

Abstract
Members of the Gadd45 family play central roles in the cellular response to genotoxic stress, and have been implicated in several human cancers including hepatocellular carcinomas. Chronic infection by hepatitis C virus (HCV) is a major risk factor for the onset and development of primary hepatocellular tumors, although the underlying mechanisms are unclear. Here, we demonstrate a novel link between diminished Gadd45β expression and HCV infection. Inhibited Gadd45β expression was observed in both non-tumoral and tumoral tissues from infected individuals, and in cell lines harboring an HCV replicon and the infectious HCV strain JFH1. Decreased Gadd45β expression was confirmed in vivo in a transgenic murine model expressing the entire HCV open reading frame. Mechanistically, hypermethylation of the Gadd45β promoter in the presence of HCV is responsible for this defect. Diminished Gadd45β expression leads to aberrant cell cycle arrest and diminished DNA excision repair. Together, these results provide a novel insight into the mechanisms involved in HCV-associated hepatocellular carcinomas, showing that reduced Gadd45β expression may play a contributory role to this process, and providing evidence that HCV may interfere with epigenetic gene expression by altering promoter methylation.
Keywords: hepatocellular carcinoma, HCV, Gadd45β
Keywords: Adult; Aged; Animals; Antigens, Differentiation; Blotting, Western; Carcinoma, Hepatocellular; Cell Cycle; Cells, Cultured; DNA Methylation; DNA Repair; Down-Regulation; Female; Hepacivirus; Hepatitis C; Hepatocytes; Humans; Liver ; Liver Neoplasms; Luciferases; Male; Mice; Mice, Inbred C57BL; Mice, Transgenic; Middle Aged; Promoter Regions, Genetic; RNA, Messenger; RNA, Small Interfering; pharmacology; Reverse Transcriptase Polymerase Chain Reaction; Virus Replication
INTRODUCTION
Infection by the hepatotropic hepatitis C virus (HCV) is a leading cause of chronic liver disease, with more than 170 million chronically infected individuals worldwide. Chronic HCV infection is associated with the development of chronic hepatitis, fibrosis and cirrhosis, and is a major risk factor for the onset and progression of hepatocellular carcinoma (HCC) (1, 2). The molecular mechanisms surrounding liver tumorigenesis in HCV-infected patients remain unclear, although cirrhosis appears to be an important, but non-specific, determinant of HCC occurrence (3). Nevertheless, several studies have suggested that HCV itself (i.e. HCV protein expression in hepatocytes) plays a role in hepatocarcinogenesis. For example, the HCV core protein has been reported to interact with the p53 and promyelocytic leukemia (PML) tumor suppressors in hepatoma cell culture, to facilitate transformation of murine fibroblasts, and to promote oncogenesis in mice (4–9). However, the reported effects of the core-p53 interaction are inconsistent. Another HCV protein, nonstructural protein 5A (NS5A), has been reported to interact with p53 and the DNA damage effector ataxia telangiectasia mutated (ATM) kinase in hepatoma cell lines (10–12). Moreover, the expression of HCV proteins has been linked with up-regulated production of reactive oxygen species (ROS) (13, 14), and a consequent increase in levels of ROS-specific DNA lesions (15). Despite these findings, the requirement for hepatoma cell lines in the cultivation and study of both sub-genomic HCV replicons and infectious viral variants of HCV (e.g. the genotype 2a JFH1 strain) represents a major barrier to the study of liver tumorigenesis in the context of HCV infection.
The growth arrest and DNA damage (Gadd45) gene family encodes three highly conserved nuclear proteins that contribute to cellular homeostasis in response to a number of stressors (16). Several lines of evidence suggest that these proteins fulfill similar functions in cellular survival, cell cycle control, apoptosis and repair of DNA damage (17). Gadd45β has been demonstrated to interact with several key cellular regulators including cyclin B1, p21, proliferating cell nuclear antigen (PCNA) and mitogen-activated protein kinase 7 (MKK7) (16, 18–21). The cellular function of Gadd45β is dependent upon its interacting partner. Notably, Gadd45β is able to suppress G2/M progression in response to genotypic stress via its ability to interact with, and suppress the kinase activities, of the cyclinB1/cell division cycle 2 (cdc2) complex (20). Accordingly, RNA silencing of Gadd45β expression impairs G2/M checkpoint activity. It remains to be determined whether interactions between Gadd45β and p21 play a role in G1 arrest. Gadd45β has also been suggested to function in DNA excision repair through its interaction with PCNA (20, 22). This interaction may contribute towards the proposed role for Gadd45β in epigenetic gene activation (23, 24). Furthermore, several lines of evidence implicate Gadd45β in apoptosis, particularly in response to transforming growth factor β (TGF-β) (25, 26).
Such observations have implicated Gadd45 proteins, including Gadd45β, as crucial sensors of genotoxic stress. In line with this proposal, Gadd45β−/− mice display increased susceptibility to ionizing radiation and chemical carcinogenesis, and accelerated melanoma growth compared to wild-type (wt) littermates (27, 28). Accordingly, in human hepatocellular tumors, and in several hepatoma cell lines, Gadd45β expression is diminished (29). This diminution has been attributed to hypermethylation of the Gadd45β promoter, as treatment with DNA methyltransferase inhibitors restores Gadd45β expression to non-tumoral levels (30). Moreover, down-regulation of Gadd45β is closely associated with the degree of malignancy in tumoral samples. Further studies demonstrated that treatment of tumoral cell lines with the hepatoprotector S-adenosylmethione could stimulate nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB)-mediated Gadd45β transcription (31). Therefore, Gadd45β downregulation may play an important role in hepatocarcinogenesis.
Transgenic mice of the FL-N/35 lineage expressing the entire open reading frame (ORF) of a genotype 1b isolate of HCV have previously been shown to develop hepatic steatosis and hepatocellular carcinomas in the absence of cirrhosis (32, 33). These animals also exhibit impaired viral clearance and decreased expression of bid, a pro-apoptotic B-cell leukemia/lymphoma 2 (Bcl2)-homology domain 3 (BH3)-only protein which has also been implicated in the response to DNA damage (34, 35). Unlike other experimental systems for HCV infection, the entire complement of viral proteins is expressed at low levels, resembling those found in the livers of chronically-infected patients (32) and does not trigger non-specific ER stress (33). Moreover, viral protein expression in this model does not elicit an immune response, allowing the role of HCV proteins in tumorigenesis to be studied in the absence of the chronic inflammation usually observed during HCV infection.
In this context, we asked whether Gadd45β expression was specifically affected by HCV protein expression. To that end, we characterized Gadd45β expression in human tissue samples, in vitro systems of HCV replication and infection in hepatoma cell lines, and the FL-N/35 murine model of HCV protein expression.
MATERIALS AND METHODS
Human liver tissues
Tumoral and adjacent non-tumoral liver tissues from 7 patients with chronic HCV infection, including 4 with cirrhosis, were examined. Similar tissues from 5 uninfected patients and 5 patients infected with hepatitis B virus (HBV) were used as controls. The patients’ characteristics are detailed in Table 1.
Table 1.
Clinical and pathological features of patients with HCCs.
| Patient number | Sex | Age (years) | Etiology | HCV genotype | Activity/Fibrosis* | Number of tumors | Size of largest tumor (cm) | Differentiation‡ | Gadd45β protein expressionΔ |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 50 | HCV | ND | A2/F4 | 2 | 6 | Pd | 0 |
| 2 | M | 48 | HCV | 3a | F4 | 5 | 3 | Md/Pd | 0 |
| 3 | M | 67 | HCV | ND | F4 | 7 | 2 | Pd | 0 |
| 4 | M | 51 | HCV | 3a | F4 | 2 | 6 | Pd | 0 |
| 5 | M | 75 | HCV+alcohol | 1b | A2/F3 | 1 | 3 | Md | + |
| 6 | F | 71 | HCV | 1b | A1/F1 | 1 | 12 | Wd | + |
| 7 | M | 69 | HCV+alcohol | 2a | A1/F2 | 1 | 3 | Pd | 0 |
| 8 | M | 61 | HBV | NA | A1/F1 | 1 | 4 | Md | +++ |
| 9 | M | 57 | HBV | NA | A1/F2 | 2 | 3 | Pd | ++ |
| 10 | M | 45 | HBV | NA | A2/F2 | 1 | 13 | Md | ++ |
| 11 | M | 60 | HBV | NA | A1/F3 | 1 | 3 | Pd | ++ |
| 12 | M | 50 | HBV | NA | F3/F4 | 2 | 3 | Md | +++ |
| 13 | M | 78 | ND | NA | A0/F3 | 10 | 6 | Md | +++ |
| 14 | M | 73 | alcohol | NA | A1/F2 | 6 | 13 | Pd | +++ |
| 15 | M | 71 | alcohol | NA | F1 | 1 | 7 | Pd | + |
| 16 | M | 67 | alcohol | NA | F1 | 1 | 10 | Md | +++ |
| 17 | M | 33 | ND | NA | ND | 1 | 10 | Wd | +++ |
HCV: hepatitis C virus; HBV: hepatitis B virus; ND: not determined; NA: not applicable
According to Metavir scoring: A = activity: absent (A0); mild (A1); moderate (A2); severe (A3); F = fibrosis: absent (F0); mild (F1), moderate (F2), severe (F3); cirrhosis (F4)
According to Edmonson grade: Wd (well-differentiated); Md (moderately differentiated); Pd (poorly differentiated)
Gadd45β protein expression in tumoral tissues as determined by densitometric analysis of immunoblots. Scores: 0 = no expression; + = weak expression; ++ = moderate expression; +++ = strong expression.
Cell culture models of HCV replication and infection
Huh7.5 cells, and Huh7.5 cells harboring the genotype 1b bicistronic HCV subgenomic replicon I389-neo/NS3-3′/5.1, kindly provided by Dr. Ralf Bartenschlager (University of Heidelberg, Heidelberg, Germany), were maintained as previously described (36). Uninfected HuAP cells, and cells infected with the HCV infectious strain JFH-1, were a kind donation from Dr Czeslaw Wychowski (Institut de Biologie de Lille, CNRS-UMR 8161, Lille, France).
HCV transgenic mice
Wild-type C57/Bl6 mice and mice transgenic for the entire HCV open reading frame (FL-N/35 lineage (32)), were bred and maintained as previously described (34). Eighteen-month-old male littermates were used for transcriptional and proteomic studies. Ten-month-old females were used for primary hepatocyte cultures. For benzo[a]pyrene treatment, mice received a single intraperitoneal injection of 55 μg benzo[a]pyrene (Sigma-Aldrich, Saint Louis, Missouri) per gram of body weight, diluted in corn oil. Treated mice were euthanized 36 hours post treatment, the livers extracted and RNA isolated as described below.
Primary cell culture
Murine hepatocytes from both HCV transgenic and non-transgenic animals were isolated by portal vein perfusion of collagenase. Freshly isolated hepatocytes were cultured in DMEM supplemented with 10 % fetal calf serum, 10 U/mL penicillin, 10 μg/mL streptomycin, 10 μg/mL insulin, 5.5 μg/mL transferrin and 5 ng/mL sodium selenite. Four hours post perfusion, media was removed and fresh media, supplemented with 0.1 μM dexamethasone (Sigma-Aldrich) and 50 ng/mL epidermal growth factor, was added.
Plasmids and antibodies
Plasmid pGL2, containing firefly luciferase under the control of the SV40 promoter, was from Promega (Madison, Wisconsin), whilst pmaxGFP was from Lonza (Basel, Switzerland). A plasmid encoding the human Gadd45β promoter (−1604 to +141; pGL2-Gadd45β) upstream of firefly luciferase was kindly provided by Dr. Mary Goldring (Hospital for Special Surgery, Laboratory for Cartilage Research, New York) (37). The antibodies used in this study are outlined in Supplementary Materials.
RNA isolation and quantitative real-time PCR analysis
Total RNA was purified using the PARIS purification kit (Ambion, Austin, Texas). RNA integrity and quantity were determined using an Agilent Bioanalyser, with samples displaying an RNA integrity number of 6.8–8.1. Complementary DNA was synthesized using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, California). Quantitative PCR were performed with an Applied Biosystems 7300 Thermal Cycler using Taqman reagents. Primer information is supplied in Supplementary Materials. Statistical significance of the results was determined using a Mann-Whitney nonparametric test.
Methylation-specific PCR
Genomic DNA was isolated from murine livers by phenol chloroform extraction, and bisulphite treated (38). Amplification of the Gadd45β promoter in methylated and unmethylated states was performed by methylation-specific PCR using the primers described in Supplemental Materials.
Cell fractionation, tumor samples and western blotting
Nuclear fractions of murine livers were prepared using the Pierce NE-PER Nuclear Cytoplasmic extraction kit (ThermoFisher Scientific, Waltham, Massachusetts). Crude extracts of human liver samples and of cultured hepatocytes were prepared by homogenization in PBS containing 10 mM EDTA and protease inhibitors. In all cases, proteins were quantified using the BCA assay (ThermoFisher Scientific). For protein analysis, polypeptides from nuclear fractions or crude extracts were separated on 4–12% SDS-polyacrylamide gels, transferred to polyvinylidene fluoride membranes (GE Healthcare, Chalfont St. Giles, UK), and proteins were detected by immunoblotting. Where appropriate, relative protein expression was quantified using National Institutes of Health ImageJ software.
Luciferase reporter gene assays
Primary murine hepatocytes were co-transfected with 1 μg pmaxGFP and either 2 μg pGL2-Gadd45β or 2 μg pGL2, using Lipofectamine LTX (Invitrogen, Carlsbad, California). Four hours post transfection, monolayers were either mock treated or exposed to 10 J/m2 UV-C using a UV-C Crosslinker (GE Healthcare). Luciferase activity was assayed 40 hours post treatment using a Mithras LB 940 plate reader (Berthold Technologies, Bad Wilbad, Germany), and normalized to the expression of green fluorescent protein (GFP). Data are presented as the mean±standard error (SEM) of three experiments.
Mitotic index analysis
Freshly isolated murine hepatocytes plated on Permanox slides (ThermoFisher Scientific) were either mock treated or exposed to 5 J/m2 UV-C 4 hours post plating. Six hours post treatment, cells were fixed and processed for immunofluorescent detection of phospho-histone H3 as previously described (39). Images were captured using a Zeiss Axioskop 40 microscope in conjunction with a Zeiss MRc5 Axiocam and Axiovision LE software. The percent of phospho-histone H3 positive (mitotic) nuclei was calculated for each field of view at 40× magnification. Data represent the mean±SEM mitotic index of at least 300 cells from two independent cultures. Statistical significance was determined using a Mann-Whitney nonparametric test.
RNA silencing
Gadd45β expression in primary murine hepatocytes was suppressed by transfection with siRNA (Silencer Gadd45β siRNA s70735 and nonsilencing control 4398043, Applied Biosystems) by electroporation using a Mouse Hepatocyte Nucleofector kit (Lonza) prior to plating. Twenty hours after transfection, cells were left untreated, or treated with 5 J/m2 UV-C, and processed for mitotic index analysis or quantitative transcript analysis as appropriate.
5-azacytidine treatment
Attached primary hepatocytes were treated with 50 μM 5-azacytidine (Sigma-Aldrich) for 12 hours, before treatment with 5 J/m2 UV. Cells were harvested for quantitative transcript analysis or were examined for the presence of phospho-histone H3 by immunofluorescence.
Plasmid host cell reactivation assays
The ability of primary hepatocytes cultures to repair a UV-C damaged reporter was assayed using a host cell reactivation assay (40, 41). Briefly, cultures of attached primary hepatocytes were cotransfected with UV-C-damaged (15 KJ/m2) pGL2, together with pGFP to control for transfection efficiency. Luciferase activity was assayed 48 hours post-transfection, and normalized to GFP expression. Relative DNA repair efficiency was calculated, and is presented as the mean±SEM of three experiments.
RESULTS
Gadd45β expression is inhibited in tumoral and non-tumoral liver tissues from HCV-infected patients
We measured Gadd45β expression in tumoral and adjacent non-tumoral liver tissues from patients with HCC and chronic HCV infection with or without cirrhosis. Patients infected with HBV only and non-infected patients were used as controls (Table 1). Immunoblotting revealed that Gadd45β was equivalently expressed in non-tumoral tissues from both uninfected and HBV-infected patients. In contrast, Gadd45β was undetectable in non-tumoral tissues from both cirrhotic and non-cirrhotic HCV-infected patients (Fig. 1A and 1B). In tumoral tissues, Gadd45β expression was nearly undetectable in uninfected and HBV-infected patients, and remained undetectable in HCV-infected patients (Fig. 1A and 1B).
Fig. 1. Gadd45β expression in tumoral and non-tumoral tissues from HCV-infected patients.

(A) Gadd45β protein expression in tumoral (T) and adjacent non-tumoral (NT) liver samples from uninfected (HBV−/HCV−), HBV-infected (HBV+) or HCV-infected (HCV+) patients. Tissue samples were analyzed by western blotting, and bands corresponding to 18 kDa Gadd45β or 45 kDa actin are indicated. (B) Densitometric quantification of multiple western blot analyses of tumoral and non-tumoral tissues from HBV−/HCV− (n=5), HBV+ (n=5) and HCV+ (n=7) patients. Gadd45β protein levels were normalized to the expression of actin, and mean±SEM densities are displayed in relation to non-tumoral non-infected (HBV−/HCV−) tissues. (C) Gadd45β mRNA expression in non-tumoral and tumoral liver tissues from HBV−/HCV− (n=5), HBV+ (n=5) and HCV+ (n=7) patients. Expression levels of Gadd45β and SFRS4 transcripts were determined by real-time PCR. Data were normalized to expression of SFRS4, and expressed as the mean±SEM mRNA expression. (D) Gadd45α and Gadd45γ mRNA expression in non-tumoral and tumoral liver tissues from HBV−/HCV− (n=5), HBV+ (n=5) and HCV+ (n=7) patients. Gadd45α, Gadd45γ and SFRS4 transcripts were quantified by real-time PCR. Data were normalized to SFRS4 expression, and expressed as the mean±SEM mRNA expression for each gene. None of the comparisons showed significant differences.
Gadd45β transcripts were quantified by quantitative RT-PCR in the same samples. As shown in Fig. 1C, Gadd45β was similarly expressed in non-tumoral tissues from both uninfected and HBV-infected patients. It was reduced at least two-fold on average (p=0.013) in tumors from these individuals when compared to adjacent non-tumoral tissues. In HCV-infected patients, Gadd45β mRNA levels were significantly lower in non-tumoral tissues than in uninfected and HBV-infected controls (p=0.02). Gadd45β mRNA levels were also lower in tumors from HCV-infected patients than in the controls (p=0.02; Fig. 1C). In contrast, the levels of neither Gadd45α nor Gadd45γ were significantly affected by the presence of HCV in either tumoral or non-tumoral tissues (Fig. 1D). Decreased Gadd45β expression was also observed in HCV-infected patients with no history of HCC (data not shown).
Correlations between Gadd45β protein expression and patients’ clinicopathological features (Table 1) were examined using Spearman’s non-parametrical test. Such analyses verified that reduced Gadd45β expression strongly correlated (p=0.007) with HCV infection, and confirmed previous observations (29) that Gadd45β expression was associated with differentiation (p=0.02). No relationship was observed between Gadd45β expression and either tumor size or frequency. Together, these data indicate that Gadd45β expression is down-regulated in liver tissues from HCV-infected individuals.
Gadd45β expression is inhibited in cultured hepatocytes harboring a sub-genomic replicon or the infectious HCV strain JFH1
The expression of the Gadd45β protein was next examined in cultured hepatoma cell lines harboring a replicating genotype 1b subgenomic replicon or the genotype 2a infectious HCV strain JFH1. Immunoblotting analysis revealed that Gadd45β expression was reduced in both models compared to uninfected cells (Fig. 2A and 2B, respectively).
Fig. 2. Gadd45β expression in hepatocyte cell lines harboring an HCV genotype.
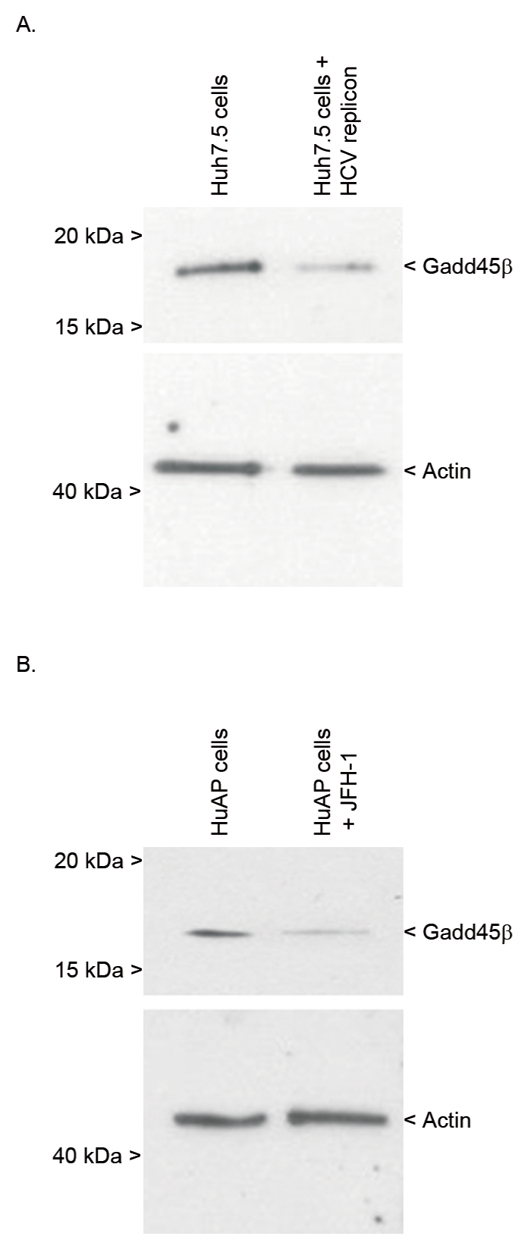
1b replicon or infected with the HCV genotype 2a JFH1 strain. (A) Immunoblotting analysis of Gadd45β expression in Huh7.5 cells in the absence and presence of an HCV genotype 1b subgenomic replicon. (B) Immunoblotting analysis of Gadd45β expression in mock-infected HuAP cells and cells infected with the full length HCV infectious variant JFH1. In each case, whole cell lysates of cultured cells were analyzed by western blotting, with bands corresponding to 18 kDa Gadd45β or 45 kDa actin indicated.
Expression of the entire HCV ORF inhibits Gadd45β expression in transgenic mice
In order to assess the direct role of HCV protein expression in Gadd45β down-regulation in HCV-infected patients, we set out to confirm our previous observations in the absence of HCV replication and local inflammation in the FL-N/35 mouse lineage, which expresses the entire HCV ORF in a liver-specific fashion. Analysis of liver-specific Gadd45β protein and mRNA expression revealed a two-fold decrease in Gadd45β expression in FL-N/35 mice compared to their wt littermates (p=0.016; Fig. 3A and 3B). Gadd45β expression reduction was liver-specific (data not shown).
Fig. 3. Gadd45β expression in FL-N/35 mice transgenic for the entire HCV open reading frame.

(A) Western blot analysis of Gadd45β expression in FL-N/35 and wt mice using representative samples from each genotype. Nuclear extracts of isolated livers were analyzed for Gadd45β and lamin A/C expression, and bands corresponding to these proteins are indicated. (B) Liver-specific Gadd45β mRNA expression in untreated and benzo[a]pyrene-treated wt and FL-N/35 mice. The expression levels of Gadd45β, GusB, GAPDH and HPRT1 were determined by quantitative real-time PCR. The results were normalized to expression of GAPDH, GusB and HPRT1, and the mean±SEM mRNA quantities are expressed in relation to untreated wt controls. Statistical significance was analyzed using a Mann-Whitney nonparametric test. (C) Gadd45β promoter activity in FL-N/35 hepatocytes. Isolated hepatocytes from wt or FL-N/35 animals were cotransfected with pmaxGFP and a plasmid expressing firefly luciferase under the control of either the human Gadd45β promoter (pGL2-Gadd45β-luc) or the SV40 promoter (pGL2), and either mock irradiated or treated with UV-C. The mean±SEM luciferase levels (normalized to GFP expression) from three independent experiments are shown relative to those obtained in untreated wt hepatocytes. The statistical significances of the data were analyzed with a Mann-Whitney nonparametric test.
Both wt and FL-N/35 mice were then treated with a single intraperitoneal injection of benzo[a]pyrene, which induces mutagenic DNA lesions repaired by nucleotide excision repair (42). Gadd45β mRNA levels increased after benzo[a]pyrene treatment in both wt and FL-N/35 mice, but to a markedly lesser extent in FL-N/35 mice (3.5-fold vs 5.5-fold in the wt mice) (Fig. 3B). Expression of other Gadd family members in this model was not affected by the expression of the HCV transgene, even in the presence of benzo[a]pyrene (data not shown). Collectively, these findings demonstrate that HCV protein expression is responsible for Gadd45β expression inhibition in vivo.
Gadd45β promoter activity is decreased in primary hepatocytes from FL-N/35 mice
In order to understand the mechanisms underlying inhibition of Gadd45β expression by the HCV proteins, we sought to determine whether Gadd45β promoter activity was affected in this context. Isolated hepatocytes from FL-N/35 mice or their wt littermates were transfected with a reporter plasmid expressing luciferase under the control of either the SV40 promoter (pGL2) or the human Gadd45β promoter (pGL2-Gadd45β-luc). As shown in Fig. 3C, basal activity of the Gadd45β promoter was significantly decreased in hepatocytes isolated from FL-N/35 mice compared to wt controls. Moreover, although acute UV-C treatment stimulated promoter activity in both wt and FL-N/35 hepatocytes, such activity in UV-treated FL-N/35 cells remained significantly reduced compared to UV-treated wt controls. Expression of the HCV proteins had no effect on basal activity of the SV40 promoter of pGL2, indicating that the HCV proteins do not induce global promoter repression. These data imply that the observed reduction in Gadd45β expression is a consequence of impaired promoter activity in the presence of HCV.
The Gadd45β promoter is hypermethylated in HCV transgenic mice
Decreased Gadd45β gene expression in tumoral cell lines has previously been attributed to hypermethylation of the Gadd45β promoter. This hypermethylation can be alleviated by in vitro treatment with the DNA methyltransferase inhibitor 5-azacytidine (30). In order to investigate whether decreased Gadd45β promoter activity in FL-N/35 hepatocytes could be due to its hypermethylation, isolated hepatocytes from wt and FL-N/35 animals were treated with 5-azacytidine for 12 hours, and then either mock-irradiated or treated with UV-C. Cells were harvested six hours post UV-treatment and analyzed for expression of Gadd45β mRNA (Fig. 4A). Isolated hepatocytes from FL-N/35 mice displayed reduced Gadd45β levels in both the presence and absence of acute UV-C treatment. However, 5-azacytidine treatment restored Gadd45β mRNA expression to wt levels in both mock irradiated and UV-irradiated FL-N/35 cells. To confirm these data, methylation-specific PCR was performed on hepatocyte DNA from wt or FL-N/35 mice. Methylation of the Gadd45β promoter was observed solely in FL-N/35 mice (Fig. 4B). Collectively, these results demonstrate that inhibition of Gadd45β promoter activity by HCV is due to promoter hypermethylation.
Fig. 4. Gadd45β promoter hypermethylation in FL-N/35 mice.

(A) Expression of the Gadd45β transcript in mock irradiated or UV-irradiated primary hepatocytes from wt or FL-N/35 animals in the presence and absence of 5-azacytidine. Gadd45β and actin mRNA expression levels were determined by quantitative RT-PCR. Data from three independent experiments were normalized to the expression of β-actin, and are expressed relative to untreated wt controls. The statistical significance of differences was analyzed with a Mann–Whitney nonparametric test. (B) Analysis of Gadd45β promoter methylation in hepatocyte DNA from wt or FL-N/35 animals by methylation-specific PCR. U indicates unmethylated PCR products, M denotes hypermethylated PCR products.
Down-regulation of Gadd45β expression by HCV proteins leads to defective cell cycle arrest
Gadd45β is involved in the induction of G2/M arrest and increased Gadd45β expression is associated with a decreased mitotic index (20, 43). Thus, we assessed whether reduced Gadd45β expression in hepatocytes isolated from HCV transgenic FL-N/35 mice was associated with defective cell cycle arrest and increased G2/M progression in the presence of DNA damage. To this end, isolated hepatocytes from FL-N/35 or wt animals were mock-treated or exposed to 5 J/m2 UV-C to induce transient cell cycle arrest. Six hours post treatment, cells undergoing mitosis were identified by immunofluorescence using an antibody to phosphorylated histone H3 (Ser-10) (Fig. 5A), and the mitotic index was calculated.
Fig. 5. Mitotic index and DNA repair analyses in primary wt or FL-N/35 hepatocytes.

(A) Immunofluorescent detection of phosphorylated histone-H3. Cells were analyzed for the presence of phosphorylated histone-H3 (left panel), and nuclei were counterstained with DAPI (right panel). A representative image (40X magnification) of phosphorylated histone-H3 and DAPI signals is shown. Scale bar = 10 μm. (B) Quantitative analysis of mitotic indices. Isolated cells receiving the indicated siRNA or a non-silencing control were subsequently either mock treated or treated with 5-azacytidine for 12 hours. Monolayers were then mock irradiated or treated with UV-C, and the presence of phosphorylated histone-H3 was analyzed six hours after irradiation. For each field of view (40X magnification), phosphorylated histone-H3-positive and DAPI-positive cells were quantified. Data are expressed as the percentage of phospho-histone H3 positive cells per field of view, and represent the analysis of at least 300 cells from two independent experiments. The statistical significance of differences was analyzed with a Mann-Whitney nonparametric test (denoted in text). (C) DNA excision repair ability of primary wt and FL-N/35 hepatocytes. Primary hepatocytes isolated from wt or FL-N/35 animals were cotransfected with pmaxGFP and UV-C-irradiated pGL2. Luciferase levels were assayed 48 hours post transfection, and normalized to GFP expression. DNA repair efficiency was calculated relative to wt control cultures, and mean±SEM repair efficiency from three independent experiments is shown. Statistical significance was analyzed with a Mann-Whitney nonparametric test.
Mitotic index analyses revealed no significant difference between basal mitotic indices in hepatocytes from wt and FL-N/35 mice (Fig. 5B). Upon UV-C treatment, the percentage of mitotic wt cells decreased significantly (p=0.001), denoting cell cycle arrest to allow repair of DNA damage. Inhibition of Gadd45β expression by RNA silencing completely abrogated UV-induced cell cycle arrest, with cells displaying mitotic indices close to those observed in untreated cells (not significant compared to untreated cultures; Fig. 5B). In FL-N/35 mice hepatocytes, the percentage of mitotic cells also decreased upon UV-C treatment, although to a lesser extent than in wt cells. Overall, a significantly greater percentage of mitotic FL-N/35 hepatocytes were observed after exposure to UV-C compared to wt cultures (p=0.001; Fig. 5B), suggesting that cell cycle arrest in response to DNA damage in HCV transgenic cells was defective. RNA silencing of Gadd45β did not significantly increase mitotic indexes in UV-C treated FL-N/35 mice (Fig. 5B), suggesting that the previously observed decrease in Gadd45β expression was sufficient to perturb temporary cell cycle arrest in response to UV-C.
In order to affirm the role of Gadd45β promoter hypermethylation in altering cell cycle arrest in HCV transgenic cells, UV-C exposed wt and FL-N/35 cells were treated with 5-azacytidine. Reactivation of Gadd45β expression by 5-azacytidine in FL-N/35 cells was able to restore UV-C-induced cell cycle arrest to wt levels (p=0.001; Fig. 5B). Cells in which Gadd45β expression was inhibited by RNA silencing prior to 5-azacytidine treatment displayed abrogated cell cycle arrest, with mitotic indices close to mock irradiated controls, suggesting that the observed restorative effect of 5-azacytidine was specifically due to increased Gadd45β expression (Fig 5B). These experiments demonstrate that inhibited hepatic Gadd45β expression in the context of HCV protein expression is due to hypermethylation of the Gadd45β promoter, and is associated with impaired cell cycle arrest in response to DNA damage.
Down-regulation of Gadd45β expression by HCV proteins adversely affects repair of damaged DNA
As Gadd45β is important for efficient nucleotide excision repair of damaged DNA, we examined whether decreased Gadd45β expression in FL-N/35 hepatocytes was associated with reduced DNA excision repair using a host cell reactivation assay. This assay permits the indirect analysis of the ability of cells to perform nucleotide excision repair of UV-C-induced TT or TC dimers in a reporter plasmid by measuring transcription of the corresponding reporter gene (40, 41).
Hepatocytes isolated from wt and FL-N/35 mice were transfected with UV-irradiated pGL2, and relative DNA repair calculated for three independent experiments from the resulting luciferase activity. As demonstrated in Fig. 5C, relative levels of luciferase activity, and thus DNA repair, were consistently and significantly (p=0.02) lower in cells isolated from FL-N/35 mice compared to wt controls. These data suggest that, in addition to impaired cell cycle arrest, HCV-mediated hypermethylation of the Gadd45β promoter also has a deleterious impact on repair of damaged DNA.
DISCUSSION
Despite advances in our knowledge of HCV-host cell interactions, the basis for HCV-associated hepatocarcinogenesis remains unclear. In infected humans, cirrhosis appears to play a major role as very few non-cirrhotic HCV-infected patients develop primary liver tumors. However, it is not clear whether the carcinogenetic process is triggered by the expression of HCV viral proteins. Gadd45 family members play a central role in cell cycle regulation and DNA repair, and have been linked with hepatocarcinogenesis (27, 29, 30, 44). In the present study, we demonstrate a previously unknown link between HCV infection and diminished Gadd45β expression. Firstly, we show, in liver biopsies from HCV-infected patients, significantly lessened Gadd45β mRNA and protein expression in both tumoral and non-tumoral tissues, in the presence and absence of cirrhosis. Gadd45β expression was also diminished in hepatoma cell lines harboring either an HCV subgenomic replicon or the infectious JFH1 strain. However, hepatoma-based cell culture models are not ideal for the study of HCV-related HCC development, as they are tumor-derived, and conflicting results have been reported (45). Importantly, similar reductions in Gadd45β expression were apparent in non-tumoral tissues from a transgenic mouse model expressing the entire HCV open reading frame, a model that develops hepatocellular carcinogenesis in the absence of chronic inflammation (32), suggesting that our observations were a direct consequence of HCV protein expression. Although Gadd45γ mRNA expression was reduced in tumoral human tissues compared to adjacent controls, we observed no effect of HCV on the expression of either Gadd45α or Gadd45γ in non-tumoral tissues. Thus, we provide the first evidence that Gadd45β expression is specifically disrupted by HCV protein expression.
Mechanistically, the Gadd45β promoter was shown to be hypermethylated in the presence of HCV. Treatment with the DNA methyltransferase suicide inhibitor 5-azacytidine restored both Gadd45β expression and cell cycle arrest. The effects of such treatment could be completely abrogated by RNA silencing of Gadd45β expression, confirming that the effect of 5-azacytidine in this context was confined to Gadd45β. Methylation-specific PCR confirmed hypermethylation of the Gadd45β promoter in HCV transgenic mice. The mechanisms by which HCV alters gene methylation during infection remain to be explored. In addition, our data suggests that alternative mechanisms are responsible for hypermethylation of the Gadd45β and Gadd45γ promoters during tumorigenesis in the presence or absence of HCV.
Previously, it was demonstrated that inhibition of Gadd45β expression by RNA interference perturbed G2/M checkpoint activity (20). Accordingly, we demonstrated that Gadd45β silencing abrogated UV-induced cell cycle arrest in murine primary hepatocytes. Consistent with these data, hepatocytes isolated from HCV transgenic mice exhibited not only diminished Gadd45β expression, but also decreased Gadd45β promoter activity, defective cell cycle arrest and DNA repair. Aberrant cell cycle arrest contributes to the development of somatic mutations, and therefore may be a contributing factor in the onset and progression of hepatocellular carcinoma. Moreover, deregulation of Gadd45β expression by HCV may directly provoke further changes in methylation patterns of several genes, since Gadd45β has itself been implicated in epigenetic regulation (23). Since epigenetic inactivation of tumor suppressors and cycle inhibitors has been implicated in the development of several human cancers including HCCs (46–48), our data suggest that similar mechanisms might play an important role in the onset and progression of HCV-associated HCC. It should be emphasized that Gadd45β downregulation in our transgenic mouse model occurred in the absence of chronic inflammation or cirrhosis, suggesting that expression of the viral proteins contribute directly to the development of HCC.
In conclusion, our data indicate that HCV proteins have a direct effect on liver tumorigenesis by promoting the hypermethylation of the Gadd45β promoter in the absence of chronic inflammation, impacting DNA repair, epigenetic gene control and cell cycle regulation. Further study is underway to determine the effect of the HCV proteins on the promoter methylation of other genes implicated in carcinogenesis.
Acknowledgments
Financial support: M. Higgs was the recipient of a Post-doctoral Fellowship and a Grant from the Agence Nationale de Recherche sur le SIDA et les Hépatites Virales (ANRS). H. Lerat was supported by a Grant from ANRS/Fondation de France, by INSERM (Young Investigator Program), and by a Grant from the European Community 6th Framework Programme (Marie Curie International Reintegration Grant).
We thank Aurore Gaudin for technical assistance; Philippe Chouteau for manuscript comments; Czeslaw Wychowski and Mary Goldring for JFH-1 infected cultures and pGL2-Gadd45β-luc, respectively; Paulette Bioulac-Sage for patient biopsies; and Ralf Bartenschlager for the HCV genotype 1b subgenomic replicon, established in our laboratory by Abdelhakim Ahmed-Belkacem.
Footnotes
Conflicts of interest: none.
References
- 1.El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36(5 Suppl 1):S74–83. doi: 10.1053/jhep.2002.36807. [DOI] [PubMed] [Google Scholar]
- 2.Saito I, Miyamura T, Ohbayashi A, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87(17):6547–9. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simonetti RG, Camma C, Fiorello F, et al. Hepatitis C virus infection as a risk factor for hepatocellular carcinoma in patients with cirrhosis. A case-control study. Ann Intern Med. 1992;116(2):97–102. doi: 10.7326/0003-4819-116-2-97. [DOI] [PubMed] [Google Scholar]
- 4.Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4(9):1065–7. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida I, Oka K, Hidajat R, Nagano-Fujii M, Ishido S, Hotta H. Inhibition of p21/Waf1/Cip1/Sdi1 expression by hepatitis C virus core protein. Microbiol Immunol. 2001;45(10):689–97. doi: 10.1111/j.1348-0421.2001.tb01303.x. [DOI] [PubMed] [Google Scholar]
- 6.Kwun HJ, Jang KL. Dual effects of hepatitis C virus Core protein on the transcription of cyclin-dependent kinase inhibitor p21 gene. J Viral Hepat. 2003;10(4):249–55. doi: 10.1046/j.1365-2893.2003.00434.x. [DOI] [PubMed] [Google Scholar]
- 7.Kao CF, Chen SY, Chen JY, Wu Lee YH. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene. 2004;23(14):2472–83. doi: 10.1038/sj.onc.1207368. [DOI] [PubMed] [Google Scholar]
- 8.Herzer K, Weyer S, Krammer PH, Galle PR, Hofmann TG. Hepatitis C virus core protein inhibits tumor suppressor protein promyelocytic leukemia function in human hepatoma cells. Cancer Res. 2005;65(23):10830–7. doi: 10.1158/0008-5472.CAN-05-0880. [DOI] [PubMed] [Google Scholar]
- 9.Sato Y, Kato J, Takimoto R, et al. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut. 2006;55(12):1801–8. doi: 10.1136/gut.2005.070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan KH, Sheu ML, Hwang SJ, et al. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene. 2002;21(31):4801–11. doi: 10.1038/sj.onc.1205589. [DOI] [PubMed] [Google Scholar]
- 11.Majumder M, Ghosh AK, Steele R, et al. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology. 2002;294(1):94–105. doi: 10.1006/viro.2001.1309. [DOI] [PubMed] [Google Scholar]
- 12.Ariumi Y, Kuroki M, Dansako H, et al. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J Virol. 2008;82(19):9639–46. doi: 10.1128/JVI.00351-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006;80(14):7199–207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishina S, Hino K, Korenaga M, et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008;134(1):226–38. doi: 10.1053/j.gastro.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Farinati F, Cardin R, Bortolami M, et al. Hepatitis C virus: from oxygen free radicals to hepatocellular carcinoma. J Viral Hepat. 2007;14(12):821–9. doi: 10.1111/j.1365-2893.2007.00878.x. [DOI] [PubMed] [Google Scholar]
- 16.Liebermann DA, Hoffman B. Gadd45 in the response of hematopoietic cells to genotoxic stress. Blood Cells Mol Dis. 2007;39(3):329–35. doi: 10.1016/j.bcmd.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cretu A, Sha X, Tront J, Hoffman B, Liebermann DA. Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther. 2009;7(A):268–76. [PMC free article] [PubMed] [Google Scholar]
- 18.Vairapandi M, Balliet AG, Fornace AJ, Jr, Hoffman B, Liebermann DA. The differentiation primary response gene MyD118, related to GADD45, encodes for a nuclear protein which interacts with PCNA and p21WAF1/CIP1. Oncogene. 1996;12(12):2579–94. [PubMed] [Google Scholar]
- 19.Vairapandi M, Azam N, Balliet AG, Hoffman B, Liebermann DA. Characterization of MyD118, Gadd45, and proliferating cell nuclear antigen (PCNA) interacting domains. PCNA impedes MyD118 AND Gadd45-mediated negative growth control. J Biol Chem. 2000;275(22):16810–9. doi: 10.1074/jbc.275.22.16810. [DOI] [PubMed] [Google Scholar]
- 20.Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol. 2002;192(3):327–38. doi: 10.1002/jcp.10140. [DOI] [PubMed] [Google Scholar]
- 21.Tornatore L, Marasco D, Dathan N, et al. Gadd45 beta forms a homodimeric complex that binds tightly to MKK7. J Mol Biol. 2008;378(1):97–111. doi: 10.1016/j.jmb.2008.01.074. [DOI] [PubMed] [Google Scholar]
- 22.Smith ML, Ford JM, Hollander MC, et al. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol. 2000;20(10):3705–14. doi: 10.1128/mcb.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135(7):1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma DK, Guo JU, Ming GL, Song H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle. 2009;8(10):1526–31. doi: 10.4161/cc.8.10.8500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoo J, Ghiassi M, Jirmanova L, et al. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278(44):43001–7. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 26.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95(4):521–30. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 27.Liebermann DA, Hoffman B. Gadd45 in stress signaling. J Mol Signal. 2008;3:15. doi: 10.1186/1750-2187-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ju S, Zhu Y, Liu L, et al. Gadd45b and Gadd45g are important for anti-tumor immune responses. Eur J Immunol. 2009;39(11):3010–8. doi: 10.1002/eji.200839154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu W, David D, Zhou B, et al. Down-regulation of growth arrest DNA damage-inducible gene 45beta expression is associated with human hepatocellular carcinoma. Am J Pathol. 2003;162(6):1961–74. doi: 10.1016/s0002-9440(10)64329-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu W, Zhou B, Zou H, et al. Hypermethylation of growth arrest DNA damage-inducible gene 45 beta promoter in human hepatocellular carcinoma. Am J Pathol. 2004;165(5):1689–99. doi: 10.1016/s0002-9440(10)63425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu W, Zhou B, Chu PG, Luh F, Yen Y. The induction of growth arrest DNA damage-inducible gene 45 beta in human hepatoma cell lines by S-adenosylmethionine. Am J Pathol. 2007;171(1):287–96. doi: 10.2353/ajpath.2007.070121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122(2):352–65. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 33.Lerat H, Kammoun HL, Hainault I, et al. Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. J Biol Chem. 2009;284(48):33466–74. doi: 10.1074/jbc.M109.019810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Disson O, Haouzi D, Desagher S, et al. Impaired clearance of virus-infected hepatocytes in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology. 2004;126(3):859–72. doi: 10.1053/j.gastro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Simonin Y, Disson O, Lerat H, et al. Calpain activation by hepatitis C virus proteins inhibits the extrinsic apoptotic signaling pathway. Hepatology. 2009;50(5):1370–9. doi: 10.1002/hep.23169. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed-Belkacem A, Ahnou N, Barbotte L, et al. Silibinin and Related Compounds are Direct Inhibitors of Hepatitis C Virus RNA-Dependent RNA Polymerase. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.11.053. [DOI] [PubMed] [Google Scholar]
- 37.Ijiri K, Zerbini LF, Peng H, et al. A novel role for GADD45beta as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J Biol Chem. 2005;280(46):38544–55. doi: 10.1074/jbc.M504202200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1(5):2353–64. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]
- 39.Dmitrieva NI, Bulavin DV, Fornace AJ, Jr, Burg MB. Rapid activation of G2/M checkpoint after hypertonic stress in renal inner medullary epithelial (IME) cells is protective and requires p38 kinase. Proc Natl Acad Sci U S A. 2002;99(1):184–9. doi: 10.1073/pnas.231623498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol. 1998;72(1):266–72. doi: 10.1128/jvi.72.1.266-272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80(6):875–9. doi: 10.1002/(sici)1097-0215(19990315)80:6<875::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 42.Maher VM, Curren RD, Oulette LM, McKormick JJ. In: De Serres FJ, editor. Role of DNA repair in the cytotoxic and mutagenic action of physical and chemical carcinogens; In Vitro Metabolic Activation in Mutagenesis Testing: Proceedings of the Symposium on the Role of Metabolic Activation in Producing Mutagenic and Carcinogenic Environmental Chemicals; Research Triangle Park, North Carolina. February 9–11, 1976; Amsterdam: Elsevier/North-Holland; 1976. pp. 313–6. [Google Scholar]
- 43.Mak SK, Kultz D. Gadd45 proteins induce G2/M arrest and modulate apoptosis in kidney cells exposed to hyperosmotic stress. J Biol Chem. 2004;279(37):39075–84. doi: 10.1074/jbc.M406643200. [DOI] [PubMed] [Google Scholar]
- 44.Sun L, Gong R, Wan B, et al. GADD45gamma, down-regulated in 65% hepatocellular carcinoma (HCC) from 23 chinese patients, inhibits cell growth and induces cell cycle G2/M arrest for hepatoma Hep-G2 cell lines. Mol Biol Rep. 2003;30(4):249–53. doi: 10.1023/a:1026370726763. [DOI] [PubMed] [Google Scholar]
- 45.Walters KA, Syder AJ, Lederer SL, et al. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009;5(1):e1000269. doi: 10.1371/journal.ppat.1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1(1):11–9. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 47.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 48.Roncalli M, Bianchi P, Bruni B, et al. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma. Hepatology. 2002;36(2):427–32. doi: 10.1053/jhep.2002.34852. [DOI] [PubMed] [Google Scholar]