

Abstract
Recent proteomics studies have revealed that protein acetylation is an abundant and evolutionarily conserved post-translational modification from prokaryotes to eukaryotes. Although an astonishing number of acetylated proteins have been identified in those studies, the acetyltransferases that target these proteins remain largely unknown. Here we characterized MSMEG_5458, one of the GCN5-related N-acetyltransferases (GNAT’s) in Mycobacterium smegmatis, and show that it is a protein acetyltransferase (MsPat) that specifically acetylates the ε-amino group of a highly conserved lysine residue in acetyl-CoA synthetase (ACS) with a kcat/Km of nearly 104 M−1 s−1. This acetylation results in the inactivation of ACS activity. Lysine acetylation by MsPat is dependent on 3′,5′-cyclic adenosine monophosphate (cAMP), an important second messenger, indicating that MsPat is a downstream target of the intracellular cAMP signaling pathway. To the best of our knowledge, this is the first protein acetyltransferase in mycobacteria that both is dependent on cAMP and targets a central metabolic enzyme by a specific post-translational modification. Since cAMP is synthesized by adenylate cyclases (AC’s) that sense various environmental signals, we hypothesize that the acetylation and inactivation of ACS is important for mycobacteria to adjust to environmental changes. In addition, we show that Rv1151c, a sirtuin-like deacetylase in Mycobacterium tuberculosis, reactivates acetylated ACS through an NAD+-dependent deacetylation. Therefore, Pat and the sirtuin-like deacetylase in mycobacteria constitute a reversible acetylation system that regulates the activity of ACS.
Protein acetylation was first discovered in histones almost 50 years ago (1). In the following four decades, it became clear that the histone acetyltransferases (HAT’s) were part of a very large enzyme superfamily referred to as the GCN5-related N-acetyltransferases (GNAT’s). The GNAT superfamily catalyzes acetyl transfer from acetyl coenzyme A (AcCoA) to a primary amine within a small molecule or a protein and is one of the largest superfamilies with over 30,000 members in all kingdoms of life (2). However, the biological functions and/or substrates for a majority of the GNAT’s are largely unknown, with only 5 of the 26 GNAT’s in Escherichia coli having known biological functions. But it is clear that some of the pro- and eukaryotic GNAT’s must be capable of acetylating proteins other than histones. In the last five years, several research groups have identified over 2,000 proteins acetylated at the e-amino group of a lysine residue in human cells (3–5), and almost 200 in E. coli and Salmonella enterica (6–8). In addition to protein phosphorylation, protein lysine acetylation is now recognized as a ubiquitous and evolutionarily conserved protein modification from prokaryotes to eukaryotes. Both of these post-translational modifications are known to be reversible, with protein Ser/Thr and Tyr phosphatases and protein deacetylases (sirtuins) found in all organisms containing the corresponding kinases and acetyltransferases. In the case of protein acetylation, many of the enzymes involved in glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle and fatty acid metabolism were found to be acetylated, implying an extensive role of acetylation in the regulation of intracellular metabolism (3–8). Despite the remarkable number of proteomically identified acetylated proteins, the effect of acetylation on the activity of most of the “identified” enzymes, or the particular GNAT responsible for acetylation, remains largely unknown.
In Mycobacterium tuberculosis, the causative agent of tuberculosis, only five out of twenty predicted GNAT’s have been biochemically, functionally or structurally characterized (9–14). Two other enzymes, Rv0995 (RimJ) and Rv3420c (RimI) are predicted to acetylate the N-terminus of ribosomal proteins S5 and S18, respectively (2). Of the twenty GNAT’s, Rv0998 exhibits a unique structural feature with a C-terminal GNAT domain fused to an N-terminal cyclic nucleotide (cNMP) binding domain. The GNAT domain exhibits highest sequence identity to a protein N-acetyltransferase identified in S. enterica (15), while the N-terminal cNMP binding domain is most similar to the cNMP domain of eukaryotic protein kinases. The orthologue of Rv0998 in Mycobacterium smegmatis, MSMEG_5458, which shares 56% identity and 70% similarity based on the protein sequence analysis (Figure S1), has recently been shown to catalyze the acetylation of the universal stress protein (USP) using a glutathione-S-transferase pull-down assay (13). However, the lack of USP orthologues in other mycobacteria, including M. tuberculosis, suggests that Rv0998 and MSMEG_5458 have an alternative substrate that is unknown, and that acetylation of this substrate may be cNMP-dependent.
Cyclic nucleotides are universal “second messengers” in both pro- and eukaryotic organisms, and cAMP was identified in extracts of several mycobacterial species in 1976 (16). While most prokaryotes contain a single adenylate cyclase (AC, e.g. E. coli) and some contain none (e.g. Bacillus species), mycobacteria, and in particular M. tuberculosis, contain over a dozen genes identified as AC’s, with many of these biochemically characterized in M. tuberculosis (reviewed in (17)). In M. tuberculosis, several of the AC’s have been shown to be specifically responsive to nitrogen and carbon limitation, pH and bicarbonate levels (18–21). Upon phagocytosis by macrophages, cAMP levels in M. tuberculosis have been shown to increase dramatically (22, 23). Restoration of basal cAMP levels is the result of hydrolysis of cAMP to 5′-AMP by cAMP phosphodiesterases, and secretion. In M. tuberculosis, the single annotated cAMP phosphodiesterase is only weakly active towards 3′,5′-cAMP and much more active with 2′,3′-cAMP (24), suggesting efflux as a major mechanism. Reports that cAMP efflux by M. tuberculosis into macrophages may produce immunosuppressive effects and reduction in cytokine production by infected macrophages have appeared (22).
In this report, we attempted to identify a physiologically relevant substrate for MSMEG_5458 that would link production of cAMP to the acetylation of a protein substrate whose activity could be implicated in carbohydrate and amino acid metabolism. In addition, we sought the corresponding deacetylase (sirtuin), since it is universally recognized that post-translational protein modification coupled to activity activation/inactivation is a reversible phenomenon.
Materials and Methods
Materials
Oligonucleotides and the EnzChek pyrophosphate assay kit were purchased from Invitrogen. Restriction enzymes and competent cells were supplied by New England Biolabs. Genomic DNA from M. tuberculosis H37Rv and M. smegmatis mc2155 were provided by ATCC. Other chemicals and reagents were obtained from Sigma-Aldrich, unless otherwise noted.
Cloning, overexpression and purification of MSMEG_5458, MSMEG_6179, Rv0998, Rv3667 and Rv1151c
The five genes were amplified by PCR from the genomic DNA of M. smegmatis mc2155 or M. tuberculosis H37Rv using the oligonucleotides shown in Table S1. After restriction digest, MSMEG_5458 (MsPat) and MSMEG_6179 (MsACS) were cloned into pET23a, while Rv0998 (MtPat), Rv3667 (MtACS) and Rv1151c were ligated into pET28a. The constructs were confirmed to be mutation free by DNA sequencing. Then the plasmids were transformed into T7 Express lysY/Iq competent E. coli cells. A single colony was selected to start a 10 mL overnight culture, which was then used to inoculate 1 L Luria-Bertani medium supplemented with 200 μg/mL ampicillin (MsPat and MsACS) or 30 μg/mL kanamycin (Rv0998, Rv1151c and MtACS). The cells were grown to mid-exponential phase at 37 °C, and then induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside at 25 °C for 15 hours.
Cells were harvested by centrifugation, resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole, pH 8.0) containing protease inhibitors (Roche) and 0.1 mg/mL lysozyme, and incubated on ice for 30 minutes. The cells were disrupted by sonication, and cell debris removed by centrifugation at 36,000 × g for 1 hour. The resulting supernatant was loaded onto a 5 mL Ni-NTA agarose column (Qiagen) that was pre-equilibrated with the lysis buffer, and incubated at 4 °C for 1 hour. After discarding the flow through, the column was washed with 50 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole, pH 8.0), and bound proteins were eluted using a linear gradient from 20 to 250 mM imidazole in 50 mM NaH2PO4 and 300 mM NaCl, pH 8.0. The fractions were analyzed by SDS-PAGE, and those containing the desired protein were pooled and dialyzed against buffer A (50 mM Tris and 150 mM NaCl, pH 7.5). The protein was then concentrated using Amicon Ultra-4 30K cut-off centrifugal device (Millipore). The protein concentration was determined by the BCA method using bovine serum albumin as the standard (25).
Purification of MsPat under denaturing conditions
The purification procedure was identical to that described above, except that 8 M urea was added to all the buffers. The eluted fractions containing MsPat after Ni-NTA chromatography were collected and dialyzed stepwise into buffer A containing 4 M, 2 M, 1 M, 0.5 M and no urea over 3 days. The refolded protein was concentrated, and the concentration was determined as described above.
Site-directed mutagenesis and purification of the MtACS mutant K617A
The mutation K617A was introduced into the pET28a(+)::acs plasmid using the Quikchange mutagenesis kit (Stratagene) with the primers listed in Table S1. The mutation was confirmed by DNA sequencing. The expression and purification of the mutant were performed following the same procedure described above for the wild type ACS.
UV-Visible spectrum and HPLC analysis of the tightly bound ligand of MsPat
60 nmol of recombinant MsPat was denatured by placing the enzyme solution in a boiling water bath for 5 minutes. The precipitated protein was then removed by centrifugation followed by ultrafiltration with Amicon Ultra-0.5 3K cut-off centrifugal device (Millipore). The absorbance spectrum of the filtrate was obtained using a UVIKON XL spectrophotometer. The filtrate was further analyzed at 260 nm by reverse phase HPLC using a Gemini-NX C18 column (250 mm × 4.6 mm, Phenomenex). HPLC solvents are listed below: solvent A: 0.2% trifluoroacetic acid in water; solvent B: 0.2% trifluoroacetic acid in acetonitrile. Gradient elution was performed at 1 mL/min and increasing solvent B from 0–100% in 30 minutes.
Substrate profiling of MsPat using a bi-substrate inhibitor approach
M. smegmatis mc2155 was grown in 7H9 medium supplemented with 0.2% glucose, 0.2% glycerol and 0.05% Tween-80 at 37 °C until OD600 reached 0.2. The cells were harvested by centrifugation and resuspended in buffer B (50 mM HEPES and 150 mM NaCl, pH 7.5). Cells were disrupted by sonication and centrifuged at 20,000 × g for 25 minutes at 4 °C. The protein concentration of the supernatant was measured by the BCA method (25). In the substrate profiling assay, a 2 mL reaction mixture contained the cell lysate (0.8 mg/mL protein), 1 μM MsPat (6×His-tagged), 500 μM ClAcCoA, 1mM cAMP and 0.25 mM TCEP. After 45 minutes incubation at room temperature, 2 mL of 200 mM TAPS buffer (pH 8.6) containing 2.4 mM CoA and 2 mM TCEP was added to the reaction mixture and further incubated at room temperature for 5 hours, converting the substrate of MsPat into a bi-substrate inhibitor that binds tightly to MsPat. After dialysis against 50 mM NaH2PO4 buffer containing 300 mM NaCl (pH 8.0), the reaction mixture was then loaded to a 0.2 mL Ni-NTA agarose column and washed by 2 mL binding buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole, pH 8.0) and 2 mL wash buffer (50 mM NaH2PO4, 300 mM NaCl, and 25 mM imidazole, pH 8.0). The proteins tightly bound to MsPat were eluted off the column with 1.2 mL of the wash buffer containing 8 M urea. The control experiment was carried out by incubating the cell lystate with MsPat and CoA instead of ClAcCoA at room temperature for 45 minutes. The urea eluates were concentrated and analyzed by SDS-PAGE.
Mass spectrometric analysis of the substrate of MsPat
The mass spectrometry analysis was performed at the Laboratory for Macromolecular Analysis and Proteomics (LMAP) of the Albert Einstein College of Medicine. The substrate of MsPat was purified from M. smegmatis cell lysate as described above. The protein band and the corresponding position in the control lane were excised from the protein gel and analyzed individually. After trypsin digestion, the resulting digests were analyzed by LC-MS/MS using a LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) equipped with a TriVersa NanoMate nanoelectrospray source (Advion BioSciences, Ithaca, NY). HPLC was performed with the UltimatePlus nano-HPLC system using a PepMap C18 column (2 μm, 100 Å, 75 μm × 15 cm, Dionex). After 15 minutes of desalting with solvent C (2% acetonitrile, 0.1% formic acid in water), a gradient elution was carried out at 300 nL/min and increasing solvent D (80% acetonitrile, 0.1% formic acid in water) from 0–35% in 40 minutes, 35–50% in 5 minutes, 50–90% in 5 minutes, and then held at 90% D for 5 minutes. The 10 most intense ions having a charge state between +2 to +4, determined from an initial survey scan from 300 to 1800 m/z, were selected for fragmentation (MS/MS). MS/MS was performed using an isolation width of 2 m/z and a normalized collision energy of 35%.
The raw MS/MS data were converted into DTA files, which were then merged and searched against the nonredundant NCBI database for proteins from M. smegmatis using the Mascot search engine (Matrix Science). Mass tolerance was set at 3.5 Da for precusor ions and 0.6 Da for fragment ions. Carbamidomethylation (Cys), deamidation (Asn and Gln), pyro-glu (Glu and Gln), and oxidation (Met) were specified as variable modifications. Up to 2 missed trypsin cleavage sites were allowed. All the identified peptides were manually verified.
Activity-based labeling of ACS by MsPat
Chloroacetyl CoA (ClAcCoA) and aminoethanethiolated 5(6)-carboxy-tetramethylrhodamine (TAMRA cysteamine) were synthesized and purified as described previously (26). Typically, the labeling assays consist of two steps, chloroacetylation and thiol quenching. Chloroacetylation was carried out in 50 μL of buffer B containing 20 μM ACS, 100 μM ClAcCoA, 1 mM cAMP, and 1 μM MsPat at room temperature for 2 hours, and the reaction mixture was quenched by the addition of an equal volume of 200 mM Tris buffer (pH 8.6) containing 0.6 mM TAMRA cysteamine. The excess fluorescent probe was removed by gel filtration. The labeled protein was then concentrated, and analyzed by SDS-PAGE. The protein gel was scanned using a Storm Scanner 845 (GE Life Sciences) to visualize the fluorescence, and then stained for protein with Coomassie brilliant blue.
Mass spectrometric analysis of the modified ACS
ACS was fluorescently labeled as described above. In addition, 20 μM ACS was acetylated by incubating with 1 mM cAMP, 50 μM AcCoA and 1 μM MsPat at room temperature for 2 hours. The modified ACS was separated from MsPat by SDS-PAGE, and then analyzed by mass spectrometry as described above. The MS/MS data were searched against MtACS with carbamidomethylation (Cys), deamidation (Asn and Gln), pyro-glu (Glu and Gln), oxidation (Met) and TAMRA (Lys) or acetylation (Lys) as variable modifications using Thermo Bioworks 3.3.1. Putative modifications identified from the search were further analyzed by manual assignments of the MS/MS spectra.
Deacetylation of ACS by Rv1151c
20 μM ACS was first incubated with 0.4 μM MsPat, 50 μM ClAcCoA and 1 mM cAMP at room temperature for 2 hours. After chloroacetylation, ACS was then treated with 1 μM or 2 μM Rv1151c in the presence or absence of 1 mM NAD+ for 2 hours. The reaction mixture was then quenched with 0.6 mM TAMRA cysteamine and analyzed by SDS-PAGE as described above.
Time-dependent inactivation of ACS by MsPat and reactivation by Rv1151c
The activity of ACS was monitored during the acetylation reaction where 20 μM ACS was incubated in 100 μL of buffer B with 1 mM cAMP/cGMP, 50 μM AcCoA and 50 nM MsPat at room temperature. 5 μL aliquots were withdrawn at different time intervals after initiating the acetylation by the addition of MsPat, and the residual activity of ACS was measured immediately by the EnzChek pyrophosphate assay where pyrophosphate liberated from the ACS reaction together with 2-amino-6-mercapto-7-methyl-purine ribonucleoside is converted into ribose 1-phosphate and ribose 2-amino-6-mercapto-7-methylpurine, causing an absorbance increase at 360 nm. A typical ACS activity assay contained 1 mM MgSO4, 100 μM CoA, 10 mM sodium acetate, 1 mM ATP, 100 μM MESG, 0.03 unit of inorganic pyrophosphatase, 0.5 unit of purine ribonucleoside phosphorylase and 200 nM ACS from the acetylation reaction. The absorbance at 360 nm was monitored, and the ACS activity (pyrophosphate release) was calculated using the molar extinction coefficient (Δε360 = 18.6 mM−1 cm−1). For the reactivation, 20 μM ACS was incubated with 1 mM NAD+ and 2 μM Rv1151c after a 3-hour acetylation by MsPat. The residual activity of ACS was measured in the same manner as described above. The control assays were performed omitting each one of the reaction components, respectively. The percentage activity of ACS was plotted against the time (minutes) after acetylation or deacetylation using SigmaPlot 11. Each data point is the average of two independent assays.
Steady-state kinetic assays
In order to monitor the acetylation reaction continuously, we utilized a coupled enzymatic assay in which CoA, a product of the acetylation reaction, was used by α-ketoglutarate dehydrogenase to convert NAD+ to NADH, resulting in an absorbance increase at 340 nm. The assay was carried out in buffer B with 0.05 unit of α-ketoglutarate dehydrogenase, 400 μM NAD+, 1.5 mM α-ketoglutarate, 2 mM MgSO4, 400 μM thiamin diphosphate, 1mM cAMP, 400 nM MsPat and various amounts of AcCoA and ACS. The initial velocities were measured at varying concentrations of one substrate while maintaining a fixed saturating concentration of the other one. The data were the fitted into the Michaelis-Menten equation using Sigma Plot 11 to obtain Km and kcat.
To measure the kinetic parameters of the deacetylation, the acetylated ACS, which was purified by gel filtration after incubating 200 nmol ACS with 1 μmol AcCoA and 0.5 nmol MsPat at room temperature for 3 hours, was used in the deacetylation reaction. A typical deacetylation reaction mixture contained 0.5 μM Rv1151c, varying amounts of one substrate and a saturating amount of the other in 100 μL of buffer B. The reaction was initiated with the addition of Rv1151c. 0.2 nmol of ACS species (acetylated and nonacetylated) were withdrawn from the reaction mixture after 10 minutes and tested for the ACS activity as described above. The initial velocities of the deacetylation reaction were calculated based on the amount of active ACS generated in 10 minutes. Each data point is the average of two identical assays. The data were fitted into the Michaelis-Menten equation as listed above to obtain the Km and kcat values.
Results
cAMP is tightly bound to the purified MsPat
MsPat was overexpressed in E. coli, and purified to >95% homogeneity by Ni-NTA chromatography (Figure S2). Since there is a gene encoding an adenylate cyclase in E. coli (27), it was possible that the purified MsPat contains bound cAMP. To determine if any ligand was bound to the protein, MsPat was precipitated by boiling, and removed by centrifugation and ultrafiltration. The UV-Visible spectrum of the filtrate displayed a maximum absorbance at 260 nm (Figure S3A). In addition, the filtrate exhibited a single peak on HPLC with a retention time identical to the cAMP standard (Figure S3B), suggesting that cAMP was bound to the purified MsPat. The amount of bound cAMP was calculated from a HPLC-based standard curve (Figure S4), and approximately 20% of the recombinant purified MsPat contained bound cAMP. In order to determine the effect of cAMP on acetyltransferase activity, it is necessary to obtain the apo enzyme. In a previous study, apo MsPat was purified after expression in an E. coli strain lacking the lone adenylyl cyclase (cya) gene (13). We obtained the apo enzyme by purifying under denaturing conditions. The denatured apo MsPat was then renatured by multi-step dialysis, and used in the following studies.
Acetyl-CoA synthetase is a bona fide substrate of MsPat
To explore the physiologically relevant substrate of MsPat, we performed a substrate profiling experiment using a GNAT activity-based method previously developed in our laboratory with slight modifications (26). Recombinant MsPat was incubated with M. smegmatis cell lysate along with chloroacetyl CoA (ClAcCoA), an analogue of AcCoA. MsPat can transfer the chloroacetyl group from ClAcCoA to its substrate, which is then converted to a bi-substrate inhibitor of MsPat by the addition of CoA (Figure 1A). It has been reported that these bi-substrate inhibitors have nanomolar affinity for their corresponding GNATs (28–30). Since the recombinant MsPat added to the cell lysate contains a hexa histidine-tag, the tightly bound substrate would be co-purified with MsPat by Ni-NTA chromatography. After extensive washing to remove nonspecifically bound proteins, the substrate of MsPat was eluted off the column by 8 M urea, which disrupts the noncovalent interactions between MsPat and the bisubstrate inhibitor. As indicated by an arrow in Figure 1B, one additional protein band was observed in the elution sample when ClAcCoA was used (lane 1), compared to the control sample (lane 2, Figure 1B). The mass spectrometric analysis of this band shows 21% sequence coverage of acetyl-CoA synthetase from M. smegmatis (MsACS) (Figure 1C, Table S2), while MsACS was not identified in the corresponding position of the control lane. This result strongly suggests that MsACS is a substrate of MsPat. This result is consistent with the protein sequence alignment analysis (Figure 2), which reveals that the GNAT domain of MsPat is 27% identical to the corresponding domain of the protein acetyltransferase in S. enterica (SePat) that has been reported to acetylate a conserved lysine residue in SeACS (15). In addition, the residue Glu173 in yeast GCN5 that serves as a general base to deprotonate the lysine residue of the acetyl acceptor (31) is conserved in MsPat and its M. tuberculosis orthologue Rv0998, while an Asp residue is present in the corresponding position in SePat, as indicated in Figure 2.
Figure 1. Substrate profiling of MsPat by using a bi-substrate inhibitor approach.

(A) Substrate profiling method. GNAT catalyzes the transfer of chloroacetyl group to its substrate. The chloroacetylated substrate is then quenched with coenzyme A, forming a bi-substrate inhibitor. (B) Substrate profiling of MsPat. The M. smegmatis cell lysate was incubated with MsPat and ClAcCoA (sample 1) or CoA (sample 2). Then the reaction was quenched with the addition of CoA. The resulting bi-substrate inhibitor of MsPat would bind tightly to MsPat, and be co-purified with MsPat by Ni-NTA chromatography. The bound inhibitor was eluted off using 8 M urea, and then analyzed by SDS-PAGE. (C) Mass spectrometric analysis of MsPat substrate. A search against nonredundant NCBI protein database, yielding a hit of MsACS with 21% sequence coverage. The peptides identified from the mass spectrometric analysis are highlighted in yellow.
Figure 2. Sequence alignment of GNAT domains.

Multiple sequence alignment of the GNAT domains from yeast GCN5, SePat, Rv0998 and MSMEG_5458 was performed using ClustalW. The Glu residue in GCN5, shown in bold, is the general base that deprotonates the acetyl acceptor. Glu or Asp is found in the corresponding positions of other GNAT’s.
We attempted to obtain recombinant ACS of M. smegmatis and M. tuberculosis. However, we were unable to express MsACS in E. coli. We then purified recombinant MtACS to confirm that ACS is a substrate of MsPat using the GNAT activity-based labeling method (26), since MtACS and MsACS share an extremely high sequence identity (76%) and similarity (88%). As described previously, GNAT’s can transfer the chloroacetyl group from ClAcCoA to their substrates, which can react subsequently with the highly fluorescent reporter molecule, TAMRA cysteamine (Figure 3A). The fluorescently labeled protein can then be visualized using fluorescent scanning. In the upper panel of Figure 3B, ACS was fluorescently labeled in the presence of ClAcCoA, cAMP, and MsPat (lane 3). Much weaker labeling of ACS was observed when cAMP was replaced with cGMP (Figure 3B, lane 2), indicating that cAMP is the preferred activator of MsPat. ACS is not fluorescently labeled (Figure 3B, lane 4–6) in the absence of any one of the three components (ClAcCoA, cAMP and MsPat), suggesting that ACS is a protein substrate for MsPAT whose activity is allosterically regulated by the cAMP-binding domain.
Figure 3. Activity-based labeling of ACS by MsPat.
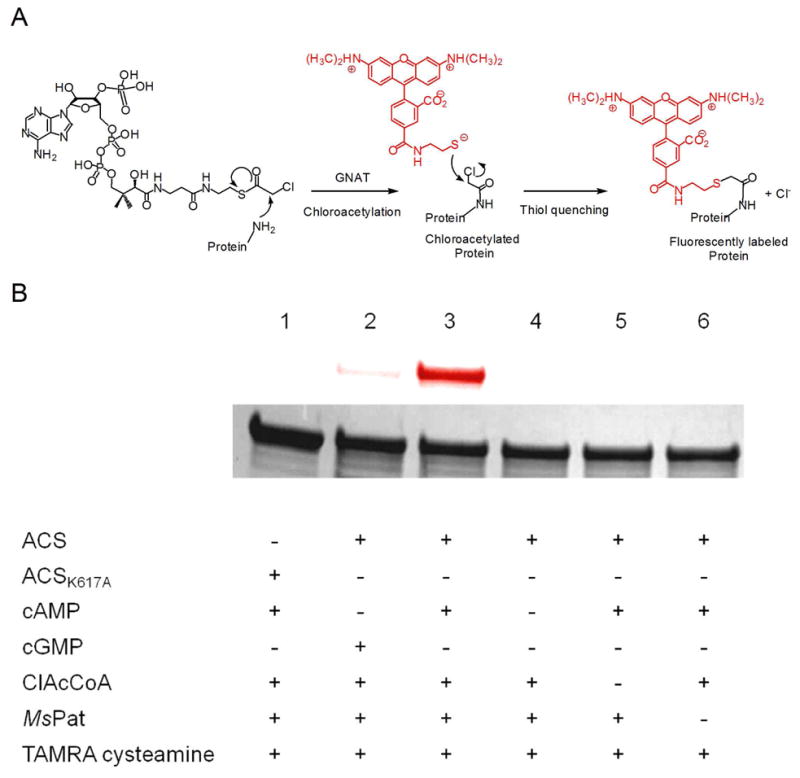
(A) GNAT activity-based labeling method. GNAT catalyzes the transfer of chloroacetyl group from ClAcCoA to its substrate. Then the chloroacetylated substrate is quenched with TAMRA cysteamine, forming a fluorescently labeled substrate. (B) Fluorescent labeling of ACS by MsPat. Wild type ACS or ACS K617A mutant was incubated with various components as indicated, followed by thiol quenching with TAMRA cysteamine. The samples were then analyzed with SDS-PAGE. Top, fluorescence image of the protein gel; bottom, coomassie blue staining image. The assays were performed twice and an identical labeling pattern was obtained.
Although we were able to express recombinant Rv0998, the protein did not show the expected GNAT activity with MtACS. Due to the high sequence homology between Rv0998 and MSMEG_5458 as well as the robust activity observed with MtACS and MSMEG_5458, the lack of the recombinant Rv0998 activity is likely to be caused by misfolding.
To determine the acetylation site, the fluorescently labeled ACS was digested by trypsin, and then analyzed by LC-MS/MS. The MS/MS data were searched against the sequence of M. tuberculosis ACS with possible TAMRA modifications on lysine residues, yielding a single hit with the sequence SGKIMR that contains a modified lysine 617 residue. The fragmentation of this peptide (triply charged, m/z of 408.11) results in a series of y fragment ions (y1, y2, y4 and y5) as well as b2 to b5 fragment ions, as shown in Figure 4A. All but three of the major fragmentation ions in the spectra were readily assigned to the SGK*IMR sequence, suggesting that K617 is the acetylation site. In addition, no fluorescent labeling was observed with the K617A mutant of ACS (Figure 3B, lane 1), further supporting the identification of K617 as the sole acetylation site. ACS was also analyzed by mass spectrometry after acetylation using AcCoA by MsPat. The MS/MS spectra of a triply charged tryptic peptide TRSGKAcIMR (Figure 4B) agrees well with acetylation at K617 due to the presence of the fragmentation ions bearing the acetylated lysine (y4, y6, y7, b5 and b6) as well as other y or b ions.
Figure 4. Identification of the modification site of ACS by MS/MS.

(A) MS/MS spectra of a triply charged tryptic peptide from ACS (SGK*IMR) bearing a lysine modified by TAMRA. The modified lysine residue is indicated as K*. (B) MS/MS spectra of a triply charged tryptic peptide from ACS (TRSGKAcIMR) bearing an acetylated lysine. The acetylated lysine residue is indicated as KAc. Most of the major fragmentation ions in the mass spectra match the predicted b or y ions. The ions containing the modified lysine residue are shown in bold.
In order to measure the kinetic parameters of the MsPat-catalyzed acetylation reaction, we used a coupled enzymatic assay to monitor the acetylation reaction continuously. The results are shown in Table 1. MsPat acetylates ACS with a kcat of 0.06 s−1, slightly lower compared to other characterized GNATs that acetylate proteins (26, 31–33). The Km value for AcCoA is 3 μM, about 8 fold lower than GCN5 and AcuA, a lysine acetyltransferase in Bacillus subtilis (31, 34), indicating that MsPat is active even at low intracellular levels of AcCoA. The low Km value of ACS (10 μM) and relatively high kcat/Km (6 × 103 M−1 s−1) suggests that ACS acetylation by MsPat is kinetically efficient. Similar kcat and Km values were obtained when using the MsPat purified under native conditions, indicating that the apo MsPat was refolded properly and its activity was not affected.
Table 1.
Kinetic parameters of acetylation and deacetylation on ACS
| Enzyme | Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| MsPat | ACS a | 0.06 ± 0.002 | 10.2 ± 1.1 | (6.0 ± 0.7) × 103 |
| Acetyl CoA b | 3.1 ± 0.2 | (1.8 ± 0.2) × 104 | ||
|
| ||||
| Rv1151c | Acetylated ACS c | 0.02 ± 0.001 | < 4 | > 5.0 × 103 |
| NAD+ d | 424 ± 35 | (4.8 ± 0.3) × 102 | ||
Kinetic parameters were measured in the presence of 200 μM AcCoA,
100 μM ACS,
2 mM NAD+ and
20 μM acetylated ACS.
MsPat has been shown to acetylate USP with an extremely low Km of 338 nM from a Western blot based assay, although the kcat value was not reported (13).
Acetylated ACS is deacetylated by Rv1151c
Two distinct protein deacetylases have been reported in bacteria so far (35, 36), CobB (a sirtuin-like deacetylase in S. enterica) and AcuC (an NAD+-independent deacetylase in Bacillus subtilis). There is no homologue of AcuC in mycobacteria, while only one homologue of CobB in M. tuberculosis, Rv1151c, was found. To test if Rv1151c can deacetylate ACS, we used the same fluorescent labeling assay described above with minor modifications. ACS was first chloroacetylated using MsPat, and then treated with Rv1151c, followed by thiol quenching using TAMRA cysteamine. In Figure 5, ACS is much less fluorescently labeled when incubated with 2 μM Rv1151c and 1 mM NAD+ (lane 3), compared to that treated with buffer (lane 1) or with only NAD+ or Rv1151c (lane 4 and 5), suggesting that the chloroacetyl group was removed from chloroacetylated ACS by Rv1151c only in the presence of the deacetylase and NAD+. The deacetylation by Rv1151c is inhibited by 2 mM nicotinamide, a known sirtuin inhibitor (Figure 5, lane 6) further supporting Rv1151c’s role as a sirtuin-like deacetylase. The extent of deacetylation is dependent on the concentration of Rv1151c added (Figure 5, lane 2 and 3). In addition, kinetic studies demonstrate that Rv115c exhibits a low Km value for acetylated ACS (< 4 μM, Table 1), ruling out the possibility of non-specific deacetylation at a high concentration of Rv1151c.
Figure 5. Deacetylation of ACS by Rv1151c.

ACS was first chloroacetylated using MsPat, and then incubated with various components as indicated. Sample 1: blank; sample 2: 1 mM NAD+ and 1 μM Rv1151c; sample 3: 1 mM NAD+ and 2 μM Rv1151c; sample 4: 1 mM NAD+; sample 5, 2 μM Rv1151c; sample 6: 1 mM NAD+, 2 μM Rv1151c and 5 mM nicotinamide. After 2-hour incubation, samples 1 to 6 were quenched with TAMRA cysteamine, and then analyzed by SDS-PAGE and fluorescence imaging. Top, fluorescence image of the protein gel; bottom, coomassie blue staining image. a: 1 μM Rv1151c; b: 2 μM Rv1151c. The assays were performed twice and an identical labeling pattern was obtained.
Interestingly, there are two sirtuin homologues in M. smegmatis, MSMEG_4620 and MSMEG_5175 (Figure S5), but only MSMEG_5175 exhibited robust activity with acetylated ACS in vitro (data not shown).
ACS is inactive upon acetylation of K617
Acetylation of lysine residues has been reported to modulate the functions of various proteins (37–39). To investigate the effects of acetylation, the activity of ACS (pyrophosphate release) was monitored using a coupled assay. In Figure 6A, ACS gradually lost its activity during acetylation by MsPat. We also found that the ACS K617A mutant is inactive in our pyrophosphate release assay. Therefore, acetylation or alanine mutation of K617 inactivates ACS, consistent with the essential role of K617 in ACS for the formation of acetyl-AMP intermediate (36). In contrast, the ACS activity was not affected when any of the three components required for the acetylation reaction (AcCoA, cAMP and MsPat) was absent (Figure 6A). In addition, ACS inactivation was much more rapid in the presence of cAMP than with cGMP. Together with the fluorescent labeling results, this supports our conclusion that upon activation by cAMP, MsPat acetylates ACS and abolishes its activity. MsPat represents a novel downstream target in the cAMP signaling pathway in mycobacteria.
Figure 6. Effects of acetylation and deacetylation on MtACS activity.

(A) Time-dependent inactivation of ACS by acetylation. ACS activity was measured using the EnzChek pyrophosphate assay kit at different time intervals during the incubation of ACS with various components. ○: 50 nM MsPat, 1 mM cAMP, and 100 μM AcCoA; ●: 50 nM MsPat, 1 mM cGMP, and 100 μM AcCoA; ▼: 1 mM cAMP and 100 μM AcCoA;△: 50 nM MsPat and 1 mM cAMP; ■: 50 nM MsPat and 100 μM AcCoA. (B) Time-dependent reactivation of acetylated ACS by deacetylation. After acetylated by MsPat, ACS was further treated with Rv1151c and/or NAD+. ●: 2 μM Rv1151c, 1 mM NAD+; ○: 2 μM Rv1151c; ▼: 1 mM NAD+. The ACS activity was then monitored using the EnzChek pyrophosphate assay kit. Each data point is the average from two identical assays.
Acetylated ACS was then used in the reactivation assays, where ACS activity was monitored after it was treated with Rv1151c and NAD+. In Figure 6B, the inactive, acetylated ACS regains activity when incubated with Rv1151c and NAD+, while it remains inactive if only Rv1151c or NAD+ is present. The results support our conclusion that Rv115c is an NAD+-dependent deacetylase and can use monoacetylated ACS as a substrate. Therefore, we have demonstrated that Pat and Rv1151c form a reversible acetylation system that regulates the catalytic activity of ACS in mycobacteria.
Discussion
There have been a number of recent reports identifying non-histone proteins that are post-translationally acetylated in both pro- and eukaryotes (3–8). The majority of these rely on the immunoprecipitation of cell extracts using antibodies generated to Nε-acetyl-L-lysine followed by MS/MS identification of peptide sequences and assignment to individual proteins. Our own approach uses activity-based methods and chloroacetylCoA as a substrate (26). This method was used in the present study not only to identify the substrate of MsPat from cell lysates, but also to confirm it in vitro using purified recombinant proteins.
Our substrate profiling data reveal that MsACS is a substrate of MsPat. ACS (AMP-forming) catalyzes the conversion of acetate, ATP and CoA to form AcCoA, an essential central carbon metabolite involved in numerous enzymatic reactions. To date, two different acetyltransferases with no sequence homology have been reported to acetylate ACS in bacteria. AcuA, a single GNAT domain-containing enzyme, acetylates ACS in B. subtilis (34), while in S. enterica, SePat, comprised of a GNAT domain fused to an ACS-like domain, acetylates the ε-NH2 group of lysine 609 of SeACS (15). However, the enzymatic function of the ACS-like domain has not yet been demonstrated. Consistent with the substrate profiling results, MsPat possesses a C-terminal GNAT domain exhibiting 27% sequence identity to that of SePat. In addition, MsPat has a unique N-terminal cAMP-binding domain that has not been identified in any other bacterial homologues.
We further confirm that ACS is the substrate of MsPat using the GNAT-activity based method with recombinant M. tuberculosis ACS (Rv3667). Our data shows that MsPat can mono-acetylate M. tuberculosis ACS, and that acetylation results in inactivation of the enzymatic activity. In addition, the acetyltransferase activity of MsPat is tightly controlled by cAMP binding, similar to eukaryotic protein kinase A, whose activity is regulated by a cAMP-binding regulatory subunit (40). Structural studies on several other cAMP-dependent enzymes indicate no common regulatory mechanism (41–44).
An essential biochemical hallmark of post-translational protein modification is the reversibility of the modification, whether it is phosphorylation or acetylation. We thus identified and purified Rv1151c, which is homologous to eukaryotic sirtuins, and showed that the protein deacetylates ACS in the presence of NAD+. This deacetylation is accompanied by reactivation of ACS. Orthologues of Rv1151c are also found in other mycobacteria. Thus, Pat and the deacetylase constitute a reversible acetylation system in mycobacteria that post-translationally controls the activity of ACS, affecting the de novo production of AcCoA from acetate, CoA and ATP.
The requirement of cAMP for ACS acetylation makes the regulation of the process more complicated. 3′,5′-Cyclic AMP, which is synthesized by multiple AC’s in different mycobacterial species, is an important second messenger (17). Notably, the activities of mycobacterial AC’s are known to be increased under various physiological conditions, including pH, bicarbonate, and fatty acids levels (18–21). However, the known targets of any downstream network of cAMP signaling pathway in mycobacteria was limited to Rv3676, a cAMP receptor protein that regulates the transcription of a large number of genes involved in cell wall synthesis, central metabolic pathways and antibiotic resistance (45). Our data suggests that MsPat is the first downstream enzymatic target of cAMP identified in mycobacteria, and regulates intracellular metabolism by post-translational modification of a central metabolic enzyme.
A burst of intracellular cAMP was recently reported following the infection of macrophages by M. tuberculosis (22, 23). The phagolyosome is an acidic, nutrient poor and oxidatively hostile environment, and the engulfed bacteria need to slow their metabolism to survive. By elevating intracellular cAMP levels, one consequence will be the activation of the mycobacterial Pat and inactivation of de novo AcCoA synthesis by ACS. As oxygen tension decreases, concentrations of NADH will build up, and pyruvate and isocitrate dehydrogenases will be inhibited, further decreasing the AcCoA levels and those of the TCA cycle intermediates. Under these conditions, the only source of AcCoA will be fatty acid oxidation, and the generation of glycolytic intermediates would require the glyoxylate shunt, which is essential for mycobacterial persistence (46). Whether the inactivation of ACS is sufficient for the organism to establish a non-replicative state leading to persistence, is under examination, but appears unlikely.
Recently it has been suggested that SePat also regulates the activities of three additional central metabolic enzymes, isocitrate dehydrogenase kinase/phosphatase, isocitrate lyase and glyceraldehydes 3-phosphate dehydrogenase through lysine acetylation in S. enterica, thus controlling flux through glycolysis versus gluconeogenesis as well as the branching between the TCA cycle and glyoxylate bypass (6). It is possible that the mycobacterial Pat also acetylates the orthologues of the three enzymes in mycobacteria, and plays additional roles in regulating the central carbon metabolic pathways.
In summary, our biochemical results demonstrate that MSMEG_5458 is a cAMP-dependent protein acetyltransferase that inactivates ACS through acetylation of a single, specific lysine residue, and that Rv1151c is an NAD+-dependent deacetylase that reactivates ACS. This acetylation/deacetylation system in mycobacteria is likely to sense the extracellular environment through cAMP levels, and also key intracellular metabolites, including NAD+ and AcCoA, since cAMP, AcCoA and NAD+ are required for acetylation and deacetylation, respectively. The mycobacterial ACS is the first M. tuberculosis enzyme that has been biochemically demonstrated to be inactivated by acetylation, and the reversibility of the acetylation suggests an important regulatory role for the enzyme. The number and types of other enzymes that may be similarly regulated is under investigation.
Supplementary Material
Acknowledgments
We are grateful to Myrarol Callaway and Edward Nieves in Albert Einstein College of Medicine for assistance in mass spectrometry.
Abbreviations
- AC
adenylate cyclase
- AcCoA
acetyl CoA
- ACS
acetyl CoA synthetase
- cAMP
3′,5′-cyclic adenosine monophophate
- ClAcCoA
chloroacetyl CoA
- GNAT
GCN5-related N-acetyltransferase
- NAD+
nicotinamide adenine dinucleotide
- Pat
protein acetyltransferase
- TAMRA
5(6)-carboxy-tetramethylrhodamine
Footnotes
This work was supported by NIH grant AI60899 (to J.S.B.).
SUPPORTING INFORMATION AVAILABLE
Oligonucleotides used in molecular cloning (Table S1), mass spectrometric analysis of a substrate of MsPat (Table S2), sequence alignment of MSMEG_5458 and Rv0998 (Figure S1), SDS-PAGE analysis of the purified proteins (Figure S2), characterization of the bound ligand of MsPat (Figure S3), the cAMP standard curve (Figure S4) and sequence alignment of sirtuin-like deacetylases (Figure S5). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vetting MW, LP SdC, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 4.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 5.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. The diversity of lysine-acetylated proteins in Escherichia coli. J Microbiol Biotechnol. 2008;18:1529–1536. [PubMed] [Google Scholar]
- 8.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Card GL, Peterson NA, Smith CA, Rupp B, Schick BM, Baker EN. The crystal structure of Rv1347c, a putative antibiotic resistance protein from Mycobacterium tuberculosis, reveals a GCN5-related fold and suggests an alternative function in siderophore biosynthesis. J Biol Chem. 2005;280:13978–13986. doi: 10.1074/jbc.M413904200. [DOI] [PubMed] [Google Scholar]
- 10.Errey JC, Blanchard JS. Functional characterization of a novel ArgA from Mycobacterium tuberculosis. J Bacteriol. 2005;187:3039–3044. doi: 10.1128/JB.187.9.3039-3044.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frankel BA, Blanchard JS. Mechanistic analysis of Mycobacterium tuberculosis Rv1347c, a lysine Nepsilon-acyltransferase involved in mycobactin biosynthesis. Arch Biochem Biophys. 2008;477:259–266. doi: 10.1016/j.abb.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hegde SS, Javid-Majd F, Blanchard JS. Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2′-N-acetyltransferase (AAC(2′)-Ic) from Mycobacterium tuberculosis. J Biol Chem. 2001;276:45876–45881. doi: 10.1074/jbc.M108810200. [DOI] [PubMed] [Google Scholar]
- 13.Nambi S, Basu N, Visweswariah SS. cAMP-regulated protein lysine acetylases in mycobacteria. J Biol Chem. 2010;285:24313–24323. doi: 10.1074/jbc.M110.118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vetting MW, Roderick SL, Yu M, Blanchard JS. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci. 2003;12:1954–1959. doi: 10.1110/ps.03153703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Padh H, Venkitasubramanian TA. Cyclic adenosine 3′, 5′-monophosphate in mycobacteria. Indian J Biochem Biophys. 1976;13:413–414. [PubMed] [Google Scholar]
- 17.Shenoy AR, Visweswariah SS. Mycobacterial adenylyl cyclases: biochemical diversity and structural plasticity. FEBS Lett. 2006;580:3344–3352. doi: 10.1016/j.febslet.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 18.Abdel Motaal A, Tews I, Schultz JE, Linder JU. Fatty acid regulation of adenylyl cyclase Rv2212 from Mycobacterium tuberculosis H37Rv. FEBS J. 2006;273:4219–4228. doi: 10.1111/j.1742-4658.2006.05420.x. [DOI] [PubMed] [Google Scholar]
- 19.Cann MJ, Hammer A, Zhou J, Kanacher T. A defined subset of adenylyl cyclases is regulated by bicarbonate ion. J Biol Chem. 2003;278:35033–35038. doi: 10.1074/jbc.M303025200. [DOI] [PubMed] [Google Scholar]
- 20.Dass BK, Sharma R, Shenoy AR, Mattoo R, Visweswariah SS. Cyclic AMP in mycobacteria: characterization and functional role of the Rv1647 ortholog in Mycobacterium smegmatis. J Bacteriol. 2008;190:3824–3834. doi: 10.1128/JB.00138-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tews I, Findeisen F, Sinning I, Schultz A, Schultz JE, Linder JU. The structure of a pH-sensing mycobacterial adenylyl cyclase holoenzyme. Science. 2005;308:1020–1023. doi: 10.1126/science.1107642. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal N, Lamichhane G, Gupta R, Nolan S, Bishai WR. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature. 2009;460:98–102. doi: 10.1038/nature08123. [DOI] [PubMed] [Google Scholar]
- 23.Bai G, Schaak DD, McDonough KA. cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol Med Microbiol. 2009;55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keppetipola N, Shuman S. A phosphate-binding histidine of binuclear metallophosphodiesterase enzymes is a determinant of 2′,3′-cyclic nucleotide phosphodiesterase activity. J Biol Chem. 2008;283:30942–30949. doi: 10.1074/jbc.M805064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 26.Yu M, de Carvalho LP, Sun G, Blanchard JS. Activity-based substrate profiling for Gcn5-related N-acetyltransferases: the use of chloroacetyl-coenzyme A to identify protein substrates. J Am Chem Soc. 2006;128:15356–15357. doi: 10.1021/ja066298w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang JK, Epstein W. Purification and characterization of adenylate cyclase from Escherichia coli K12. J Biol Chem. 1983;258:3750–3758. [PubMed] [Google Scholar]
- 28.Gao F, Yan X, Baettig OM, Berghuis AM, Auclair K. Regio- and chemoselective 6′-N-derivatization of aminoglycosides: bisubstrate inhibitors as probes to study aminoglycoside 6′-N-acetyltransferases. Angew Chem Int Ed Engl. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 29.Khalil EM, De Angelis J, Ishii M, Cole PA. Mechanism-based inhibition of the melatonin rhythm enzyme: pharmacologic exploitation of active site functional plasticity. Proc Natl Acad Sci U S A. 1999;96:12418–12423. doi: 10.1073/pnas.96.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams JW, Northrop DB. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase I. J Antibiot (Tokyo) 1979;32:1147–1154. doi: 10.7164/antibiotics.32.1147. [DOI] [PubMed] [Google Scholar]
- 31.Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 32.Miao L, Fang H, Li Y, Chen H. Studies of the in vitro Nalpha-acetyltransferase activities of E. coli RimL protein. Biochem Biophys Res Commun. 2007;357:641–647. doi: 10.1016/j.bbrc.2007.03.171. [DOI] [PubMed] [Google Scholar]
- 33.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem. 2000;275:22048–22055. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 34.Gardner JG, Escalante-Semerena JC. Biochemical and mutational analyses of AcuA, the acetyltransferase enzyme that controls the activity of the acetyl coenzyme a synthetase (AcsA) in Bacillus subtilis. J Bacteriol. 2008;190:5132–5136. doi: 10.1128/JB.00340-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD(+) involvement in Bacillus subtilis. J Bacteriol. 2006;188:5460–5468. doi: 10.1128/JB.00215-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 37.Crosby HA, Heiniger EK, Harwood CS, Escalante-Semerena JC. Reversible N epsilon-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol Microbiol. 2010;76:874–888. doi: 10.1111/j.1365-2958.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lammers M, Neumann H, Chin JW, James LC. Acetylation regulates cyclophilin A catalysis, immunosuppression and HIV isomerization. Nat Chem Biol. 2010;6:331–337. doi: 10.1038/nchembio.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trosky JE, Li Y, Mukherjee S, Keitany G, Ball H, Orth K. VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J Biol Chem. 2007;282:34299–34305. doi: 10.1074/jbc.M706970200. [DOI] [PubMed] [Google Scholar]
- 40.Krebs EG. Protein kinases. Curr Top Cell Regul. 1972;5:99–133. [PubMed] [Google Scholar]
- 41.Das R, Esposito V, Abu-Abed M, Anand GS, Taylor SS, Melacini G. cAMP activation of PKA defines an ancient signaling mechanism. Proc Natl Acad Sci U S A. 2007;104:93–98. doi: 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher DT, Smith N, Kim SK, Robinson H, Reddy PT. Profound asymmetry in the structure of the cAMP-free cAMP Receptor Protein (CRP) from Mycobacterium tuberculosis. J Biol Chem. 2009;284:8228–8232. doi: 10.1074/jbc.C800215200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 44.Reddy MC, Palaninathan SK, Bruning JB, Thurman C, Smith D, Sacchettini JC. Structural insights into the mechanism of the allosteric transitions of Mycobacterium tuberculosis cAMP receptor protein. J Biol Chem. 2009;284:36581–36591. doi: 10.1074/jbc.M109.041343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rickman L, Scott C, Hunt DM, Hutchinson T, Menendez MC, Whalan R, Hinds J, Colston MJ, Green J, Buxton RS. A member of the cAMP receptor protein family of transcription regulators in Mycobacterium tuberculosis is required for virulence in mice and controls transcription of the rpfA gene coding for a resuscitation promoting factor. Mol Microbiol. 2005;56:1274–1286. doi: 10.1111/j.1365-2958.2005.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.