported previously for a set of only 12 classical strains7 (Supplementary Figure 10).
Although our approach of recombination intervals cannot directly be extended to wild-derived strains we have used a fixed window approach to determine the level of haplotype diversity and IBD among these strains. This analysis demonstrates that, as expected, there is much more diversity in wild-derived strains than in classical strains (Figure 2b-e) and, therefore, opportunities to optimize genetic research. Analysis of the frequency distribution of genotype identity in pairwise comparisons between wild-derived strains provides insight into the natural history of these strains and the populations from which they were derived. In contrast with comparison to classical inbred strains these distributions are typically unimodal in intrasubspecific comparisons (Supplementary Figure 6b). However, we observe also a strong signature of IBD in several pairwise comparisons. Some of the strongest cases involve pairs of strains derived from mice trapped in geographically close localities (Supplementary Table 1). Excess IBD can be explained by the presence of introgression from classical inbred strains that are themselves IBD for significant fraction of their genome (Supplementary Figure 6). There are some strains that are connected to several cliques creating a complex network. Finally, all M. m. molossinus wild-derived strains (Supplementary Table 1) have very high levels of IBD (∼34%). This observation and the unusually high level of genotype identity between the M. m. molossinus haplotypes present in classical strains and wild-derived M. m. molossinus strains strongly suggest a recent population bottleneck in this hybrid subspecies.
In summary, our observation of residual heterozygosity among inbred mouse strains, the striking local differences in the level of genetic similarity between substrains, the identification of large deletions of different ages and prevalence of contamination emphasizes the importance of deep, unbiased and frequent genetic characterization of laboratory stocks. Our genome browser provides access to the trees and links between recombination intervals, local trees, and the maps for subspecific origin and haplotype diversity. Our analysis demonstrates that classical inbred strains are in fact mosaics of a handful of haplotypes present in the founder fancy mice population. The genetic divergence among these haplotypes varies widely both locally and across the genome. Furthermore, the contribution of subspecies other than M. m. domesticus is limited and its distribution highlights the complex population structure in these strains. On the other hand, wild-derived laboratory strains represent a deep reservoir of genetic diversity untapped in classical strains and are in many cases analogous to three-way intersubspecific hybrids that classical inbred strains were thought to be. Our previous work7,21 combined with the results of the deep survey of mouse resources presented here demonstrates that the laboratory mouse represents an unparalleled model for genetic studies in mammals.
Methods
Sample preparation and Genotyping
Most DNA samples were prepared at the University of North Carolina and all were genotyped using the Mouse Diversity Array17 at The Jackson Laboratory. The processed arrays were computationally genotyped using MouseDivGeno (http://cgd.jax.org/tools/mousedivgeno.shtml), a genotyping software written in R language specifically designed for the Mouse Diversity array. Genotyping of the samples involved three steps: normalization of the intensity variation due to restriction fragment lengths in the genome amplification step and the C+G content of probe sequences; genotype calling using a combined maximum likelihood and hierarchical clustering algorithm; and identification of VINOs, as described below. We excluded 73,525 SNPs out of a total of 623,124 based on poor performance among our samples. We identified thousands of previously unknown genetic variants using an algorithm designed for mutation discovery in the Affymetrix platform. VINOs are characterized by a distinct clustering of samples with low hybridization intensity and designated by the genotype “V”. The genotype of the target SNP in a sample with a VINO call is missing. To confirm that VINOs do indeed represent novel genetic variation, we selected 15 SNP probes with VINO calls and for each probe we selected at least four samples of each genotype (homozygous for allele A, homozygous B, or VINO) for targeted sequencing. Strains for resequencing were selected to maximally sample across subspecies and strain-type (classical or wild-derived). Primers were designed approximately 200 bp proximal and distal to each probe using PrimerQuest (Integrated DNA Technologies, Coralville, IA). Probe regions were amplified by Polymerase Chain Reaction (PCR) and sequenced by automated Sanger sequencing at UNC. Sequences were aligned using Sequencher 4.9 (Gene Codes). Supplementary Table 4 lists all probes, strains and primer sequences. All sequences have been submitted to GenBank under accession numbers GU992455-GU992863. All homozygous SNP genotype calls were confirmed (211/211) as were most of the VINO (14/15). Unconfirmed VINO calls could be explained by polymorphisms outside of the sequenced region that, for example, alter the cut sites for the enzymes used for genome-wide amplification. Thus 100% validation was not expected.
We mapped regions of heterozygosity in each laboratory strain by calculating the frequency of heterozygous calls in 500kb windows with 250kb overlaps and applied a Hidden Markov Model (HMM) with strain specific noise level. We found that most heterozygous calls (H) in inbred strains reflect genotype calling errors that are randomly distributed throughout the genome, whereas in truly heterozygous regions H calls occur in clusters. Array probe design was based on the reference C57BL/6J genome which is mainly M. m. domesticus. Thus genotype error rates are higher in strains that do not share common subspecific origin with C57BL/6J. All heterozygous calls (H) in laboratory strains outside of heterozygous regions were replaced by no calls (N).
We identified large deletions that resulted in hybridization failures (VINOs) in multiple consecutive probes by calculating the VINO frequency in 500kb windows with 250kb overlap. Using an HMM we identified contiguous intervals in which VINO frequencies were higher than the strain-specific noise level. We visually mapped the start and end of deletions and designated genotypes in these regions as “D”. We validated nine of the putative deletions using PCR to amplify markers within and flanking the deletions in DNA samples with or without the deletions. There is 100% concordance between our predictions and the results of this test.
All genotypes are available at http://cgd.jax.org/datasets/popgen.shtml.
Identification of SNPs and VINOs with diagnostic alleles
We used 10 M. m. domesticus, 16 M. m. musculus, and 10 M. m. castaneus wild caught mice to identify informative SNPs and VINOs. For each subspecies we identified SNPs and VINOs for which all mice from the remaining two subspecies share the same allele and denoted the alternative allele as diagnostic. For instance, if all M. m. domesticus mice have an A allele, and all M. m. musculus and all M. m. castaneus mice have a B allele at a SNP, then the A allele at that SNP is a fully informative and diagnostic for M. m. domesticus. We assigned fully informative SNPs a score of 1. In addition, there are cases where the A allele occurs in only one subspecies but is not fixed in that subspecies. These partially informative SNPs are assigned a score that is the fraction of mice with homozygous A genotype over the total number of mice in the subspecies. We allowed for up to two misclassifications due to genotyping errors (typically H calls), homoplasy or gene flow in the determination of diagnostic alleles and penalized the score by a factor of 0.5 (one genotype error) or 0.3 (two genotyping errors). No calls and VINOs were ignored in this procedure. We then applied the same rule to find fully and partially informative VINOs based on dichotomized genotypes - VINO or no VINO.
Assignment of subspecific origin
We assigned subspecific origin based on diagnostic alleles and scores from a given subspecies in each region of a sample. An HMM was used to identify the boundaries, and subspecific origin based on the cumulative scores within these regions.
Recombination intervals and perfect phylogeny trees
The genome of classical inbred strains was partitioned into overlapping intervals that show no evidence of recombination using the four-gamete test. Maximal intervals were computed by a left-to-right scan, adding successive SNPs to an interval until one is not four-gamete compatible with any SNP in that interval. The starting point of the next interval is found by removing SNPs from the left side until all incompatibilities have been removed, and left-to-right scan resumes. All resulting intervals are maximal, and cannot be extended in either direction. A minimal subset of these intervals is found that covers the entire genome while maximizing their overlap. This is computed by finding the longest path in a k-partite graph22. For each such compatible interval there exists a “perfect” phylogenetic tree, in which each node correspond to an haplotype and each edge to SNPs with the same strain distribution.
Identity by descent
To identify IBD regions in classical strains, we first performed pairwise comparisons, and then expanded the IBD strain set using a clique finding algorithm. IBD regions were defined based on the compatible intervals framework described above. The sizes of the compatible intervals were often too small to calculate robust statistics, thus we merged consecutive compatible intervals for pairs of strains sharing the same terminal leaf node of consecutive perfect trees. Based on the merged intervals, we calculated a pairwise genotype similarity score as the proportion of matching variants (SNPs and VINOs) in that interval. After we assigned the score to each pair in each compatible interval, we identified the cliques in each interval. We connected pairs of strains with similarity scores >0.99. To accommodate poorly performing samples and noise, we implemented a clique extension algorithm, and generated a single clique if at least 80% of edges were connected and the mean average similarity is >0.99. Strains belonging to the same clique in an interval were considered IBD over that interval. The reliability of this IBD analysis depends on the number of variants used to calculate the similarity score. Thus to estimate the degree of reliability in each clique, we calculated a clique penalty score. First, we calculated Pij = log10(number of variants used to calculate the similarity score) for every pair of strains and we capped the number of variants per interval at 100. Then, the penalty score is calculated as a variance of Pij. The logarithmic transformation inflates the variance from pairs with small number of variants. If the number of variants from all pairs of strains are bigger than 100, the penalty is zero. We flagged cliques with less than 20 variants, or less than 40 variants with high clique penalty score. We excluded regions with very low SNP density from the IBD analyses. Excluded regions are listed in Supplementary Table 5. Finally, we excluded a single region with a pattern consistent with structural variation (Supplementary Table 6).
To identify regions of IBD in comparisons involving wild-derived strains we calculated the genotype similarity in pairwise comparisons using 1Mb non-overlapping intervals. We declared regions to be IBD based on a threshold of 0.98 identity but we also considered the overall shape of the frequency distribution.
Distance trees
Each distance tree is based on the mean score of strains belonging to the same clique, and provides a quantitative measure of difference among strains belong to different cliques. In each compatible interval, we generated a similarity clique score matrix M of size N×N, where N is the number of cliques, and each element M[i,j] was a mean similarity between strains belonging to clique i and clique j. We built a neighbor-joining tree based on this matrix.
Clique coloring
Using eight pastel colors, we assigned unique colors to each haplotype in an interval such that the total color change across all intervals was minimized. For the first interval, colors were assigned arbitrarily to each haplotype. If there were more than eight haplotypes in an interval, the least frequent were not assigned colors and remain white. For each subsequent interval, every haplotype was assigned a color such that the total number of color transitions in each interval was minimized. There were no constraints on the color differences among intervals that were not adjacent, so this method does not ensure that large blocks of identity, perhaps punctuated by a discordant interval, are of a consistent color.
Web browser
The Mouse Phylogeny Viewer (MPV, http://msub.csbio.unc.edu/PhylogenyTool.html) is intended to provide visual summaries of the results of this study and to allow downloading of the relevant information for selected strains in selected regions of the genome. A tutorial and the LAMP capabilities and meaning of the different analysis is provided online. The complete set of genotypes are available at http://cgd.jax.org/datasets/popgen.shtml.
Supplementary Material
Acknowledgments
This work was supported by NIGMS Centers of Excellence in Systems Biology program, grant GM-076468, by an NIH grant to MWN (R01 GM74245), by a grant to FB (ISEM 2010-141) and by a Czech Science Foundation grant to JP (206-08-0640). JPD was partially supported by NIH Training Grant Number GM067553-04, UNC Bioinformatics and Computational Biology Training Grant. JPD, RJB and TAB are partially supported by an NIH grant to FP-MV (P50 MH090338). We also wish to thank Fredmarie Oyola for help annotating the samples genotyped in this study.
Footnotes
Authors Contribution: F.P.-M.V., G.A.C. and H.Y. conceived the study design and wrote the paper. H.Y., J.W., J.P.D., L.M. and C.W. carried out the bioinformatics analyses. J.P.D., T.A.B. and R.J.B prepared the samples and conducted the targeted PCR amplification and sequencing and F.B., P.B. A.H-T.Y., M.N., J.P. and P.T. provided biological samples. All authors contributed to the interpretation of the results and the writing of the manuscript.
References
- 1.Boursot P, Auffray JC, Britton-Davidian J, Bonhomme F. The evolution of the house mice. Annual Review of Ecology and Systematics. 1993;24:119. [Google Scholar]
- 2.Geraldes A, Bassett P, Gibson B, Smith KL, Harr B, Yu HT, Bulitova N, Siv Y, Nachman MW. Inferring the history of speciation in house mice from autosomal, X-linked, Y-linked and mitochondrial genes. Mol Ecol. 2008;17:5349–5363. doi: 10.1111/j.1365-294X.2008.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teeter KC, Payseur BA, Harris LW, Bakewell MA, Thibodeau LM, O'Brien JE, Krenz JG, Sans-Fuentes MA, Nachman MW, Tucker PK. Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Res. 2008;18:67–76. doi: 10.1101/gr.6757907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yonekawa H, Takahama S, Gotoh O, Miyashita N, Moriwaki K. Genetic diversity and geographic distribution of Mus musculus subspecies based on the polymorphism of mitochondrial DNA. In: Moriwaki K, Shiroishi T, Yonekawa H, editors. Genetics in Wild Mice Its application to Biomedical Research. Japan Scientific Societies Press; Tokyo and Karger, Basel: 1994. pp. 25–40. [Google Scholar]
- 5.Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM. Genealogies of mouse inbred strains. Nat Genet. 2000;24:23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- 6.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nature Genetics. 2007;39:1100–1107. doi: 10.1038/ng2087. [DOI] [PubMed] [Google Scholar]
- 8.Guénet JL, Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/s0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 9.Mouse Genome Sequencing Consortium. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigó R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 10.Sudbery I, Stalker J, Simpson JT, Keane T, Rust AG, Hurles ME, Walter K, Lynch D, Teboul L, Brown SD, Li H, Ning Z, Nadeau JH, Croniger CM, Durbin R, Adams DJ. Deep short-read sequencing of chromosome 17 from the mouse strains A/J and CAST/Ei identifies significant germline variation and candidate genes that regulate liver triglyceride levels. Genome Biol. 2009;10:R112. doi: 10.1186/gb-2009-10-10-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chesler EJ, Miller DR, Branstetter LR, Galloway LD, Jackson BL, Philip VM, Voy BH, Culiat CT, Threadgill DW, Williams RW, Churchill GA, Johnson DK, Manly KF. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19:382–389. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan C, Ye C, Yang X, Gao J. A review of current large-scale mouse knockout efforts. Genesis. 2010;48:73–85. doi: 10.1002/dvg.20594. [DOI] [PubMed] [Google Scholar]
- 13.Szatkiewicz JP, Beane GL, Ding Y, Hutchins L, Pardo-Manuel de Villena F, Churchill GA. An imputed genotype resource for the laboratory mouse. Mamm Genome. 2008;19:199–208. doi: 10.1007/s00335-008-9098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harr B. Genomic islands of differentiation between house mouse subspecies. Genome Res. 2006;16:730–737. doi: 10.1101/gr.5045006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boursot P, Belkhir K. Mouse SNPs for evolutionary biology: beware of ascertainment biases. Genome Res. 2006;16:1191–1192. doi: 10.1101/gr.5541806. [DOI] [PubMed] [Google Scholar]
- 16.White MA, Ané C, Dewey CN, Larget BR, Payseur BA. Fine-scale phylogenetic discordance across the house mouse genome. PLoS Genet. 2009;5(11):e1000729. doi: 10.1371/journal.pgen.1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen BJ, Graber JH, de Villena FP, Churchill GA. A customized and versatile high-density genotyping array for the mouse. Nat Methods. 2009;6:663–666. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagamine CM, Nishioka Y, Moriwaki K, Boursot P, Bonhomme F, Lau YF. The musculus-type Y chromosome of the laboratory mouse is of Asian origin. Mamm Genome. 1992;3:84–91. doi: 10.1007/BF00431251. [DOI] [PubMed] [Google Scholar]
- 19.Tucker PK, Lee BK, Lundrigan BL, Eicher EM. Geographic origin of the Y chromosomes in “old” inbred strains of mice. Mamm Genome. 1992;3:254–261. doi: 10.1007/BF00292153. [DOI] [PubMed] [Google Scholar]
- 20.Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science. 2009;323:373–375. doi: 10.1126/science.1163601. [DOI] [PubMed] [Google Scholar]
- 21.Ideraabdullah FY, de la Casa-Esperón E, Bell TA, Detwiler DA, Magnuson T, Sapienza C, de Villena FP. Genetic and haplotype diversity among wild-derived mouse inbred strains. Genome Res. 2004;14:1880–1887. doi: 10.1101/gr.2519704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Moore KJ, Zhang Q, Pardo-Manuel de Villena F, Wang W, McMillan L. Genome-wide compatible SNP intervals and their properties. Proceedings of ACM International Conference on Bioinformatics and Computational Biology; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.