

Abstract
Metabolomics provides a readout of the state of metabolism in cells or tissue and their responses to external perturbations. For this reason, the approach has great potential in clinical diagnostics. Clinical metabolomics using stable isotope resolved metabolomics (SIRM) for pathway tracing represents an important new approach to obtaining metabolic parameters in human cancer subjects in situ. Here we provide an overview of the technology development of labeling from cells in culture and mouse models. The high throughput analytical methods NMR and mass spectrometry, especially Fourier transform ion cyclotron resonance, for analyzing the resulting metabolite isotopomers and isotopologues are described with examples of applications in cancer biology. Special technical considerations for clinical applications of metabolomics using stable isotope tracers are described. The whole process from concept to analysis will be exemplified by our on-going study of nonsmall cell lung cancer (NSCLC) metabolomics. This powerful new approach has already provided important new insights into metabolic adaptations in lung cancer cells, including the upregulation of anaplerosis via pyruvate carboxylation in NSCLC.
Introduction
Lung cancer
Lung cancer remains the biggest killer of men and women of all cancers in the West, and imposes huge health and economic burdens. There are approximately 170,000 deaths annually from all lung cancers in the United States alone (ACS, 2008). As for many other carcinomas, the seeds of the disease are sown early, but the clinical presentation is delayed many years (the median age is about 63 years for both men and women). Therefore, despite the decrease in smoking incidence over the last 20–30 years, the number of cases is still high. As in the early stages the disease largely asymptomatic, most patients present with advanced and incurable disease. The 5-year survival rate is only 17% overall, and has changed little over the past 4 decades. Unfortunately, screening modalities including sputum analysis and chest roentgenography or computed tomography of the chest have yet to demonstrate improved overall survival in study populations (http://www.cancer.gov/cancertopics/factsheet/detection/spiral-ct-lung). Nevertheless, for those subjects whose disease is detected early (stage 1) and who are suitable for surgical resection, have a much higher 5-year survival rate (ca. 70–80%), although recurrence after that period remains substantial (ACS, 2008). Hence, a crucial and urgent challenge to reducing the fatality rate of lung cancers is the development of routine methodologies that not only enable early detection (Boyer et al., 1998) but also distinguish malignant from benign tumors. This has been a major obstacle to realizing the benefit of early detection for improving survival for lung cancer and other cancers alike (Editorial, 2009).
Therefore improved diagnostic tools for early detection of lung cancer are urgently needed, as are much better therapeutic strategies for treating advanced or recurrent disease. In an attempt to get a deeper understanding of the fundamental biochemistry of lung cancer, we have been applying a stable isotope tracer approach to discovering the functional differences between normal lung tissue and lung cancer at different stages.
Metabolic requirements for cancer growth
Uncontrolled growth is a universal trait of tumor cells, which requires profound changes in cellular metabolism to sustain the additional energy and biosynthetic precursor demands of proliferation. Accelerated aerobic glycolysis is one such correlate of malignant transformation, which was first described more than 80 years ago by Warburg (1956). Despite the dramatic upregulation of glycolysis in many cancer cells (DeBerardinis et al., 2007; Fan et al., 2005, 2008; Thornburg et al., 2008; Wong et al., 2004), this process alone is insufficient to provide the necessary precursors for anabolic metabolism; they must be supplied by additional metabolic processes. Several of the Krebs cycle metabolites, citrate, oxaloacetate/aspartate, and α-ketoglutarate/glutamate are respectively precursors for the biosynthesis of fatty acids, nucleic acids, and proteins (Nelson and Cox, 2005), all of which are required for growth. As some of these metabolites (e.g., oxaloacetate and α-ketoglutarate) are maintained at low cellular concentration, they must be replenished via anaplerosis to sustain both the Krebs cycle and biosynthetic activities. The two principal anaplerotic pathways involve pyruvate carboxylation (Fan et al., 2008) and glutaminolysis (DeBerardinis et al., 2008; Mazurek and Eigenbrodt, 2003; Mazurek et al., 2000, 2005; Wong et al., 2004). The relative importance of these two pathways appears to be tissue specific (DeBerardinis et al., 2007; Fan and Lane, 2008; Fan et al., 2009; Portais et al., 1993).
Terminally differentiated cells maintain a basal metabolism devoted largely to cellular homeostasis, repair, and transport of nutrients and waste products into and out of the cell. Transport makes use of ion gradients, which are ultimately driven by the hydrolysis of ATP. For a cell to divide, it must double its biomass, which is roughly 65% protein, 19% lipid, 13% carbohydrate, and 3% nucleic acid. The assembly of biopolymers is highly endergonic; energy metabolism be upregulated; 1 mg biomass is equivalent to roughly 33 μmol carbon from proteins, 11 μmol from lipids, 4 μmol from carbohydrates, and 0.9 μmol carbon from nucleic acids. In mammalian cells, approximately 1 mg biomass comprises 30–35 μmol carbon such as is available from glucose (i.e., 5–6 μmol) or anabolic amino acids. Using textbook values for the number of ATP equivalents needed to synthesize biopolymers (Nelson and Cox, 2005), the creation of 1 mg biomass requires at least 30 μmol carbon from biosynthetic routes and at least 30 μmol ATP to drive the anabolic reactions, although complete oxidation of 1 μmol glucose to CO2 can generate up to 32 μmol ATP (using P:O = 2.5 and 1.5 for NADH and FADH2, respectively) including two from glycolysis and five from the glycolytically generated NADH. In fact, tumor cells typically convert more than one-third or more of the glucose they consume into excreted lactate (DeBerardinis et al., 2007; Fan et al., 2008), which removes not only the carbon that could have been oxidized by the mitochondria, but also the NADH generated by glycolysis. In practise, this means that oxidation of glucose produces much less ATP than the theoretical maximum of 32 mol/mol glucose. Amino acids and fatty acids can also be oxidized if the mitochondria are functional. Fatty acid oxidation can provide copious quantities of ATP, but the carbons cannot be used for anabolic purposes. Glutamine via glutaminolysis is an important source of anabolic carbon (Mazurek and Eigenbrodt, 2003), and depending on the details of how it is oxidized, may provide modest amounts of ATP. Note that such oxidations absolutely require functioning mitochondria, even at low oxygen tension. Fortunately for cancer cells, the terminal oxygen acceptor cyt c oxidase, has a high affinity for oxygen (Gnaiger et al. 1998).
Why stable isotopes?
The variation in concentration of metabolites inside cells in response to changes in conditions is generally relatively small (“metabolic homeostasis”), largely as a consequence of the extensive interacting network of reactions that are coupled by cycles of feedback and feed-forward regulation (Gunawardena, 2010; Nelson and Cox, 2005). Furthermore, owing to compartmentation in eukaryotic cells, the actual pool sizes of metabolites in the different compartments are neither known nor routinely measured. Only the total amount of metabolites is generally measured, which is normalized to some standard proportional to tissue quantity.
Figure 1 shows a simplified metabolic scheme that represents a truncated form of glycolysis in which there are two inputs (glucose or G and B) and multiple outputs (A, C, and lactate, or Lac). This simple scheme immediately shows why concentration measurements alone are inadequate, as they provide no information about flux, and as the same metabolites are used in multiple “pathways” it is not possible to determine the origins of the metabolite. At steady state, for this metabolic scheme, the rates of production of the products are as follows:
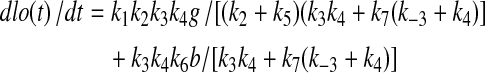 |
(1A) |
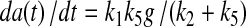 |
(1B) |
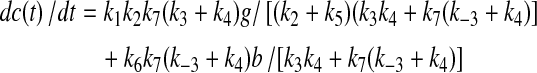 |
(1C) |
FIG.1.

Simple metabolic scheme. G represents extracellular glucose, which is immediately phosphorylated by hexokinase to G6P in the cell. Laco and Laci represent extracellular (excreted) and intracellular lactate, respectively. The apparent rate constants, k, for metabolic transformations are functions of enzyme concentrations. In the limit of Michaelis-Menten kinetics, ki = (kcat/Km)iei[1−p/sKeq], where ei is the free enzyme concentration, p and s are the product and substrate concentrations catalyzed by the enzyme and Keq is the equilibrium constant for the reaction (Roberts et al., 1985).
Under constant supply of G and B, the time courses of the products are linear. However, the flux control under these conditions may be distributed and complex, and change with the conditions. If glucose depletes exponentially, g(t) = g(0)exp(−k1t), then the terms in g also decrease exponentially over time, whereas those in b remain constant; this can lead to a switch in the metabolic profile. Furthermore, the concentrations of products now vary nonlinearly with time.
This problem is compounded in organisms where there is communication between cells within a tissue and between different organs. However, if the system is minimally perturbed chemically, by using tracers, then not only can fluxes be measured from the changes in isotopic enrichment, but also which segments of a network are directly being utilized. In principle, the tracer can be any appropriate version of a natural metabolite that can be independently measured; traditionally this was achieved using radioisotopes. Stable isotopes offer several advantages, not only via their biocompatibility and nonhazardous nature, but also because stable isotopes such as 13C or 15N are readily distinguishable from their more abundant natural isotopes (12C and 14N) both by NMR and by mass spectrometry. These two techniques are sensitive, selective and can provide an enormous amount of information about the degree and which atoms are enriched from a particular precursor, even within a complex mixture (Fan and Lane, 2008; Fan et al., 1986; Lane and Fan, 2007; Lane et al., 2008a, 2009a). By using multidimensional techniques with isotope editing (NMR), or ultrahigh mass resolution, it is not necessary to separate the components from the rich mixture of compounds present in a cell extract or biofluid. The resolution is provided by the analytical tool itself, and thus avoids the many complications of chromatographic separations (Fan et al., 2008; Lane et al., 2008a).
Applications
Cells
In studies of cells in culture, the simplest assumption is that the inputs are constant (i.e., do not deplete) and that there is no change in the number of cells. In practice, one usually measures the total amount of metabolite X, so that if the cell mass increases during the experiment, then the number of enzyme molecules present also increases. In exponential growth phase, the rate constants are also exponential functions of time. Furthermore, rapidly proliferating cancer cells that have a high demand for glucose deplete glucose significantly over the time course of the experiment. Chemostats (Wick et al., 2002; Zhong et al., 2004) have not been developed for mammalian cells.
Figure 2 shows the transformation of 13C glucose into uniformly 13C-labeled lactate (13C3-lactate) by cultured cancer cells, each subjected to four different treatment conditions, control, 5 μM methylseleninic acid (MSA), 6.25 μM selenite (SeO3), and 500 μM selenomethionine (SeM). Relative to the control, all three Se compounds suppressed the concentration of intracellular 13C3-lactate after 24 h of treatment, with the MSA treatment having the most pronounced effect. Because 13C3-lactate is largely derived from 13C glucose via glycolysis, these results indicate that Se compounds inhibit glycolysis to a varying degree.
FIG. 2.

Lactate production from 13C glucose and release into the medium was altered by different Se treatments. A549 cells were grown in RPMI containing 0.2% [U-13C]-glucose in the absence or presence of 5 μM methylseleninic acid (MSA), 6.25 μM selenite (SeO3), or 500 μM selenomethionine (SeM) for 24 h. Media metabolites were measured by 1D 1H NMR. Methyl region showing the central resonance of 1H attached to 12C and the two 13C satellites of lactate. The splitting pattern of the 13C satellites indicates the dominance of the uniformly 13C-labeled lactate isotopomer (13C3-lactate). MSA treatment caused a greater reduction in the release of 13C3-lactate than the other two Se treatments, relative to the control.
Figure 3 shows the utilization of 13C glucose and the excretion of 13C lactate from cultured cancer cells over a period of more than three cell doublings. During this period, the glucose concentration decreased approximately 80% (Fig. 3A). The time dependence of the concentration of glucose in the medium can be approximated according to:
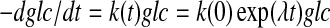 |
(2) |
FIG. 3.

Time course of glucose consumption and lactate secretion into the medium. MDAMB231 cells were grown in RPMI containing 0.2% [U-13C]-glucose. Media metabolites were measured by 1D 1H NMR. (A) • 13C glucose; ▪ 13C Lac; □ 12C Lac; ○ valine. The continuous lines are regression fits to equations described in the text with specific growth rates (λ) of 0.07 h−1. R2 for glucose and 13C lactate were 0.996 and 0.998, respectively. The rate of change of valine was not significantly different form zero, and the rate of 13C lactate production was 0.01 mM h−1. (B) Fraction of 13C labeled lactate in the medium (○) and the fraction of 13C glucose consumed that was converted to excreted 13C lactate (•).
here λ is related to the (exponential) growth rate of the cells, rendering the glucose uptake/utilization rate constant an exponential. The solution to Eq. (2) is simply:
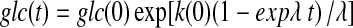 |
(3) |
Furthermore, the rate of production of 13C lactate is not linear but increases with time (the time course is concave up) (Fig. 2B). Such time dependences of sources are directly relevant to in vivo experiments, where holding the source concentration constant is not always possible (e.g., in bolus administration).
Other information available from this experiment, where just glucose input and lactate excretion are concerned, is the 13C enrichment of the excreted lactate, and the fraction of the glucose that was consumed converted into lactate (Fig. 3B). The enrichment into lactate in this experiment reached a plateau value of about 92%, implying that most of the lactate produced by these cells derived directly from glucose, and not from other unlabeled sources (collectively only 8%). The fraction of glucose consumed that was converted to lactate reached about 38 % (Fig. 2B) in the MDA-MB-231 cells, which is comparable to other cancer cell lines we have studied (Fan et al., 2008; Lane et al., 2009a, 2009b). Without labeling this information is impossible to obtain.
Measuring input and output alone is not sufficient to delineate specific intracellular network dynamics. The cells can be harvested at various time points post labeling, and the fraction enrichment in 13C can be determined in numerous metabolites in the same way. To improve the reliability of identification and of quantifying positional isotopomers, we have developed a set of tools based on proton detection of carbon for quantitative purposes (Fan and Lane, 2008; Lane and Fan, 2007; Lane et al., 2008a, 2009b).
Figure 4 shows a 2D TOCSY experiment in which 13C-labeled metabolites are identified from the patterns of crosspeaks in the spectrum, and the degree of enrichment at specific atoms can be calculated from the volumes of the “satellite” peaks, that are due to the presence of 13C. 13C glucose led to the production of labeled lactate, Ala, Glu, the Glu of reduced glutathione, and the ribose moiety of adenine (5′AXP) and uracil nucleotides (5′ UXP) (Fig. 4A). The pattern of satellite crosspeaks for Glu/GSH is typical of scrambling through the Krebs cycle (Fan et al., 2008; Lane and Fan, 2007; Lane et al., 2008a, 2008b), implying functional mitochondria in these cancer cells. When 13C Gln was the source (Fig. 4B), intracellular Lac was not significantly labeled, Ala and the ribose moiety of AXP and UXP were not at all labeled, and Glu/GSH were labeled very strongly. This is very different from the case with 13C-glucose as the source. Furthermore, the excreted lactate showed no significant labeling compared to the 13C glucose experiment (data not shown). The scrambling in the Glu peaks was minor, showing that Gln is a preferred carbon source for Glu and GSH, but glucose is the preferred carbon source for Lac and Ala and ribose. Thus, glucose is essential for de novo biosynthesis of the ribose moiety of nucleotides and that gluconeogenesis was not functional in these cells. In contrast, although 13C glucose provides some carbon for pyrimidine ring biosynthesis, 13C Gln labeled the 5,6 position of the uracil ring much more heavily, indicating a major anabolic role for Gln.
FIG. 4.

TOCSY Spectra: effect of 13C source. A549 cells were cultured in RPM1 containing either 10 mM [U-13C]-glucose + 2 mM 12C Gln or 10 mM 12C glucose + 2 mM [U-13C]-Gln. The cells were harvested and the polar metabolites were extracted. TOCSY spectra were recorded at 14.1 T and 293 K using an isotropic mixing time of 50 ms. (A) 13C glucose; (B) 13C Gln. Green boxes connect the 13C satellite peaks and red boxes trace the 1H covalent linkages of various metabolites.
SIRM in the Mouse Model
Cells are very valuable for developing technology under controlled conditions and for discovering the repertoire of biochemical responses that are available to a particular cell type. In living tissue, however, cells are in contact with neighboring cells that may be of the same type (e.g., in epithelial sheets) or of fundamentally different types such as supporting fibroblasts, immune cells, blood, or lymph vessel endothelial cells, and so forth. Cells communicate with one another; the metabolic cooperation between neurons and astrocytes is well established (Fan et al., 2010). Therefore, it is necessary also to study the cells in a more realistic tissue context, for which the xenografted human tumor in the severe combined immunodeficiency (SCID) mouse is a good example. Human tumors or cell lines can be propagated in SCID mice either ectopically (e.g., as a subcutaneous tumor), or better in an orthotopic environment. We have xenografted human lung tumor cells in mouse lungs. Although the xenograft is not in an identical environment as in the human case, nevertheless the tumor mass is three dimensional with a full complement of cell types, less well vascularized than surrounding tissue, and relies on blood for nutrient supply. The degree of experimental control in the mouse model is less than in cell culture but considerably greater than in human subjects. Therefore, the mouse model can be regarded as a surrogate for the human tumor microenvironment, which can translate cell culture details to understanding of tumor metabolism in the microenvironmental context. It is our contention that measurements on tumors cannot be unambiguously interpreted unless the biochemical behavior of the cells is understood.
Figure 5 shows NMR spectra of extracts of different tissues from SCID mice 15 min after tail vein injection of [U-13C]-glucose. These spectra are 13C-edited, and thus detect only those protons attached to 13C derived from the bolus of 13C glucose: it thus represents a snapshot of tissue-specific metabolism in the 15-min time period. As expected, the 13C metabolic profiles of the various tissues are distinct, and thus reflects the very different metabolic activities various cell types in these tissues.
FIG. 5.

Mouse U-13C glucose. SCID mice were injected with [U-13C]-glucose via the tail vein, and tissues were harvested 15 min later. 1H-{13C} HSQC spectra were recorded at 14.1 T 20°C. The spectra show the different relative labeling patterns of metabolites derived from glucose within 15 min. Left blood plasma at 1and 30 min showing mainly glucose (1 min) and glucose plus lactate (30 min). Right: tissues as shown on the figure.
SIRM Analysis in Human NSCLC
We have extended the application of such 13C-tracer and isotopomer approaches to human lung cancer patients (Fan et al., 2009; Lane et al., 2008a, 2009b). The stable isotope tracer [U-13C]-glucose was administered intravenously into recruited patients prior to surgical resection of the primary tumor and surrounding noncancerous tissues. The 13C tracer is fully compatible with human experimentation—there are no readily detectable biological effects of the heavy isotope, and the amount of glucose injected is about 20% of the amount that is used in the glucose tolerance test. Table 1 summarizes the statistics of subjects in the SIRM study of NSCLC at Louisville.
Table 1.
Statistics of NSCLC Samples Collected for Metabolomics at Louisville
| Factor | Number or % |
|---|---|
| Total N (surgical) | 73 |
| N (13C) | 43 |
| N (no 13C) | 30 |
| Gender | 54% M, 46% F |
| Median, mean age (years) | 62; 63 ± 9 |
| Ethnicity | 70% Caucasian, 25% African American, 5% other |
| Stage | 67% I, 19% II, 14% III + IV |
| subtype | 29% AdenoC, 42% SquamousCC, 4% BAC, 25% large cell + adenosquamous |
| Plasma from healthy controls | 24 |
In our protocol, developed over 4 years, a consented subject with operable NSCLC is infused with [U-13C]-glucose preoperatively approximately 3 h prior to the time of resection. A blood sample is collected immediately prior to and following glucose infusion (which takes 5–6 min including a saline flush). The blood samples are immediately placed on ice, and then centrifuged to separate red cells from plasma. Aliquots of whole blood and plasma samples are flash frozen in lN2 for storage, within 30 min of venipuncture. Figure 6 shows stages in the tissue harvesting in the operating room. To minimize ischemia, tumorous tissue is wedge resected whenever compatible with patient care. The tumor is exposed and for NSCLC the margins can be identified visually by the surgeon. Two to three small slices of the tumor are blotted, and then freeze clamped in lN2 (Fig. 6). Slices of nontumorous lung from the same lobe, at least 2 cm from the tumor margins, are similarly treated. Additional pieces of tissue are blotted, and placed in formalin, OPT or fresh frozen for subsequent pathological examination.
FIG. 6.

Resecting a nonsmall cell lung tumor. Upper left: stapling the blood vessels; upper right: collection into an endo bag; middle left taking tumor tissue (white) and nontumor lung tissue from the excised lung; middle right: cutting tumor for pathology samples and freezing; lower left: freeze clamping tumor slice in liquid nitrogen; lower right: blotting tissue for pathology samples. The time between resection and freezing was <5 min.
The time between resection and tissue freezing is less than 5 min, which preserves the metabolic state of the tissue. Frozen tissue can be stored essentially indefinitely at lN2 temperature. Postoperative blood and urine samples are taken and treated as for the preoperative blood, to provide an overview of the glucose uptake and utilization in terms of 13C lactate production, in the plasma and 13C glucose excretion in urine.
Differences in metabolic pathways between paired noncancerous lung and cancer tissues were profiled using NMR and MS. This approach enabled analysis of distinct metabolic traits of cancer tissues without interferences from either intrinsic (e.g., genetic) or external environmental factors (e.g., diet) because the patient's own noncancerous tissue served as internal control. Figure 7 summarizes the flow of 13C atoms from 13C glucose through glycolysis into the Krebs cycle, and the fate of the precursors as they are incorporated into observable metabolites. Compared with noncancerous lung tissues, lung tumors demonstrated an enhanced capacity for glycolysis while displaying other distinct metabolic activities either unexpected or previously unknown. In particular, lung tumor tissues exhibited an altered but full Krebs cycle activity and enhanced pyruvate carboxylation, which could play a pivotal role in replenishing anabolic precursors required by tumor growth (Fan et al., 2009). This anabolic trait could also be fundamental to other human cancers (Fan et al., 2008).
FIG. 7.

Flow of 13C atoms from [U-13C]-glucose into detectable metabolites. 13C atoms from glucose (red) enter pyruvate and alanine by glycolysis, and then into Krebs cycle makers via the pyruvate dehydrogenase or pyruvate carboxylase reactions, and ultimately into macromolecules.
Plasma lipids from NSCLC patients
In addition to the polar metabolites, the lipid species can also be readily examined by both NR and MS. Methanol or chloroform/methanol mixtures extract the polar and nonpolar lipids, respectively, which can be analyzed as a mixture by both NMR and FT-ICR-MS (Lane et al., 2009b), again with no chromatographic separation. Mass isotopologue analysis by direct infusion FT-ICR-MS analysis combined with appropriate software (Precalculated Exact Mass Isotopologue Search Engine or “PREMISE”) for identifying the thousands of species present makes it possible to determine the synthesis and turnover of the acyl chains and backbones of the PL simultaneously (Lane et al., 2008a, 2009b). Lipid vesicles of various kinds are present at higher concentrations in cancer-bearing patients than healthy individuals (Rabinowits et al., 2009; Taylor et al., 2008). Figure 8 shows FT-ICR MS spectra of lipids extracted from lipid vesicles present in the plasma of adults with or without lung cancer (Fig. 8B), compared with lipid extracts from lung cancer and non tumors lung tissue (matched pairs) from three NSCLC patients (Fig. 8A). The ultrahigh resolution makes it possible to discriminate between lipids of very similar mass (e.g., #1 and #2 in Fig. 8A) Lipids 1 and 2 clearly differ between the cancerous and noncancerous tissues from the same patients. The same lipids in the plasma microvesicle fraction show even more radical differences between NSCLC and healthy individual (Fig. 8B).
FIG. 8.

FT-ICR MS spectra. Lipids were solvent extracted from microvesicles of plasma and lung tissue, freeze dried, and redissolved in methanol. FT-ICR-MS spectra were recorded at 7 T on a Thermo LTQ-FT MS spectrometer by direct infusion. (A) Expanded region of the phospholipids extracted from normal and NSCLC tissue from three patients. (B) Lipids extracted from microvesicles obtained from the plasma of the same lung cancer patients and three control subjects (no history of cancer).
Conclusions and Future Directions
The following points can be made about the value of stable isotope resolved metabolomics (SIRM), and for the future of clinical metabolic analysis. (1) stable isotope resolved metabolomics (SIRM) is the key to tracing pathway and fluxes in cells, animals and humans, in situ. (2) Energy and anabolic metabolism are both increased in cultured lung cancer cells and in NSCLC compared with normal lung. (3) Correlation of SIRM-based metabolic differences with gene expression data, enzyme activity crossvalidated findings. This was exemplified by the novel finding of PC activation in NSCLC, which is an important anaplerotic enzyme (Fan et al., 2009) and as we have subsequently verified with a larger number of samples. (4) Further mechanistic SIRM studies are being carried out in an orthotopic SCID mouse model bearing human lung cancer cells or primary human tumors. This approach will help bridge the gap between cell culture and in situ human studies.
Correlations with clinical outcomes and pathology should lead to improved tools for diagnosis (e.g., early biomarkers) and drug response prediction, as well as discovery of new molecular targets.
Acknowledgments
The study was supported in part by NIH Grant Numbers P20RR018733 from the National Center for Research Resources, 1R01CA118434-01A2, 3R01CA118434-02S1, the National Science Foundation EPSCoR Grant #EPS-0447479, the Robert W. Rounsavall Jr, Family Foundation, the Kentucky Challenge for Excellence, and Drive Cancer Out Campaign, Louisville. We thank Jin Lian Tan, Melissa Barousse, for expert technical assistance, and Drs. Pawel Lorkiewicz and S. Arumugam for mass spectrometry and NMR.
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
- Cancer Facts and Figures—2008. American Cancer Society; Boston, MA: ACS. (2008) pp. 1–72. [Google Scholar]
- Boyer M.J. Tannock I.F. Cellular and molecular basis of chemotherapy. In: Tannock I.F, editor; Hill R.P., editor. The Basic Science of Oncology. McGraw Hill; New York: 1998. pp. 350–369. [Google Scholar]
- Deberardinis R.J. Mancuso A. Daikhin E. Nissim I. Yudkoff M. Wehrli S., et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis R.J. Sayed N. Ditsworth D. Thompson C.B. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Editorial. Early warnings. Nature. 2009;458:679–679. doi: 10.1038/458679a. [DOI] [PubMed] [Google Scholar]
- Fan T. Bandura L. Higashi R. Lane A. Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer cells. Metabolomics. 2005;1:1–15. [Google Scholar]
- Fan T.W. Lane A.N. Structure-based profiling of metabolites and isotopomers by NMR. Prog NMR Spectrosc. 2008;52:69–117. [Google Scholar]
- Fan T.W. Lane A.N. Higashi R.M. Farag M.A. Gao H. Bousamra M. Miller D.M. Altered regulation of metabolic pathways in human lung cancer discerned by 13c stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T.W.M. Higashi R.M. Lane A.N. Jardetzky O. Combined use of proton NMR and gas chromatography-mass spectra for metabolite monitoring and in vivo proton NMR assignments. Biochim Biophys Acta. 1986;882:154–167. doi: 10.1016/0304-4165(86)90150-9. [DOI] [PubMed] [Google Scholar]
- Fan T.W.-M. Kucia M. Jankowski K. Higashi R.M. Rataczjak M.Z. Rataczjak J., et al. Proliferating rhabdomyosarcoma cells shows an energy producing anabolic metabolic phenotype compared with primary myocytes. Mol Cancer. 2008;7:79. doi: 10.1186/1476-4598-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T.W.-M. Yuan P. Lane A.N. Higashi R.M. Wang Y. Hamidi A., et al. Stable isotope resolved metabolomic analysis of lithium effects on glial–neuronal interactions. Metabolomics. 2010;6:165–179. doi: 10.1007/s11306-010-0208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnaiger B. Lassnig B. Kuznetsov A. Rieger G. Margreiter R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol. 1998;201:1129–1139. doi: 10.1242/jeb.201.8.1129. [DOI] [PubMed] [Google Scholar]
- Gunawardena J. Biological systems theory. Science. 2010;328:581. doi: 10.1126/science.1188974. [DOI] [PubMed] [Google Scholar]
- Lane A.N. Fan T.W. Quantification and identification of isotopomer distributions of metabolites in crude cell extracts using 1H TOCSY. Metabolomics. 2007;3:79–86. [Google Scholar]
- Lane A.N. Fan T.W. Higashi R.M. Isotopomer-based metabolomic analysis by NMR and mass spectrometry. Biophys Tools Biol. 2008a;84:541–588. doi: 10.1016/S0091-679X(07)84018-0. [DOI] [PubMed] [Google Scholar]
- Lane A.N. Fan T.W-M. Higashi R.M. Stable isotope assisted metabolomics in cancer research. IUBMB Life. 2008b;60:124–129. doi: 10.1002/iub.17. [DOI] [PubMed] [Google Scholar]
- Lane A.N. Fan T.W. Higashi R.M. Tan J. Bousamra M. Miller D.M. Prospects for clinical cancer metabolomics using stable isotope tracers. J Exp Mol Pathol. 2009a;86:165–173. doi: 10.1016/j.yexmp.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane A.N. Fan T.W.-M. Xie X. Moseley H.N. Higashi R.M. Stable isotope analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal Chim Acta. 2009b;651:201–208. doi: 10.1016/j.aca.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek S. Eigenbrodt E. The tumor metabolome. Anticancer Research. 2003;23:1149–1154. [PubMed] [Google Scholar]
- Mazurek S. Grimm H. Oehmke M. Weisse G. Teigelkamp S. Eigenbrodt E. Tumor M2-PK and glutaminolytic enzymes in the metabolic shift of tumor cells. Anticancer Res. 2000;20:5151–5154. [PubMed] [Google Scholar]
- Mazurek S. Boschek C.B. Hugo F. Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Nelson D.L. Cox M.M. Lehninger Principles of Biochemistry. W.H. Freeman and Company; New York: 2005. [Google Scholar]
- Portais J.-C. Schuster R. Merle M. Canioni P. Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon (1–13C)glucose incubation. Eur J Biochem. 1993;217:457–468. doi: 10.1111/j.1432-1033.1993.tb18265.x. [DOI] [PubMed] [Google Scholar]
- Rabinowits G. Gercel-Taylor C. Day J.M. Taylor D.D. Kloecker G.H. Exosomal MicroRNA: a diagnostic marker for lung cancer. Clin Lung Cancer. 2009;10:42–46. doi: 10.3816/CLC.2009.n.006. [DOI] [PubMed] [Google Scholar]
- Roberts J.K.M. Lane A.N. Clark R.A. Nieman R.H. Relationships between the rate of synthesis of atp and the concentrations of reactants and products of Atp hydrolysis in maize root-tips, determined by P-31 nuclear magnetic-resonance. Arch Biochem Biophys. 1985;240:712–722. doi: 10.1016/0003-9861(85)90080-3. [DOI] [PubMed] [Google Scholar]
- Taylor D.D. Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- Thornburg J.M. Nelson K.K. Lane A.N. Arumugam S. Simmons A. Eaton J. W., et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wick L.M. Weilenmann H. Egli T. The apparent clock-like evolution of Escherichia coli in glucose-limited chemostats is reproducible at large but not at small population sizes and can be explained with Monod kinetics. Microbiology-Sgm. 2002;148:2889–2902. doi: 10.1099/00221287-148-9-2889. [DOI] [PubMed] [Google Scholar]
- Wong M.S. Raab R.M. Rigoutsos I. Stephanopoulos G.N. Kelleher J.K. Metabolic and transcriptional patterns accompanying glutamine depletion and repletion in mouse hepatoma cells: a model for physiological regulatory networks. Physiol Genomics. 2004;16:247–255. doi: 10.1152/physiolgenomics.00088.2003. [DOI] [PubMed] [Google Scholar]
- Zhong S.B. Khodursky A. Dykhuizen D.E. Dean A.M. Evolutionary genomics of ecological specialization. Proc Natl Acad Sci USA. 2004;101:11719–11724. doi: 10.1073/pnas.0404397101. [DOI] [PMC free article] [PubMed] [Google Scholar]