

Abstract
Tumor resistance to chemotherapy in advanced breast cancer is a major impediment to treatment success. Resistance can be induced by the drugs themselves or result from the action of internal factors. The role of hormones in chemoresistance has received little attention. This article focuses on two classes of hormones: lactogens and estrogens. Lactogens include prolactin, growth hormone and placental lactogen, all of which can activate the prolactin receptor. Estrogens include endogenous steroids and nonsteroidal compounds from the environment termed endocrine disruptors, all of which can activate ‘classical’ estrogen receptors (ERα and ERβ), as well as other types of receptors. Both lactogens and estrogens antagonize cytotoxicity of multiple chemotherapeutic agents through complementary mechanisms. The implications of chemoresistance by these hormones to patients with breast cancer, and the potential benefits of developing combinatorial anti-lactogen/anti-estrogen treatment regimens, are discussed.
Keywords: bisphenol A, breast cancer, chemoresistance, cisplatin, endocrine disruptors, endogenous estrogens, growth hormone, placental lactogen, prolactin, prolactin receptor
The overarching goal of therapy in breast cancer is to suppress the growth of primary tumors and eliminate cancer cells that have metastasized. Responsiveness to hormones, which is a hallmark of early-stage breast cancer, has been exploited in the successful application of antihormone therapy. However, for patients with hormone-insensitive tumors and for those with advanced metastatic disease, chemotherapy is the mainstay of treatment. Despite an initial ability of many chemotherapeutic regimens to curb tumor growth and metastases, tumor resistance presents a major obstacle that results in treatment failure. Resistance can be induced by the drugs themselves or caused by the actions of internal and external factors. Drug resistance involves multiple cellular components, including membrane transporters, detoxification enzymes, anti-apoptotic proteins, tumor-suppressor genes and DNA-repair mechanisms [1].
Both estradiol (E2) and prolactin (PRL) are well recognized for their roles in the pathogenesis of breast cancer [2,3], but their association with chemoresistance has only recently become appreciated [4-6]. PRL is a member of the family of lactogens, physiologically defined by their ability to affect the production and secretion of milk. In humans, lactogens include three closely-related protein hormones: human PRL (hPRL), human growth hormone (hGH) and human placental lactogen (hPL). Estrogens are physiologically defined by their ability to develop and maintain the female reproductive system. Estrogenic compounds include naturally occurring steroids, as well as a variety of nonsteroidal compounds from the environment, which are termed endocrine disruptors.
Although lactogens and estrogens differ in their chemical structure, receptor characteristics and primary mode of action, they have several features in common, such as multiple sites of origin and promiscuous receptors that bind to more than one ligand. There is also cross-talk between estrogen and PRL receptors [5], which inevitably affects the therapeutic applications in breast cancer. The purpose of this article is to summarize recent evidence that lactogens and estrogens confer resistance against multiple chemotherapeutic agents, compare the mechanisms by which they exert anti-cytotoxicity and discuss clinical implications for patients with breast cancer.
Human lactogens
The hPRL gene is located on chromosome 6 and is expressed in the pituitary as well as in multiple extrapituitary sites, including the decidua, myometrium, breast and prostate [7]. The GH/PL gene cluster is located on chromosome 17 and encodes five proteins with high sequence homology: GH-N (normal), GH-V (variant) and three placental lactogen variants (PL-A, PL-B and PL-L). GH-N (also called GH-1) is expressed in the pituitary, while the other four proteins are the products of the placenta [8]. Alternative splicing generates additional isoforms of each placental hormone, thereby increasing their functional diversity. Similar to PRL, there is evidence for extrapituitary GH production in several normal and malignant tissues [9].
The three lactogens have a similar tertiary structure and overlapping functions. Each protein is composed of a single polypeptide chain of 191–199 amino acids that forms a four anti-parallel helical bundle held by disulfide bridges [10]. Unique to humans, all three lactogens bind the PRL receptor (PRLR). Thus, in addition to its cognate receptor (GHR), hGH binds to the PRLR, whereas nonprimate GH binds only to the GHR. PRL in any species binds only to the PRLR but not to GHR, while hPL does not have a unique receptor and binds only to the PRLR [11]. The recent report on GHR–PRLR heterodimerization in breast cancer cells [12] raises the possibility of hPL binding to such a hybrid receptor.
Pituitary PRL expression is controlled by a proximal promoter which is sensitive to inhibition by dopamine and to stimulation by several hypothalamic and peripheral hormones. On the other hand, expression of extrapituitary PRL is driven by a super-distal promoter, located 6.5 kb upstream of the pituitary start site, whose transcriptional regulation is not well understood [11]. Consequently, exposure of breast tissue to PRL comes from the circulation as well as through local production. Expression of PRL at the mRNA and protein levels has been detected in normal breast tissue, tumors and breast cancer cell lines [13]. Within the normal human breast, PRL is produced in larger quantities by stromal adipocytes than by the epithelial cells [14], but whether local PRL production in either breast compartment increases during tumorigenesis is unclear. In rodent models, autocrine PRL stimulates the growth of mammary tumors, while in cultured breast cancer cells, it affects cell proliferation, survival and clonogenic capacity [15-17].
Similar to PRL, the control of pituitary GH expression is complex, involving both positive and negative input by hypothalamic neuropeptides and peripheral hormones. There is evidence that elevated serum GH levels are associated with a small increase in risks of breast cancer, while GH deficiency results in the opposite effects [18,19]. Overexpression of GH in MCF-7 breast cancer cells promotes cell proliferation and survival, increases epithelial–mesenchymal transition and invasion, upregulates genes associated with chemoresistance [20] and antagonizes the antiproliferative effects of tamoxifen [21]. Whether GH is actually expressed in breast carcinomas is controversial [22,23]. In large concentrations, exogenous GH protects breast cancer cells from doxorubicin cytotoxicity [24]. Although hGH may act via the ubiquitously expressed GHR, the concurrent expression of PRLR in breast cancer and the ability of hGH to activate this receptor suggest that some of its actions are mediated by the PRLR.
Human placental lactogen, also known as human chorionic somatomammotropin, is the major secretory product of the syncytiotrophoblast. By the end of gestation, the very high maternal serum hPL levels surpass those of any other protein hormone [25]. Notably, immunoreactive hPL has been detected in breast carcinomas [26], and in serum from some breast cancer patients, but not those with benign breast disease or in serum from normal men and women [27]. The exact functions of locally produced hPL are unknown, but its presence in breast cancer raises the intriguing possibility that it serves as a tumor biomarker. This is supported by the recent demonstration of amplification of the three PL genes in approximately 25% of breast carcinomas, often coinciding with amplification of c-erbB-2, which is located on chromosome 17q11.2 in close proximity to the GH/pl locus [28]. Following the detection of immunoreactive hPL in 16 out of 26 tumors, with 77% of the tumors with amplified PL genes associated with lymph node metastases, the authors suggested that PL gene amplification could be used as a prognostic factor for aggressive breast cancer [28]. Serum biomarkers have been widely used in the management of patients with ovarian, colorectal and prostate cancer, but there is a paucity of reliable biomarkers in breast cancer [29]. Ectopic production of placental hormones by tumors is not uncommon, as exemplified by the expression of human chorionic gonadotropin not only in choriocarcinomas, but also in other types of tumors [30].
The PRL receptor
The human PRLR gene is located on chromosome 5p and is comprised of ten exons spanning 100 kb [31]. The PRLR is a single-pass membrane protein belonging to the large cytokine/GH receptor superfamily. The receptor is characterized by a tripartite structure composed of an extracellular ligand-binding domain, a short transmembrane domain and an intracellular domain that couples to a variety of signaling molecules [3,32]. In addition to the long form of the receptor, which has been most extensively studied, there are several splice variants and shorter isoforms, some of which can act as dominant negatives [31]. The PRLR is expressed in the normal breast, and both the long form and several of the isoforms are variably expressed in carcinomas [33]. Amplification of the PRLR suggests its importance in the pathogenesis and/or progression of lobular carcinomas [34].
Two binding sites on the ligand are required for PRLR activation. One receptor binds to a high affinity site 1, while a second receptor binds to a lower affinity site 2. This forms an active ternary complex composed of one hormone molecule and a receptor homodimer [35]. The three lactogens have dissimilar binding affinities to the two sites. As determined by surface plasmon resonance, the Kds for hPRL ranged from 6.5 to 62 nM for site 1, and 33 μM for site 2 [36,37]. hGH has a Kd of 5.8 μM to site 1, which increases to 388 nM in the presence of zinc. In the presence of zinc, hPL binds to the PRLR at 1.5 nM. Upon using cell proliferation as the end point [38], the potency of hGH binding the PRLR at EC50 of 1.53 nM approached that of hPRL (EC50 of 0.38 nM). Consequently, the nondiscriminating nature of the PRLR makes it a key player in the transduction of signaling from three distinct ligands that impact on breast cancer.
Binding of a ligand to the PRLR induces allosteric reorganization of the intracellular domain, leading to phosphorylation of the associated Jak2 kinase and the subsequent activation of other kinases and adaptor proteins [3,32]. An activated Jak2 rapidly phosphorylates Stat proteins, which subsequently dimerize, translocate to the nucleus and bind to γ-activated site (IFN-γ-activated sequence) elements on the promoters of target genes. Receptor activation also induces expression of endogenous suppressors of cytokine signaling proteins, which rapidly terminate the PRL signaling pathway.
Although the Stat 5a/b transcription factors mediate many of PRL’s actions in normal and malignant breast cells [3], other PRL-induced pathways are also involved. One is the Ras–Raf–MAPK pathway, with ERK1/2 and c-jun N-terminal kinase being its primary mediators. PRL induces phosphorylation of ERK1/2 in both T47D and MCF-7 cells and synergizes with EGF to activate ERK [39]. PRL also activates the PI3K/Akt survival pathway, which is implicated in breast cancer progression and resistance to therapy [5]. Activation of src-family kinases in breast cancer cells by PRL [40], which may be independent of Jak2 [41], has also been reported.
Cross-talk between the PRLR and estrogen receptor (ER)-α occurs at multiple levels. For example, PRL and E2 cooperatively enhance AP-1 activity [42], E2 rapidly phosphorylates Stat 5 [43], while PRL activates the unliganded ER [44]. Accumulating evidence also points to synergistic interactions between the PRL and members of the EGF and IGF families [5]. Consequently, interactions between lactogens, estrogens and oncogenic growth factors, many of which are associated with poor prognosis and/or therapeutic resistance, should inspire the development of effective combinatorial treatments in breast cancer.
Polymorphism of the PRLR
Breast cancer has a strong polygenetic component, where susceptibility to the disease is determined by a large number of loci, each with a small effect on risk of developing the disease [45]. Breast cancer is twice as common in first-degree relatives of women with the disease versus the general population. Known susceptibility genes, such as BRCA1 and BRCA2, account for less than 25% of the familial risk of breast cancer, while the remaining 75% are determined by variants with a more moderate risk [46]. Variation in DNA sequences can affect how individuals develop a disease and/or how they respond to treatments.
A single-nucleotide polymorphism (SNP) is a single base-pair mutation that occurs at a specific site in the DNA sequence. SNPs may fall within a coding sequence, in a non-coding region or in regions between genes. Those that occur within a coding sequence do not necessarily change the amino acid sequence of the encoded protein due to the degeneracy of the genetic code, and are termed synonymous or silent mutations. SNPs that generate a different polypeptide sequence are termed nonsynonymous. On average, SNPs occur once every 1000 DNA base pairs. Approximately 50% lie in introns, 25% are synonymous, while the remaining 25% nonsynonymous SNPs result in missense mutations [47,48]. The effects of SNP can vary from mild to profound. Polymorphisms in ERα [49] and G-protein-coupled receptor 30 (GPR30) [50] are associated either with increased susceptibility to breast cancer or with more aggressive tumors.
The PRLR gene contains five nonsynonymous SNPs, three of which (Ile100Val, Ile170Leu and Glu578Gln) have recently been studied [51-53]. The Ile170Leu SNP increases the survival of MCF-7 cells in response to PRL and activates the PRL signaling cascade even in the absence of PRL. This SNP is associated with benign follicular breast adenoma [52], and approached significance in a small number of patients with breast cancer [51]. The Ile100Val SNP also shows constitutive activity, albeit lower than that of Ile170Leu. Two other SNPs, Arg477Gly and Val137Glu, have not been studied. Neither of these PRLR SNPs has been examined with respect to the severity of the disease or to responsiveness to therapy in a large cohort of patients with breast cancer. Figure 1 depicts the locations of the five SNPs throughout the long form of the hPRL receptor.
Figure 1. Schematics of the structure of the long form of the human prolactin receptor and the locations of the five single-nucleotide polymorphisms.
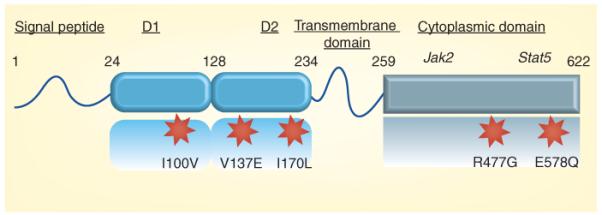
D1 and D2 are fibronectin-type III domains that are involved in ligand binding. The general locations of the docking sites of Jak2 and Stat5 within the cytoplasmic domain are also shown. This diagram is not drawn to scale.
Previous studies, which examined all PRLR SNPs in breast cancer (both synonymous and nonsynonymous) known at the time, yielded conflicting results. One study, comparing 1615 patients and 1962 controls in a multi-ethnic panel, reported no association between PRLR SNPs and breast cancer [54]. On the other hand, a study matched for ethnicity and using 441 cases versus 552 controls reported that a PRLR haplotype was associated with a decreased risk of breast cancer [55]. Both approaches were based on the Hapmap and tagged SNPs. The basis of this method ology is that recombination is not evenly spaced across the genome but tends to cluster at distinct ‘hot-spots’. Neighboring polymorphisms often exist in linkage disequilibrium with each other; common genetic variants can, therefore, be evaluated for association, using a few selected SNPs as tags for all other variants within a region of interest [56]. The problem inherent to this strategy is a bias towards the common-disease common-variant hypothesis, which is currently debated in the complex trait field. Association will not be detected if there are a number of rare alleles affecting disease. Ideally, therefore, each SNP should be examined in a large, sufficiently powered association study that is stratified by ethnicity, age, tumor stage and grade.
Chemoresistance by PRL
The reader is directed to several excellent reviews that cover the role of PRL in mammary tumorigenesis and breast carcinogenesis [3,5,11,13,32,57]. The focus of this article is on PRL and chemoresistance, a topic that has received much less attention. Epidemiological studies reveal that women with elevated blood PRL levels have increased rates of treatment failure and worse survival [57]. Hyperprolactinemic patients with metastatic breast cancer were less responsive to taxol than those with normal serum PRL levels [58], while patients treated with a combination of taxol and bromocriptine (which suppresses pituitary PRL release) showed a better outcome than those receiving taxol alone [59].
Prolactin is well recognized as an anticytotoxic factor in many cancer cell types, including ovarian, prostate and breast cancers [4]. Induction of apoptosis by cisplatin in T47D cells is enhanced by cotreatment with a hPRL antagonist, suggesting that endogenous PRL is protective [60]. A different antagonist of PRL potentiates the effects of paclitaxel and doxorubicin in breast cancer cells [17]. Furthermore, cells such as T47D and MCF-7, which is known to produce endogenous PRL, are more resistant to ceramide-induced apoptosis than cell lines producing low or no PRL [61]. PRL can also overcome growth arrest caused by γ-irradiation [62]. None of the aforementioned studies, however, has elucidated the mechanisms responsible for the protective effects of PRL.
In a recent study, low physiological doses of PRL antagonized cytotoxicity by two distinct classes of chemotherapeutic agents: those that interfere with microtubule dynamics (taxol and vinblastine), and those that cause DNA damage (doxorubicin and cisplatin) [63]. Cisplatin is a platinum-based drug that is highly effective against testicular, ovarian and lung cancers, but only has limited efficacy in breast cancer [64]. Following its uptake into the nucleus, cisplatin interacts with DNA and forms adducts via intrastrand crosslinks that cause cell cycle arrest. The DNA can either be repaired by the nucleotide excision pathway or the cell is destined to die [65]. The low efficacy of cisplatin in breast cancer raised the intriguing possibility that this might be due to protection by PRL. Subsequently, interactions between PRL and cisplatin were examined in greater detail.
As determined by mass spectrometry, exposure of breast cancer cells to PRL reduced the amount of platinum bound to DNA [63]. It was then reasoned that the reduced entry of cisplatin into the nucleus could be due either to a direct effect of PRL on membrane transporters that extrude the drug, or to PRL-induced activation of detoxification enzymes that alter the drug, resulting in its elimination from the cells. Subsequent investigation revealed that PRL activated glutathione-S-transferase (GST), a Phase II detoxification enzyme that conjugates electrophilic compounds to glutathione and facilitates their expulsion from the cells [66]. Among the various GST isozymes, PRL increased the expression of GSTμ, but not GSTπ [63]. The GSTμ-null genotype is associated with increased survival in women with advanced breast cancer who were treated with chemotherapy [67]. It remains to be determined whether GST also mediates the protective actions of PRL against other anticancer drugs, and to what extent other mechanisms, especially activation of anti-apoptotic proteins [68], come into play in response to PRL. Figure 2 compares the proposed mechanisms by which PRL and E2 confer resistance against cisplatin in breast cancer.
Figure 2. Complementary mechanisms by which estrogens (represented by estradiol) and lactogens (represented by prolactin) confer resistance against cisplatin in breast cancer.

Cisplatin diffuses into the cell and enters the nucleus, where it binds to DNA and causes interchain crosslinking. The ensuing cell cycle arrest leads to the release of mitochondrial Cyt C, which activates the caspase cascade and apoptosis. Estrogens bind either to a membrane receptor or diffuse into the cell and bind to ‘classical’ cytoplasmic/nuclear receptors. This results in increased expression of the anti-apoptotic protein Bcl-2, which blocks Cyt C release and prevents apoptosis. Binding of lactogens to the PRL receptor induces rapid phosphorylation of the associated Jak2, and the subsequent activation of the Stat5a/b and ERK1/2 signaling pathways. It is through either, or both, of these pathways that PRL increases transcription of the detoxification enzyme GST. Increased GST activity promotes the conjugation of GSH to cisplatin, enabling extrusion of the conjugate from the cell via membrane transporters. Consequently, less cisplatin is available to enter the nucleus, resulting in decreased apoptosis.
Cyt C: Cytochrome c; E2: Estradiol; GSH: Glutathione; GST: Glutathione-S-transferase; PRL: Prolactin; Pt: Cisplatin.
Estrogenic compounds
Among the naturally occurring estrogens, E2, estriol and estrone, 17β E2 is by far the most potent. During the reproductive years, the ovaries are the primary source of estrogens. After menopause, estrogens are mostly generated by aromatization of androgens, which are secreted by both the adrenals and the ovaries [69]. Similar to the situation with PRL, the breast is exposed to natural estrogens from two sources: the circulation and local production. Breast cancer expresses several sex steroid-producing enzymes, including aromatase, 17β-hydroxysteroid dehydrogenase, which catalyzes interconversion among estrogens, and steroid sulfatase, which hydrolyzes sulfated steroids to their bioactive forms [70].
Although serum E2 levels in postmenopausal women are less than 10% of those before menopause, their tumors are exposed to comparable levels of active estrogens due to local synthesis [71]. Similar to the higher production of PRL by breast stroma [14], estrogen synthesis is greater in the stroma than in the epithelium because of the high expression of aromatase in this compartment [72]. The ability of aromatase inhibitors to reduce estrogen availability at the site of the tumor has been successfully used in the clinic to block growth of ER-positive tumors, especially in postmenopausal women [73].
Interestingly, a number of industrial chemicals, pesticides, pharmaceuticals and plant-derived compounds (phytoestrogens) mimic or antagonize endogenous estrogens [74]. Human exposure to these xenoestrogens occurs through the food and water supply, household goods, some dental composites and even medical tubing in patients [75]. Many of these compounds have low molecular weight, have drug-like pharmacokinetics and, by virtue of being lipophilic, their potential storage in adipose tissue can increase their bioavailability.
The best-studied endocrine disruptors are components of plastics, such as bisphenol A (BPA), and detergents such as octyl- and nonylphenols. Chlorinated insecticides, for example, kepone, dichlorodiphenyltrichloroethane, dieldrin and methoxychlor, also have estrogen-like properties. Atrazine, a commonly used broad-leaf herbicides, has well-documented estrogen-like disrupting properties, except that it does not act at the level of the ER, but induces aromatase, leading to higher endogenous estrogen levels [76].
Potent pharmaceuticals, including diethylstilbestrol and ethinylestradiol, are used by millions of patients as contraceptives, to treat symptoms of menopause and as a palliative therapy in advanced prostatic cancer. Home toilets serve as the main source of these pharmaceuticals in wastewater [77]. Phytoestrogens, including resveratrol, daidzein, quercetin and genistein, show variable binding affinities to ERα and ERβ, exert some nongenomic actions and can also affect estrogen biosynthesis and metabolism [78]. Although consumption of soy products, which contain phytoestrogens, has been advocated as a means for reducing the risk of breast cancer, the beneficial effects of phytoestrogens to breast cancer patients have been disputed [79].
Estrogen receptors
Estrogens bind to several receptors of diverse structures that can be localized in the membrane, cytoplasm and nucleus. Two ‘classical’ cytoplasmic receptors, ERα and ERβ, are the products of distinct genes. They differ primarily in their ligand-binding domains, which accounts for the wide variations in binding affinities among estrogenic ligands [80]. Following ligand binding, classical ERs dimerize, translocate to the nucleus and bind to estrogen response elements in the promoters of target genes. Recruitment of coregulators results in the formation of large complexes that mediate transcription [81]. The plethora of cell-specific coactivators and corepressors account, in part, for the partial agonist versus antagonist activities of tamoxifen in the uterus, breast, bone and the cardiovascular system.
Similar to the differential binding dynamics of lactogens to the PRLR [82], the various estrogenic ligands can induce distinct changes in ER conformation and thus alter cofactor recruitment or receptor stability [83]. This is exemplified by the induction of rapid degradation of ERα by the pure ER antagonist ICI182780, but not by tamoxifen or E2 [84]. Activated ERs can also regulate transcription via protein–protein interactions with transcription factors such as the Fos–Jun complex [85]. Expression of ERα is higher in in situ carcinomas than in the normal breast epithelium, while the expression pattern of ERβ shows the opposite trend [86]. Given the decreased ratio of ERβ/ERα during the neoplastic process, it has been proposed that cancer progression is associated with a loss of ERβ [80].
In addition to their well-established genomic actions, estrogens can rapidly activate the MAPK and PI3K/Akt signaling pathways, traditionally associated with membrane receptors [87]. However, the nature of the receptor(s) that mediate the rapid actions of estrogens remains elusive. In neurons, pituitary and endothelial cells, G proteins, ion channels, cytoplasmic protein kinases and adaptor proteins have all been implicated as receptors for estrogens [43,88]. In breast cancer cells, there is evidence that a sub population of ERα is localized to the cell membrane. Given its lack of transmembrane or kinase domains, ERα cannot be incorporated into the cell membrane as an integral protein, but can be localized adjacent to the membrane through palmitoylation [89]. Both IGF-1 and EGF receptors are presumably involved in tethering ERα to the membrane and in the activation of the MAPK and PI3K signaling pathways. Although the tethered ER model provides a plausible explanation for some of the nongenomic actions of E2, it does not clarify how endocrine disruptors that have much lower binding affinities to ERα or -β can still be active at sub-nanomolar concentrations [90]. It is plausible, therefore, that many of these endocrine disruptors have their own unique receptor.
Another class of estrogen-binding receptors is represented by GPR30, a seven-transmembrane domain receptor that signals through G proteins [91,92]. Binding of E2 to GPR30 stimulates the cAMP pathway and induces the release of heparan-bound EGF. EGF, in turn, binds to its cognate membrane receptor and initiates downstream signaling that includes MAPK, PI3K and phospholipase C [92]. Both tamoxifen and ICI act as agonists, rather than antagonists, of GPR30. Given the high expression of GPR30 in invasive carcinoma and its association with larger tumors, GPR30 has been proposed as a predictor of a more aggressive disease [93]. The relatively high binding affinity of GPR30 to E2 (Kd of 3 nM) makes this receptor a potential mediator of estrogen actions in ER-negative breast cancer cells [94], but its role in cells that also express ERα and ERβ is unclear.
Chemoresistance by estrogens
The ability of estrogens to activate the PI3K/Akt survival pathway and to increase expression of anti-apoptotic proteins [95,96] serves as the backdrop to their involvement with resistance to chemotherapy. Most studies link estrogen-induced chemoresistance to activation of Bcl-2. For example, MCF-7 cells grown without estrogens become sensitized to doxorubicin. This is accompanied by a decrease in Bcl-2 expression, while Bcl-2 reconstitution restores resistance to the drug [97]. Induction of Bcl-2 by estrogen [96], as well as ERα overexpression [6] in breast cancer cells, causes a significant decrease in taxol-induced apoptosis. A combination of tamoxifen and TNF-related apoptosis-inducing ligand was more effective than each alone in inducing apoptosis in ERα-negative MDA-MB-231 breast cancer cells and in arresting tumor growth in xenografts [98]. This sensitization was associated with decreased Bcl-2 and increased expression of the proapoptotic protein Bax.
Recent studies reveal that low doses of E2 (0.01–10 nM) abrogated cisplatin toxicity in breast cancer cells by increasing cell proliferation as well as by decreasing apoptosis [99]. Protection by estrogen occurred in the presence of ERα and ERβ antagonists in ERα-negative MDA-MB-468 cells, and in T47D cells with ERβ knockdown, strongly suggesting an action that is independent of classical ERs. Unlike PRL, E2 does not reduce cisplatin entry into the nucleus, indicating that it acts downstream of DNA damage [99]. Indeed, E2 increases the expression of Bcl-2 in T47D cells, both in the presence and absence of cisplatin. However, given that a potent Bcl-2 inhibitor only partially abrogated protection by E2, other mediators may be involved [99]. As depicted in Figure 2, antagonism of cisplatin action by PRL and estrogen via complementary mechanisms provides a plausible explanation for the poor efficacy of this drug in breast cancer.
In addition to activating anti-apoptotic proteins, estrogen antagonizes both taxol- and radiation-induced apoptosis by altering JNK activity [100]. Another mechanism by which estrogens can increase chemoresistance is by affecting membrane drug exporters. This was demonstrated by an estrogen-induced increase in cytoplasmic P-glycoprotein in MCF-7 cells, which are resistant to doxorubicin, but not in T47D cells, which are sensitive to the drug [101].
Chemoresistance by BPA
Bisphenol A, a monomer of polycarbonate plastics, is a major constituent of consumer products such as food and beverage containers, lining of metal food cans and many epoxy-based products [102]. Migration of BPA into food or water from plastic utensils is influenced by the manufacturing process, storage conditions and heating practice by the users [103,104]. As a consequence of the exponential increase in plastic use in the last few decades, humans worldwide have been inadvertently exposed to BPA. Indeed, BPA is detectable at 0.2–10 ng/ml in serum from most individuals tested [102], concentrations that have demonstrable effects in cell culture conditions. In addition to blood and urine, lipophilic BPA was detected in breast adipose tissue [105] and in breast milk [106].
The endocrine-disrupting properties of BPA have recently been widely covered in the news media. Concerns over adverse effects of early exposure to BPA on infant development have prompted the legislative banning of the use of BPA in the manufacture of baby bottles in several countries. While this represents an initial step in the removal of an endocrine disruptor that may affect child health, it does not resolve the contentious issue of the potential health hazards of adult exposure to BPA, as discussed in the following paragraph.
Bisphenol A exerts estrogen-like properties in multiple reproductive and nonreproductive systems [102,107,108]. In breast cancer cells, BPA was reported to affect cell proliferation and activate certain sets of genes, not all of which were also induced by E2 [109]. BPA also rapidly activates nongenomic signaling, with phosphorylation of MAPK and Akt occurring in MCF-7 cells within 10 min of exposure [110]. BPA at low doses also induces a rapid influx of calcium in breast cancer cells, which was neither reversed by ICI182780 nor reproduced by E2 or diethylstilbestrol, leading the authors to conclude that classical ERs are not involved [111].
Although the binding affinity of BPA to ERα or ERβ is several orders of magnitude lower than that of E2 [112], BPA at low nanomolar and even subnanomolar doses shows similar activities to those of E2 [113,114]. To explain this discordance, it has been suggested that BPA binds differentially within the ligand-binding domain of ERs, or recruits a different set of coactivators [115]. In addition, estrogen-related receptors (ERRs) have been proposed as alternative receptors for transmitting BPA signals [116]. Although ERRs do not bind estrogen, ERR-γ binds BPA with high affinity (Ki of 5.5 nM) [117]. ERR-γ is overexpressed in 75% of breast tumors compared with the normal epithelium [118].
Similar to the actions of PRL and E2, BPA antagonizes multiple anticancer drugs, showing equimolar potency with E2 in opposing cisplatin toxicity [99]. BPA alone or in combination with doxorubicin [119] or cisplatin [99] increases Bcl-2 expression. Treatment with a Bcl-2 inhibitor completely blocks the BPA-induced antagonism of cisplatin, whereas it only partially abrogates protection by E2. This suggests that BPA and estrogen exert protection against drug cytotoxicity by somewhat different mechanisms – that is, anti-apoptosis versus mitogenesis. This notion is supported by flow cytometery and bromodeoxyuridine incorporation showing that BPA alone increases cell survival while estrogen alone increases cell proliferation [99].
Because chemoprotection by BPA does not appear to be mediated via ERα and ERβ, it raises the intriguing possibility that BPA utilizes an as-yet unidentified unique receptor. This prospect is not unprecedented, as exemplified by opioid and cannabinoid receptors, which were discovered many years after the bioactivity of their exogenous ligands was recognized. Indeed, there are still hundreds of membrane, cytoplasmic and nuclear orphan receptors without an identified ligand.
Expert commentary
The various sources of lactogens and estrogenic compounds that impinge on breast cancer are summarized in Figure 3. Several intractable issues confound the full understanding of the roles of these hormones in chemoresistance. A major problem is their multiple sites of origin, with endogenous hormones reaching breast tumors at variable levels from the systemic circulation, from the adjacent stroma and as autocrine/paracrine factors by cancer cells themselves. Consequently, determination of blood levels of lactogens or estrogens does not reveal the full extent of tumor exposure to these hormones. Yet, many logistical problems limit the clinical ability to assess the true exposure level of breast tumors to these hormones in the microenvironment of the intact human breast.
Figure 3. Different lactogenic and estrogenic compounds that affect breast cancer and their sites of origin.
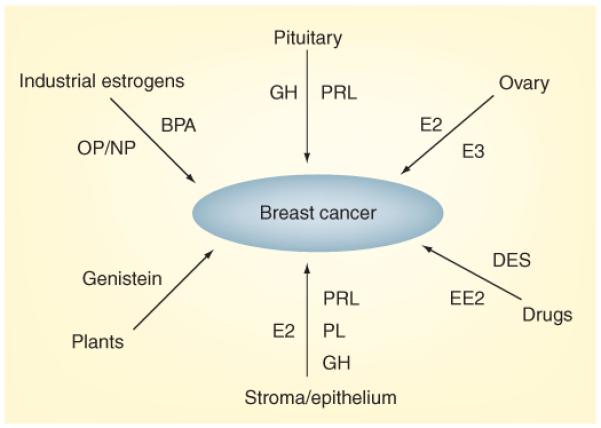
The three structurally-related lactogens, PRL, GH and PL, which bind to the PRL receptor, originate either from the pituitary or are produced locally by both stromal and epithelial breast cells. Estrogens include endogenous steroids (E2 and E3) that are either secreted by the ovary or are converted from precursors within the breast. Exogenous estrogenic compounds include pharmaceuticals such as DES and EE2, components of plastics such as BPA, detergents such as OP and NP, and phytoestrogens, such as genistein. Both lactogens and estrogens (probably via aromatization) are also produced within tumor cells themselves, where they act as autocrine/paracrine agents.
BPA: Bisphenol A; DES: Diethylstilbestrol; GH: Growth hormone; E2: Estradiol; E3: Estriol; EE2: Etinylestradiol; NP: Nonylphenol; OP: Octylphenol; PL: Placental lactogen; PRL: Prolactin.
Another confounding issue is receptor promiscuity, with ERs capable of binding both steroidal and nonsteroidal compounds, while the PRLR is capable of binding three distinct lactogens. This is further complicated by the presence of multiple receptor isoforms, with estrogens binding to several classical and nonclassical receptors, while lactogens bind to different isoforms of the PRLR that are coupled to several signaling pathways. Another poorly understood issue is crosstalk between lactogens and estrogens, which occurs downstream of their receptors. Addressing some of these issues by basic researchers would require nonconventional thinking. Clinicians should consider the implementation of combinatorial treatments, that is, antilactogens and anti-estrogens, as a means of improving the efficacy of chemotherapeutic agents.
Five-year view
Several PRLR antagonists, made by small modifications of the PRL molecule, have already been generated [120]. Their therapeutic efficacy would probably increase upon further improvements of their binding affinity, the prolongation of their serum half-life and/or by developing an effective targeted delivery to the tumors. Efforts are also underway to identify small molecules that selectively block the PRLR and can be used as oral medications [121]. Although blockade of the PRLR has not been a high-priority goal for drug discovery by the pharmaceutical industry, the aforementioned evidence on the critical role of lactogens in chemoresistance should inspire increased interest in this area.
No matter how well designed a therapy is, it is often only maximally effective in certain patient subgroups. SNPs in genes that are involved in a particular disease can predict the course of therapeutic outcome. Studies with large cohorts of patients with breast cancer are needed to search for an association of PRLR SNPs with the severity of the disease and/or with chemoresistance. In line with the ongoing progress towards personalized medicine, identification of PRLR variants should set apart those patients who could benefit most from antilactogen therapy.
There is an urgent need for serological markers in breast cancer. These could be used to independently verify diagnosis and prognosis, provide surveillance after surgery, predict tumor responsiveness to chemotherapy and assess the success of therapy in advanced cancer. Tumor-derived hPL, which is normally only detected during pregnancy, has the potential to serve as a tumor marker. To this end, efforts should be invested to validate hPL as a tumor biomarker and to develop sensitive, specific and practical assays for its analysis in patients’ blood. Once materialized, the payoff could be significant for millions of women with breast cancer.
Only a few studies so far have examined the role of endogenous estrogens in chemoresistance. This oversight is enigmatic because estrogens enhance tumor growth, not only as mitogens, but also as anti-apoptotic factors. There is a certain shortcoming in defining hormone-insensitive tumors based on the lack of expression of classical ERα/ERβ, as tumor cells can still respond to estrogenic ligands via alternative mechanisms (i.e., GPR30 or novel receptors). Future studies examining the anticytotoxic actions of estrogens should not overlook those breast cancer cells that do not express classical ERs and are, therefore, traditionally considered ‘estrogen-unresponsive’.
With increased public awareness of health hazards imposed by endocrine disruptors, regulatory agencies should become more proactive in taking measures for limiting human exposure to bioactive chemicals such as BPA. However, as was the case with dichlorodiphenyltrichloroethane, it may take decades before BPA is totally removed from landfills and the water supply. Hence, many key issues remained to be resolved, including a clear identification of the receptors involved, and a better understanding of their mode of action and the combined effects of chemicals in mixtures.
Key issues.
Chemoresistance can be induced by the drugs themselves, as well as by endogenous and exogenous factors.
Lactogens are homologous protein hormones that can bind the prolactin receptor (PRLR) and activate its signaling. Estrogens include endogenously produced steroid hormones, as well as nonsteroidal compounds from the environment, which bind to a variety of estrogen receptors.
Breast cancer is exposed to hormones from multiple sources, including the circulation, breast stroma and cancer cells themselves, as well as through exposure to endocrine-disrupting chemicals from the environment.
Cross-talk between estrogen and prolactin receptors and activation of complementary mechanisms that enhance the survival of cancer cells underlies resistance to many anticancer drugs.
The presence of PRLR variants could be exploited in the identification of patients who would benefit most from blockade of prolactin signaling. Effective blockade of the PRLR in combination with anti-estrogen therapy should increase the efficacy of certain anticancer drugs.
Acknowledgments
Financial & competing interests disclosure
This work was supported by NIH grants ES012212 and CA096613, DOD BC05725, Susan G Komen BCRT87406, and a grant from the Elsa U Pardee foundation (to Nira Ben-Jonathan), and the NIH Center for Environmental Genetics P30 ES06096 (to Eric M Jacobson). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008;34:378–390. doi: 10.1016/j.ctrv.2008.01.007. • A comprehensive review of the mechanism by which chemotherapeutic agents from different classes induce chemoresistance in breast cancer.
- 2.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 3.Clevenger CV, Gadd SL, Zheng J. New mechanisms for PRLr action in breast cancer. Trends Endocrinol. Metab. 2009;20:223–229. doi: 10.1016/j.tem.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Lapensee EW, Ben-Jonathan N. Novel roles of prolactin and estrogens in breast cancer: resistance to chemotherapy. Endocr. Relat. Cancer. 2010;17:R91–R107. doi: 10.1677/ERC-09-0253. [DOI] [PubMed] [Google Scholar]
- 5.Carver KC, Arendt LM, Schuler LA. Complex prolactin crosstalk in breast cancer: new therapeutic implications. Mol. Cell Endocrinol. 2009;307:1–7. doi: 10.1016/j.mce.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sui M, Huang Y, Park BH, Davidson NE, Fan W. Estrogen receptor α mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res. 2007;67:5337–5344. doi: 10.1158/0008-5472.CAN-06-4582. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr. Rev. 1996;17:639–669. doi: 10.1210/edrv-17-6-639. [DOI] [PubMed] [Google Scholar]
- 8.Kimura AP, Sizova D, Handwerger S, Cooke NE, Liebhaber SA. Epigenetic activation of the human growth hormone gene cluster during placental cytotrophoblast differentiation. Mol. Cell Biol. 2007;27:6555–6568. doi: 10.1128/MCB.00273-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harvey S. Extrapituitary growth hormone. Endocrine. 2010;38:335–359. doi: 10.1007/s12020-010-9403-8. [DOI] [PubMed] [Google Scholar]
- 10.Keeler C, Dannies PS, Hodsdon ME. The tertiary structure and backbone dynamics of human prolactin. J. Mol. Biol. 2003;328:1105–1121. doi: 10.1016/s0022-2836(03)00367-x. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Jonathan N, Lapensee CR, Lapensee EW. What can we learn from rodents about prolactin in humans? Endocr. Rev. 2008;29:1–41. doi: 10.1210/er.2007-0017. • A broad review on the similarities and differences between rodents and humans in the regulation and action of prolactin.
- 12.Xu J, Zhang Y, Berry PA, et al. Growth hormone signaling in human T47D breast cancer cells: potential role for a growth hormone receptor-prolactin receptor complex. Mol. Endocrinol. 2011;25(4):597–610. doi: 10.1210/me.2010-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J. Endocrinol. 2010;206:1–11. doi: 10.1677/JOE-10-0069. [DOI] [PubMed] [Google Scholar]
- 14.Zinger M, McFarland M, Ben-Jonathan N. Prolactin expression and secretion by human breast glandular and adipose tissue explants. J. Clin. Endocrinol. Metab. 2003;88:689–696. doi: 10.1210/jc.2002-021255. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Jonathan N, Liby K, McFarland M, Zinger M. Prolactin as an autocrine/paracrine growth factor in human cancer. Trends Endocrinol. Metab. 2002;13:245–250. doi: 10.1016/s1043-2760(02)00603-3. [DOI] [PubMed] [Google Scholar]
- 16.Scotti ML, Langenheim JF, Tomblyn S, Springs AE, Chen WY. Additive effects of a prolactin receptor antagonist, G129R, and herceptin on inhibition of HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 2008;111:241–250. doi: 10.1007/s10549-007-9789-z. [DOI] [PubMed] [Google Scholar]
- 17.Howell SJ, Anderson E, Hunter T, Farnie G, Clarke RB. Prolactin receptor antagonism reduces the clonogenic capacity of breast cancer cells and potentiates doxorubicin and paclitaxel cytotoxicity. Breast Cancer Res. 2008;10:R68. doi: 10.1186/bcr2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry JK, Mohankumar KM, Emerald BS, Mertani HC, Lobie PE. The contribution of growth hormone to mammary neoplasia. J. Mammary Gland Biol. Neoplasia. 2008;13:131–145. doi: 10.1007/s10911-008-9070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinberg DL, Wood TL, Furth PA, Lee AV. Growth hormone and insulin-like growth factor-I in the transition from normal mammary development to preneoplastic mammary lesions. Endocr. Rev. 2009;30:51–74. doi: 10.1210/er.2008-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry JK, Emerald BS, Mertani HC, Lobie PE. The oncogenic potential of growth hormone. Growth Horm. IGF Res. 2006;16:277–289. doi: 10.1016/j.ghir.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Mojarrad M, Momeny M, Mansuri F, et al. Autocrine human growth hormone expression leads to resistance of MCF-7 cells to tamoxifen. Med. Oncol. 2010;27:474–480. doi: 10.1007/s12032-009-9237-5. [DOI] [PubMed] [Google Scholar]
- 22.Chhabra Y, Waters MJ, Brooks AJ. Role of the growth hormone-IGF-1 axis in cancer. Exp. Rev. Endocrinol. Metab. 2011;6:71–84. doi: 10.1586/eem.10.73. [DOI] [PubMed] [Google Scholar]
- 23.Raccurt M, Lobie PE, Moudilou E, et al. High stromal and epithelial human GH gene expression is associated with proliferative disorders of the mammary gland. J. Endocrinol. 2002;175:307–318. doi: 10.1677/joe.0.1750307. [DOI] [PubMed] [Google Scholar]
- 24.Zatelli MC, Minoia M, Mole D, et al. Growth hormone excess promotes breast cancer chemoresistance. J. Clin. Endocrinol. Metab. 2009;94:3931–3938. doi: 10.1210/jc.2009-1026. [DOI] [PubMed] [Google Scholar]
- 25.Handwerger S. New insights into the regulation of human cytotrophoblast cell differentiation. Mol. Cell Endocrinol. 2010;323:94–104. doi: 10.1016/j.mce.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horne CH, Reid IN, Milne GD. Prognostic significance of inappropriate production of pregnancy proteins by breast cancers. Lancet. 1976;2:279–282. doi: 10.1016/s0140-6736(76)90731-5. [DOI] [PubMed] [Google Scholar]
- 27.Sheth NA, Suraiya JN, Sheth AR, Ranadive KJ, Jussawalla DJ. Ectopic production of human placental lactogen by human breast tumors. Cancer. 1977;39:1693–1699. doi: 10.1002/1097-0142(197704)39:4<1693::aid-cncr2820390445>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 28.Latham C, Zhang A, Nalbanti A, et al. Frequent co-amplification of two different regions on 17q in aneuploid breast carcinomas. Cancer Genet. Cytogenet. 2001;127:16–23. doi: 10.1016/s0165-4608(00)00427-1. [DOI] [PubMed] [Google Scholar]
- 29.Duffy MJ. Serum tumor markers in breast cancer: are they of clinical value? Clin. Chem. 2006;52:345–351. doi: 10.1373/clinchem.2005.059832. [DOI] [PubMed] [Google Scholar]
- 30.Butler SA, Iles RK. Ectopic human chorionic gonadotropin β secretion by epithelial tumors and human chorionic gonadotropin β-induced apoptosis in Kaposi’s sarcoma: is there a connection? Clin. Cancer Res. 2003;9:4666–4673. [PubMed] [Google Scholar]
- 31.Hu ZZ, Zhuang L, Dufau ML. Prolactin receptor gene diversity: structure and regulation. Trends Endocrinol. Metab. 1998;9:94–102. doi: 10.1016/s1043-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 32.Swaminathan G, Varghese B, Fuchs SY. Regulation of prolactin receptor levels and activity in breast cancer. J. Mammary Gland Biol. Neoplasia. 2008;13:81–91. doi: 10.1007/s10911-008-9068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginsburg E, Alexander S, Lieber S, et al. Characterization of ductal and lobular breast carcinomas using novel prolactin receptor isoform specific antibodies. BMC Cancer. 2010;10:678. doi: 10.1186/1471-2407-10-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran-Thanh D, Arneson NC, Pintilie M, et al. Amplification of the prolactin receptor gene in mammary lobular neoplasia. Breast Cancer Res. Treat. 2010 doi: 10.1007/s10549-010-1025-6. DOI: 10.1007/s10549-010-1025-6. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 35.Teilum K, Hoch JC, Goffin V, Kinet S, Martial JA, Kragelund BB. Solution structure of human prolactin. J. Mol. Biol. 2005;51:810–823. doi: 10.1016/j.jmb.2005.06.042. [DOI] [PubMed] [Google Scholar]
- 36.Jomain JB, Tallet E, Broutin I, et al. Structural and thermodynamic bases for the design of pure prolactin receptor antagonists: x-ray structure of Del1-9-G129R-hPRL. J. Biol. Chem. 2007;282:33118–33131. doi: 10.1074/jbc.M704364200. [DOI] [PubMed] [Google Scholar]
- 37.Voorhees JL, Brooks CL. Obligate ordered binding of human lactogenic cytokines. J. Biol. Chem. 2010;285:20022–20030. doi: 10.1074/jbc.M109.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Utama FE, Tran TH, Ryder A, LeBaron MJ, Parlow AF, Rui H. Insensitivity of human prolactin receptors to nonhuman prolactins: relevance for experimental modeling of prolactin receptor-expressing human cells. Endocrinology. 2009;150:1782–1790. doi: 10.1210/en.2008-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acosta JJ, Munoz RM, Gonzalez L, et al. Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol. Endocrinol. 2003;17:2268–2282. doi: 10.1210/me.2002-0422. [DOI] [PubMed] [Google Scholar]
- 40.Piazza TM, Lu JC, Carver KC, Schuler LA. SRC family kinases accelerate prolactin receptor internalization, modulating trafficking and signaling in breast cancer cells. Mol. Endocrinol. 2009;23:202–212. doi: 10.1210/me.2008-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fresno Vara JA, Carretero MV, Geronimo H, Ballmer-Hofer K, Martin-Perez J. Stimulation of c-Src by prolactin is independent of Jak2. Biochem. J. 2000;345(Pt 1):17–24. doi: 10.1042/0264-6021:3450017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutzman JH, Nikolai SE, Rugowski DE, Watters JJ, Schuler LA. Prolactin and estrogen enhance the activity of activating protein 1 in breast cancer cells: role of extracellularly regulated kinase 1/2-mediated signals to c-fos. Mol. Endocrinol. 2005;19:1765–1778. doi: 10.1210/me.2004-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids. 2009;74:622–627. doi: 10.1016/j.steroids.2008.10.014. • Different aspects of the actions of various estrogens on membrane and cytoplasmic receptors.
- 44.Gonzalez L, Zambrano A, Lazaro-Trueba I, et al. Activation of the unliganded estrogen receptor by prolactin in breast cancer cells. Oncogene. 2009;28:1298–1308. doi: 10.1038/onc.2008.473. [DOI] [PubMed] [Google Scholar]
- 45.Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed S, Thomas G, Ghoussaini M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat. Genet. 2009;41:585–590. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 48.Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anghel A, Narita D, Seclaman E, Popovici E, Anghel M, Tamas L. Estrogen receptor α polymorphisms and the risk of malignancies. Pathol. Oncol. Res. 2010;16:485–496. doi: 10.1007/s12253-010-9263-9. [DOI] [PubMed] [Google Scholar]
- 50.Giess M, Lattrich C, Springwald A, Goerse R, Ortmann O, Treeck O. GPR30 gene polymorphisms are associated with progesterone receptor status and histopathological characteristics of breast cancer patients. J. Steroid Biochem. Mol. Biol. 2010;118:7–12. doi: 10.1016/j.jsbmb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Canbay E, Degerli N, Gulluoglu BM, Kaya H, Sen M, Bardakci F. Could prolactin receptor gene polymorphism play a role in pathogenesis of breast carcinoma? Curr. Med. Res. Opin. 2004;20:533–540. doi: 10.1185/030079904125003232. [DOI] [PubMed] [Google Scholar]
- 52.Bogorad RL, Courtillot C, Mestayer C, et al. Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proc. Natl Acad. Sci. USA. 2008;105:14533–14538. doi: 10.1073/pnas.0800685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Courtillot C, Chakhtoura Z, Bogorad R, et al. Characterization of two constitutively active prolactin receptor variants in a cohort of 95 women with multiple breast fibroadenomas. J. Clin. Endocrinol. Metab. 2010;5:271–279. doi: 10.1210/jc.2009-1494. [DOI] [PubMed] [Google Scholar]
- 54.Lee SA, Haiman CA, Burtt NP, et al. A comprehensive analysis of common genetic variation in prolactin (PRL) and PRL receptor (PRLR) genes in relation to plasma prolactin levels and breast cancer risk: the multiethnic cohort. BMC Med. Genet. 2007;8:72. doi: 10.1186/1471-2350-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaclavicek A, Hemminki K, Bartram CR, et al. Association of prolactin and its receptor gene regions with familial breast cancer. J. Clin. Endocrinol. Metab. 2006;91:1513–1519. doi: 10.1210/jc.2005-1899. [DOI] [PubMed] [Google Scholar]
- 56.Hinds DA, Stuve LL, Nilsen GB, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 57.Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. J. Mammary Gland Biol. Neoplasia. 2008;13:41–53. doi: 10.1007/s10911-008-9063-y. [DOI] [PubMed] [Google Scholar]
- 58.Lissoni P, Vaghi M, Ardizzoia A, et al. Efficacy of monochemotherapy with docetaxel (taxotere) in relation to prolactin secretion in heavily pretreated metastatic breast cancer. Neuro. Endocrinol. Lett. 2001;22:27–29. [PubMed] [Google Scholar]
- 59.Lissoni P, Bucovec R, Malugani F, et al. A clinical study of taxotere versus taxotere plus the antiprolactinemic agent bromocriptine in metastatic breast cancer pretreated with anthracyclines. Anticancer Res. 2002;22:1131–1134. [PubMed] [Google Scholar]
- 60.Ramamoorthy P, Sticca R, Wagner TE, Chen WY. In vitro studies of a prolactin antagonist, hPRL-G129R in human breast cancer cells. Int. J. Oncol. 2001;18:25–32. doi: 10.3892/ijo.18.1.25. [DOI] [PubMed] [Google Scholar]
- 61.Perks CM, Keith AJ, Goodhew KL, Savage PB, Winters ZE, Holly JM. Prolactin acts as a potent survival factor for human breast cancer cell lines. Br. J. Cancer. 2004;91:305–311. doi: 10.1038/sj.bjc.6601947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakravarti P, Henry MK, Quelle FW. Prolactin and heregulin override DNA damage-induced growth arrest and promote phosphatidylinositol-3 kinase-dependent proliferation in breast cancer cells. Int. J. Oncol. 2005;26:509–514. [PubMed] [Google Scholar]
- 63.Lapensee EW, Schwemberger SJ, Lapensee CR, Bahassi EM, Afton SE, Ben-Jonathan N. Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase. Carcinogenesis. 2009;30:1298–1304. doi: 10.1093/carcin/bgp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Decatris MP, Sundar S, O’Byrne KJ. Platinum-based chemotherapy in metastatic breast cancer: current status. Cancer Treat. Rev. 2004;30:53–81. doi: 10.1016/S0305-7372(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 65.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 66.Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ambrosone CB, Sweeney C, Coles BF, et al. Polymorphisms in glutathione S-transferases (GSTM1 and GSTT1) and survival after treatment for breast cancer. Cancer Res. 2001;61:7130–7135. [PubMed] [Google Scholar]
- 68.Peirce SK, Chen WY. Human prolactin and its antagonist, hPRL-G129R, regulate bax and bcl-2 gene expression in human breast cancer cells and transgenic mice. Oncogene. 2004;23:1248–1255. doi: 10.1038/sj.onc.1207245. [DOI] [PubMed] [Google Scholar]
- 69.Jongen VH, Hollema H, Van Der Zee AG, Heineman MJ. Aromatase in the context of breast and endometrial cancer. A review. Minerva Endocrinol. 2006;31:47–60. [PubMed] [Google Scholar]
- 70.Suzuki T, Miki Y, Nakamura Y, et al. Sex steroid-producing enzymes in human breast cancer. Endocr. Relat. Cancer. 2005;12:701–720. doi: 10.1677/erc.1.00834. [DOI] [PubMed] [Google Scholar]
- 71.Pasqualini JR, Chetrite G, Blacker C, et al. Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients. J. Clin. Endocrinol. Metab. 1996;81:1460–1464. doi: 10.1210/jcem.81.4.8636351. [DOI] [PubMed] [Google Scholar]
- 72.Santen RJ, Santner SJ, Pauley RJ, et al. Estrogen production via the aromatase enzyme in breast carcinoma: which cell type is responsible? J. Steroid Biochem. Mol. Biol. 1997;61:267–271. [PubMed] [Google Scholar]
- 73.Nabholtz JM, Mouret-Reynier MA, Durando X, et al. Comparative review of anastrozole, letrozole and exemestane in the management of early breast cancer. Expert Opin. Pharmacother. 2009;10:1435–1447. doi: 10.1517/14656560902953738. [DOI] [PubMed] [Google Scholar]
- 74.Gray J, Evans N, Taylor B, Rizzo J, Walker M. State of the evidence: the connection between breast cancer and the environment. Int. J. Occup. Environ. Health. 2009;15:43–78. doi: 10.1179/107735209799449761. [DOI] [PubMed] [Google Scholar]
- 75.Wolff MS. Endocrine disruptors: challenges for environmental research in the 21st Century. Ann. NY Acad. Sci. 2006;1076:228–238. doi: 10.1196/annals.1371.009. [DOI] [PubMed] [Google Scholar]
- 76.Holloway AC, Anger DA, Crankshaw DJ, Wu M, Foster WG. Atrazine-induced changes in aromatase activity in estrogen sensitive target tissues. J. Appl. Toxicol. 2008;28:260–270. doi: 10.1002/jat.1275. [DOI] [PubMed] [Google Scholar]
- 77.Falconer IR. Are endocrine disrupting compounds a health risk in drinking water? Int. J. Environ. Res. Public Health. 2006;3:180–184. doi: 10.3390/ijerph2006030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mense SM, Hei TK, Ganju RK, Bhat HK. Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ. Health Perspect. 2008;116:426–433. doi: 10.1289/ehp.10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin JH, Crotty S, Nelson PN. Phytoestrogens: perpetrators or protectors? Future Oncol. 2007;3:307–318. doi: 10.2217/14796694.3.3.307. [DOI] [PubMed] [Google Scholar]
- 80.Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 81.Nilsson S, Makela S, Treuter E, et al. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 82.Gertler A, Grosclaude J, Strasburger CJ, Nir S, Djiane J. Real-time kinetic measurements of the interactions between lactogenic hormones and prolactin-receptor extracellular domains from several species support the model of hormone-induced transient receptor dimerization. J. Biol. Chem. 1996;271:24482–24491. doi: 10.1074/jbc.271.40.24482. [DOI] [PubMed] [Google Scholar]
- 83.Bai Z, Gust R. Breast cancer, estrogen receptor and ligands. Arch. Pharm. 2009;342:133–149. doi: 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- 84.Van Den Bemd GJ, Kuiper GG, Pols HA, Van Leeuwen JP. Distinct effects on the conformation of estrogen receptor α and β y both the antiestrogens ICI 164,384 and ICI 182,780 leading to opposite effects on receptor stability. Biochem. Biophys. Res. Commun. 1999;261:1–5. doi: 10.1006/bbrc.1999.0864. [DOI] [PubMed] [Google Scholar]
- 85.Normanno N, Di MM, De ME, et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr. Relat. Cancer. 2005;12:721–747. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 86.Khan SA, Bhandare D, Chatterton RT., Jr. The local hormonal environment and related biomarkers in the normal breast. Endocr. Relat. Cancer. 2005;12:497–510. doi: 10.1677/erc.1.00732. [DOI] [PubMed] [Google Scholar]
- 87.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 88.Manavathi B, Kumar R. Steering estrogen signals from the plasma membrane to the nucleus: two sides of the coin. J. Cell Physiol. 2006;207:594–604. doi: 10.1002/jcp.20551. [DOI] [PubMed] [Google Scholar]
- 89.Song RX, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor alpha in breast cancer. Endocr. Relat. Cancer. 2006;13(Suppl. 1):S3–S13. doi: 10.1677/erc.1.01322. [DOI] [PubMed] [Google Scholar]
- 90.Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids. 2007;72:124–134. doi: 10.1016/j.steroids.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 92.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol. Metab. 2005;16:362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 93.Filardo EJ, Graeber CT, Quinn JA, et al. Distribution of GPR30, a seven membrane- spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006;12:6359–6366. doi: 10.1158/1078-0432.CCR-06-0860. [DOI] [PubMed] [Google Scholar]
- 94.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 95.Rodrik V, Zheng Y, Harrow F, Chen Y, Foster DA. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol. Cell Biol. 2005;25:7917–7925. doi: 10.1128/MCB.25.17.7917-7925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang Y, Ray S, Reed JC, et al. Estrogen increases intracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis of human breast cancer MCF-7 cells. Breast Cancer Res. Treat. 1997;42:73–81. doi: 10.1023/a:1005777219997. [DOI] [PubMed] [Google Scholar]
- 97.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto- oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 98.Lagadec C, Adriaenssens E, Toillon RA, et al. Tamoxifen and TRAIL synergistically induce apoptosis in breast cancer cells. Oncogene. 2008;27:1472–1477. doi: 10.1038/sj.onc.1210749. [DOI] [PubMed] [Google Scholar]
- 99.Lapensee EW, Lapensee CR, Fox S, Schwemberger S, Afton S, Ben-Jonathan N. Bisphenol A and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett. 2010;290:167–173. doi: 10.1016/j.canlet.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to antiapoptosis in breast cancer. Mol. Endocrinol. 2000;14:1434–1447. doi: 10.1210/mend.14.9.0526. [DOI] [PubMed] [Google Scholar]
- 101.Zampieri L, Bianchi P, Ruff P, Arbuthnot P. Differential modulation by estradiol of P-glycoprotein drug resistance protein expression in cultured MCF7 and T47D breast cancer cells. Anticancer Res. 2002:2253–2259. [PubMed] [Google Scholar]
- 102.Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147:S56–S69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- 103.Kang JH, Kondo F, Katayama Y. Human exposure to bisphenol A. Toxicology. 2006;226:79–89. doi: 10.1016/j.tox.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 104.Le HH, Carlson EM, Chua JP, Belcher SM. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol. Lett. 2008;76:149–156. doi: 10.1016/j.toxlet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fernandez MF, Arrebola JP, Taoufiki J, et al. Bisphenol-A and chlorinated derivatives in adipose tissue of women. Reprod. Toxicol. 2007;24:259–264. doi: 10.1016/j.reprotox.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 106.Yi B, Kim C, Yang M. Biological monitoring of bisphenol A with HLPC/FLD and LC/MS/MS assays. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2010;878:2606–2610. doi: 10.1016/j.jchromb.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 107.Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr. Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. • Good discussion on the various aspects of the endocrine effects of bisphenol A.
- 108.Ben-Jonathan N, Hugo ER, Brandebourg TD. Effects of bisphenol A on adipokine release from human adipose tissue: implications for the metabolic syndrome. Mol. Cell Endocrinol. 2009;304:49–54. doi: 10.1016/j.mce.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Singleton DW, Feng Y, Chen Y, et al. Bisphenol-A and estradiol exert novel gene regulation in human MCF-7 derived breast cancer cells. Mol. Cell Endocrinol. 2004;221:47–55. doi: 10.1016/j.mce.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 110.Li X, Zhang S, Safe S. Activation of kinase pathways in MCF-7 cells by 17β-estradiol and structurally diverse estrogenic compounds. J. Steroid Biochem. Mol. Biol. 2006;8:122–132. doi: 10.1016/j.jsbmb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 111.Walsh DE, Dockery P, Doolan CM. Estrogen receptor independent rapid non-genomic effects of environmental estrogens on [Ca2+]i in human breast cancer cells. Mol. Cell Endocrinol. 2005;230:23–30. doi: 10.1016/j.mce.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 112.Kuiper GG, Lemmen JG, Carlsson B, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 113.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-α: estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005;70:364–371. doi: 10.1016/j.steroids.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 114.Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ. Health Perspect. 2008;116:1642–1647. doi: 10.1289/ehp.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Safe SH, Pallaroni L, Yoon K, Gaido K, Ross S, McDonnell D. Problems for risk assessment of endocrine-active estrogenic compounds. Environ. Health Perspect. 2002;10(Suppl. 6):925–929. doi: 10.1289/ehp.02110s6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr. Top. Med. Chem. 2006:203–215. doi: 10.2174/1568026610606030203. [DOI] [PubMed] [Google Scholar]
- 117.Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-γ. Environ. Health Perspect. 2008;116:32–38. doi: 10.1289/ehp.10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ariazi EA, Clark GM, Mertz JE. Estrogen- related receptor α and estrogen-related receptor γ associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002;62:6510–6518. [PubMed] [Google Scholar]
- 119.Lapensee EW, Tuttle TR, Fox SR, Ben-Jonathan N. Bisphenol A at low nanomolar doses confers chemoresistance in estrogen receptor-α-positive and -negative breast cancer cells. Environ. Health Perspect. 2009;117:175–180. doi: 10.1289/ehp.11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goffin V, Bernichtein S, Touraine P, Kelly PA. Development and potential clinical uses of human prolactin receptor antagonists. Endocr. Rev. 2005;26:400–422. doi: 10.1210/er.2004-0016. • A comprehensive review of the generation of human prolactin receptor antagonists by modifications of the prolactin molecule.
- 121.Jacobson EM, Hugo ER, Tuttle TR, Papoian R, Ben-Jonathan N. Unexploited therapies in breast and prostate cancer: blockade of the prolactin receptor. Trends Endocrinol. Metab. 2010;21:691–698. doi: 10.1016/j.tem.2010.08.004. • Evaluation of the screening and potential use of small molecule inhibitors of the prolactin receptor.