


Abstract
We present a new version of miRanalyzer, a web server and stand-alone tool for the detection of known and prediction of new microRNAs in high-throughput sequencing experiments. The new version has been notably improved regarding speed, scope and available features. Alignments are now based on the ultrafast short-read aligner Bowtie (granting also colour space support, allowing mismatches and improving speed) and 31 genomes, including 6 plant genomes, can now be analysed (previous version contained only 7). Differences between plant and animal microRNAs have been taken into account for the prediction models and differential expression of both, known and predicted microRNAs, between two conditions can be calculated. Additionally, consensus sequences of predicted mature and precursor microRNAs can be obtained from multiple samples, which increases the reliability of the predicted microRNAs. Finally, a stand-alone version of the miRanalyzer that is based on a local and easily customized database is also available; this allows the user to have more control on certain parameters as well as to use specific data such as unpublished assemblies or other libraries that are not available in the web server. miRanalyzer is available at http://bioinfo2.ugr.es/miRanalyzer/miRanalyzer.php.
INTRODUCTION
Short non-coding RNA molecules such as microRNAs play important roles in the regulation of gene expression (1). They have been recognized as key players in many basic pathways, and their aberrant expression is implicated in numerous diseases such as cancer (2). With the advent of high-throughput sequencing (HTS) technologies, it is now possible to rapid and inexpensively measure the expression levels of known microRNAs and to improve the prediction of new microRNAs by including the expression data into the prediction models (3). Not in vain, the number of HTS experiments aiming to study microRNA expression has rapidly increased over the past few years. For example, the number of entries for ‘(Illumina Genome Analyzer) AND microRNA’ in the GEO repository (4) are 12, 33 and 107 for 2008, 2009 and 2010, respectively. These numbers show a clear tendency that is expected to be even stronger when HTS techniques become cheaper and more accessible to everyone.
A number of algorithms have been developed in order to process these large amounts of data (5–8). Two years ago, we developed miRanalyzer (9), a tool for the detection of known and prediction of new microRNAs in HTS experiments. Here, we describe a new version of the tool, which has been completely redesigned and includes various new features. First, the alignments are now based on the ultrafast short read aligner Bowtie (10) that grants full colour space support, allows mismatches in the alignment of the read to the genome and is faster and more memory efficient than the previously implemented alignment algorithm. Second, the scope of the tool is extended to 31 species (including 6 plants) and allows to easily adding new ones. Third, the tool has no restriction on the number of input sequences for the prediction of new microRNAs, and the training of the prediction models takes into account differences between plant and animal microRNAs (11). Fourth, we have implemented a module, based on the DESeq package (12), to detect differential expression of microRNAs between two conditions. Additionally, taking advantage of the fact that multiple samples are needed for this last module, we have also implemented the computation of the consensus sequences for predicted mature and precursor microRNAs. This will help assessing the reliability of the predictions, i.e. microRNAs predicted in different samples are more likely to be functional than those predicted in just one sample. Finally, we have prepared a standalone version of the miRanalyzer tool that works with an easily customized local file-based database.
miRanalyzer UPDATED
miRanalyzer workflow
Although some features have changed, the general workflow of the current version is broadly maintained (Figure 1). Two input formats are accepted: (i) read-count files (read sequences and counts tab separated), which can be generated from sequence or colour space fastq files using a provided perl script or generated by other means by the user, and (ii) multi-fasta files (see tutorial on the web page for more details). In a first step, the tool removes all reads with ‘N’ (or other irregular bases) and those shorter than 17 bases, and reads longer than 26 bases are trimmed and regrouped. The reads are then successively aligned to the corresponding species sequences in miRBase (to detect known microRNAs), the transcriptome (to detect mRNA contamination) and the genome (to predict new microRNAs). The mapping to miRBase is done in four substeps, aligning subsequently to mature, maturestar, unobserved maturestar and hairpin sequences. After each of these steps, the mapped reads are removed from the input file so they cannot erroneously be predicted as new microRNA. The reads that did not map to miRBase are successively aligned to transcriptome libraries (RefSeq and RFam). Among the aligned reads, those which map to more than N different entities within the same library are removed, i.e. will not be used in the following analysis steps. The parameter N is fixed to 5 in the web server, but can be modified in the stand-alone version. Finally, the remaining reads are mapped to the genome, and the alignments are used to predict new microRNAs following three steps: (i) clustering reads into putative mature microRNAs (see ‘Data and Methods’, ‘Detection of read clusters’ section); (ii) extracting candidate pre-microRNAs from the genome to select the energetically best candidate (see ‘Data and Methods’, ‘Generating precursor candidates’ section); and (iii) applying five different Random Forest models to calculate the probability that a given candidate is a microRNA (see ‘Data and Methods’, ‘Prediction Models’ section). The web server reports only those candidates having been predicted by at least three out of the five models. The predicted microRNAs can be viewed within a genome context by means of links to the UCSC Genome Browser and the NGSmethDB browser (13,14). Table 1 shows a summary of the miRanalyzer parameters.
Figure 1.

General workflow of miRanalyzer. The fastq file is transformed into a read count file, which is filtered to keep only sequences from 17 to 26 bases. These reads are successively mapped to several databases in order to identify known microRNAs, discard messenger RNA contaminations and select sequences for the microRNA prediction step.
Table 1.
The default values of the parameters used in miRanalyzer are shown
| General parameters | ||
|---|---|---|
| Name | Description | Value |
| minLength | The minimum read length, all others will be removed | 17 |
| maxLength | The maximum read length, all reads will be trimmed to this length | 26 |
| Bowtie parameters | ||||
|---|---|---|---|---|
| Name | Description | miRBase | Trans. libraries | Genome |
| −k | Max. number of reported alignments | 10 | 20 | 6 |
| −l | The seed length | 17 | 20 | 17 |
| −n | The number of mismatches within the seed | 1 | 1 | 1 |
| Prediction parameters | ||
|---|---|---|
| Name | Description | Value |
| Score | The posterior probability that the candidate is a true microRNA | 0.9 |
| minNoPositives | miRanalyzer predicts using five models (five different negative sets). This parameter determines the minimum number of models which predicts a candidate to be a new microRNA (default: 3). | 3 |
The web server version allows the user to change the ‘−n’ parameter. The standalone version allows manipulating all of them. We used −l 17 to detect known microRNAs and predict new microRNAs (align to the genome) as this is the shortest microRNA length in miRBase but −l 20 for the other libraries.
Aligning the reads
The ultrafast short read aligner Bowtie (10) is used to align the reads to the different libraries and the genome, which allows, compared to the previous version, (i) the use of colour space sequences, (ii) a wider range of accepted mismatches and (iii) a gain in speed and memory efficiency. Bowtie requires a number of input parameters that define which alignments are legal and how many of them should be reported. Since no quality values do currently exist in the miRanalyzer input, the sum of the quality values at all mismatched read positions (−e/−maqerr) is set to an arbitrary value of 2000, which disables the quality values. Furthermore, we use the—best and—strata options to get only the best alignments, that is, those with least mismatches in the seed. Parameters −k (maximum number of reported alignments), −l (length of the seed) and −n (number of mismatches within the seed) are set to different values depending on the library used (Table 1). Note that in the case of alignments to the genome, we remove all reads with equal or more than −k valid best alignments. The reason is that reads with a high number of alignments are unlikely derived from microRNAs. When mapping against miRBase, transcriptome and Rfam databases, the −norc option (do not attempt to align against the reverse-complement reference strand) is used as the mappings to the reverse strand would be meaningless.
Bowtie detects the best alignments under a given seed length, to which the maximum number of allowed mismatches parameter is applied. Since microRNAs do not have a fixed size, a common seed cannot accommodate all possible length variants. In order to overcome this issue, we fix the seed to 17, the length of the shortest microRNA known, and post-process the Bowtie results to select the longest alignment that maintains the number of observed mismatches within the seed (see Figure 2 for details).
Figure 2.

Selection of longest alignments performed by miRanalyzer. The example shows the best alignments for two reads obtained with Bowtie, and the one selected (light grey square). The 17 nt seed is outlined and the longest alignment maintaining the number of observed mismatches within the seed is highlighted in red. Note that for Read2, the chosen alignment is not the one that contains the least total number of mismatches.
The range of input sequence length (17–26 nt) allows that reads can be longer than the reference sequence, i.e. the average length of known microRNAs is ∼22 nt, something that Bowtie does not allow. In order to use Bowtie, a sequence of 25 ‘Gs’ is artificially added to the known microRNA reference sequences. It might be that the seed alignment extends into this artificially added sequence. Those cases are detected and corrected (removing the matches between read bases and bases of the G-run) or removed (if after the correction the alignment is shorter than 17 nt).
Plant genomes
Although plant and animal microRNAs have a well-defined fold-back hairpin structure in common, plant pre-miRNAs have larger and more variable stem-loop structures (15). Therefore, the basic workflow is the same for plants and animals, but modifications have been introduced to take these differences into account when generating the prediction models (see ‘Data and Methods’, ‘Prediction Models’ section). In general, we found that the prediction models are much more accurate for animals compared to plants. In the training set, by means of a 10-fold cross-validation we obtain a mean recall (over all five models) and a mean precision of 0.923 and 0.892, respectively, for plants and of 0.978 and 0.965, respectively, for animals. Furthermore, given the wider range of pre-microRNA length in plants, the prediction for these organisms is more CPU intensive (many more secondary structures need to be calculated).
Differential expression
The differential expression module is based on the DESeq package, which is used to calculate the fold changes of the expression values and to assess its statistical significance (12). In order to use this module, all samples need to be processed first with miRanalyzer. The IDs assigned to each miRanalyzer job are then used to define the two groups to be compared. The DESeq input is a matrix where every entity (known and predicted microRNAs in our case) has an assigned read count for each sample. For known microRNAs, the names will be the same in each sample, which makes the matrix generation easy. However, the newly predicted microRNAs have arbitrarily assigned names such as Candidate_256 or Candidate_12, which normally do not coincide between samples, that is, Candidate_1 in sample 1 will not forcedly correspond to Candidate_1 in sample 2. This means that candidates from different samples need to be grouped using the chromosomal coordinates and the sequence of the predicted mature microRNA. Precursor sequences from different samples correspond to the same predicted microRNA if they overlap in more than 80% and if their pairwise mature microRNA sequence identity is higher than 80%. The microRNA candidates detected in over 55% of all samples are selected. The module also calculates the consensus sequences for mature and precursor microRNAs using clustalw (16). Finally, for differentially expressed microRNAs, a new process that uses TargetSpy (17) can be launched to calculate putative target sites. We have chosen TargetSpy for three reasons: (i) it can easily detect targets for newly predicted microRNAs, (ii) it does not rely on cross-species comparison (conservation) and (iii) it has been shown to work very well in a broad range of different species, which is particularly important. The functional analysis (18) of the target genes is currently being redesigned and will be available soon.
Standalone version
The miRanalyzer standalone version needs a local file-based data base that holds miRBase, mRNA and RFam libraries, genome sequences, Bowtie indexes, prediction models and all other user defined libraries. miRanalyzer relies on three programs or packages that must be installed before: the Open Source Machine Learning Software Weka (19), the Vienna RNA Package (20) and Bowtie (10). There are several advantages when using the standalone version: parameter values can be changed, customized libraries can be added and not publically available assemblies can be included.
Conversion of fastq to RC format (read count)
A perl script to convert fastq format into read-count format is provided on the miRanalyzer web page. The script allows now (i) to process colour sequence data and SCARF format, (ii) to select a maximum read length and (iii) to force all reported sequences to be present in all of the analysed samples.
OUTLOOK
We present an updated version of miRanalyzer including many new features. The introduced improvements like full colour space support, differential expression analysis and plant genomes will allow to address the needs of a wider range of users. In the future, we intend to improve the accuracy of the alignments, by adding the possibility of using sequence quality values and the efficiency of the standalone version by introducing the possibility of parallelization. Other topics will be the incorporation of gene expression values in order to infer microRNA regulatory networks.
DATA AND METHODS
Sequence data
The detection of known microRNAs is currently based on the latest miRBase version 16 (21) and will be updated as new miRBase versions are released. In this version, we also distinguish between mature, maturestar, unobserved maturestar and hairpin microRNA sequences. Unobserved maturestar are those maturestar microRNAs that are theoretically possible but that are not present in miRBase (as a consequence of not having been experimentally observed). An updated list of all available species and assemblies can be found on the miRanalyzer tutorial page. The data were obtained from UCSC Genome Browser (13) with the exception of: silkworm (Bombyx mori) genome version 2 from SilkDB (22), Arabidopsis thaliania from the Arabidopsis Information Resource—TAIR (23), maize (Zea mays) version 1, vine grape (Vitis vinifera) version 12x and rice (Oryza sativa) version 6.1 from plantGDB (24) and Medicago truncatula version 3.0 from Medicago truncatula genome project (25). NCBI reference sequences (mRNA and 3′UTR) were used whenever available (26), and the mapping to other RNA families was carried out with the RFam database version 10 (27).
Training datasets
The size of the training set has been notably increased compared to the first version. Table 2 shows the data used for training the prediction models.
Table 2.
Data sets used to train the prediction models
| Species | Tissues/Conditions | No. of microRNAs | References | GEO references |
|---|---|---|---|---|
| Animal | ||||
| H. sapiens | 16 | 10 321 | (29,30) | GSE19812, GSE20384, GSE21279, GSE20892 |
| M. musculus | 9 | 6201 | (30,31) | GSE20384, GSE19473 |
| D. melanogaster | 9 | 587 | (32) | GSE12462,GSE24314, GSE24608, SE24542, GSE24540 |
| C. elegans | 12 | 2091 | (33,34) | GSE18634, GSE13339 |
| D. rerio | 2 | 695 | (35,36) | GSE21503, GSE22068 |
| B. mori | 3 | 46 | (37) | GSE17965 |
| Plant | ||||
| A. thaliana | 4 | 295 | (38,39) | GSE20448, GSE16971 |
| O. sativa | 9 | 1302 | (40,41) | GSE23217, GSE20748 |
| Z. mays | 3 | 193 | (42) | GSE17339 |
| V. vinifera | 1 | 28 | (43) | GSE18406 |
Detection of read clusters
Once aligned to the genome, the reads that may belong to the same candidate mature microRNA are clustered. Each cluster is defined by two coordinates: (i) the start and end coordinates, that is, the start and end positions of the most upstream and downstream reads, respectively, and (ii) the start position of the most expressed read; this latter is named the ‘cluster anchor’ and is used to decide if a read belongs to a cluster or not. Clusters are constructed following these two steps:
Reads are sorted according to their read count from highest to lowest (most to least expressed).
The most expressed read defines/opens the first cluster and the following reads are added to an existing cluster if the read (i) is located on the same strand as the pre-existing cluster and (ii) falls totally inside an already opened cluster, or its start coordinate lies within a window defined by (cluster anchor −2 bp, cluster anchor + 5 bp).
If a read is not found to belong to any pre-existing cluster, a new cluster is opened being this read the most expressed one (defining the cluster anchor) in the new cluster.
Generating precursor candidates
From the clustering process described above, genome positions of the putative mature microRNAs are obtained; however, the candidate precursor sequence on which many machine-learning features are based needs yet to be defined. Since we do not know neither the arm in which the mature microRNA is located nor the length of the precursor sequence, several candidate precursor sequences with different lengths for both, the hypothetical location in 3′ and 5′, are generated, and the one with best structural criteria and binding energy is kept.
The chromosome coordinates are given as:
- For the 5′-arm (+ strand):
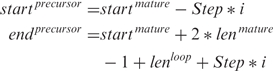
- For the 3′-arm (+ strand):
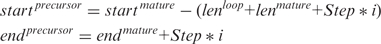
We set the mature microRNA length to 20 nt and the loop to 15 nt for both, animal and plants. The parameters Step and i are set to 5 and 8 for animals and to 7 and 10 for plants. This is because plant precursor sequences can be longer than animal ones. Applying these values to the formulas above, we get that the minimum and maximum lengths are 65 and 135 for animals and 69 and 195 for plants.
Prediction models
For all candidate microRNAs generated in the step above, we calculate several features based on both, the secondary structure and expression derived properties [see Ref. (9) for a more detailed description]. In a first step, we discard a candidate if (i) its read cluster overlaps with the loop by more than 5 bp in the 5′-arm (on the 3′-arm no overlap is allowed), (ii) it has no hairpin, (iii) it has less than 19 bindings to the putative precursor sequence and (iv) it has less than 11 bindings to the region occupied by the read cluster (putative mature microRNA sequence). For the remaining candidates, the features described in Table 3 are calculated. These features have been selected out of a large pool of possible features applying the CfsSubsetEval algorithm in Weka. Finally, the training of five Random forest models (28) for both animals and plants is performed.
Table 3.
Features used for the Random forest prediction models
| Feature | Used for kingdom |
|---|---|
| Number of bindings in read cluster sequence | Animal |
| Normalized mean free energy of precursor sequence | Plant and Animal |
| Number of bindings in precursor | Animal |
| Length of read cluster | Plant and Animal |
| The corresponding putative maturestar sequence is also present (binary value 0, 1) | Plant and Animal |
| Number of bindings in read cluster divided by the read cluster length | Plant |
| Number of reads in read cluster | Plant and Animal |
| Mean free energy of precursor sequence | Plant and Animal |
| Degree of bulb asymmetry in precursor | Animal |
| The number of bulbs in precursor secondary structure | Plant |
FUNDING
The Ministry of Innovation and Science of the Spanish Government (BIO2010-20219 to M.H.); the Junta de Andalucia (P07FQM3163 to M.H.); the ‘Juan de la Cierva’ fellowship (to M.H.); the Department of Industry, Tourism and Trade of the Government of the Autonomous Community of the Basque Country (Etortek Research Programs 2009/20011 to A.M.A.); from the Innovation Technology Department of the Bizkaia County (to A.M.A.). Funding for open access charge: Department of Industry, Tourism and Trade of the Government of the Autonomous Community of the Basque Country (Etortek Research Programs 2009/2011 to A.M.A.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We want to thank all miRanalyzer users for their valuable feedback helping us to improve the tool.
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Bushati N, Cohen SM. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Xu J, Yang D, Tan X, Wang H. Computational approaches for microRNA studies: a review. Mamm. Genome. 2010;21:1–12. doi: 10.1007/s00335-009-9241-2. [DOI] [PubMed] [Google Scholar]
- 4.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pantano L, Estivill X, Marti E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 2010;38:e34. doi: 10.1093/nar/gkp1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu E, Zhao F, Xu G, Hou H, Zhou L, Li X, Sun Z, Wu J. mirTools: microRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Res. 2010;38:W392–W397. doi: 10.1093/nar/gkq393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang PJ, Liu YC, Lee CC, Lin WC, Gan RR, Lyu PC, Tang P. DSAP: deep-sequencing small RNA analysis pipeline. Nucleic Acids Res. 2010;38:W385–W391. doi: 10.1093/nar/gkq392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronen R, Gan I, Modai S, Sukacheov A, Dror G, Halperin E, Shomron N. miRNAkey: a software for microRNA deep sequencing analysis. Bioinformatics. 2010;26:2615–2616. doi: 10.1093/bioinformatics/btq493. [DOI] [PubMed] [Google Scholar]
- 9.Hackenberg M, Sturm M, Langenberger D, Falcon-Perez JM, Aransay AM. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 2009;37:W68–W76. doi: 10.1093/nar/gkp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millar AA, Waterhouse PM. Plant and animal microRNAs: similarities and differences. Funct. Integr. Genomics. 2005;5:129–135. doi: 10.1007/s10142-005-0145-2. [DOI] [PubMed] [Google Scholar]
- 12.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–D882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackenberg M, Barturen G, Oliver JL. NGSmethDB: a database for next-generation sequencing single-cytosine-resolution DNA methylation data. Nucleic Acids Res. 2011;39:D75–D79. doi: 10.1093/nar/gkq942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturm M, Hackenberg M, Langenberger D, Frishman D. TargetSpy: a supervised machine learning approach for microRNA target prediction. BMC Bioinformatics. 2010;11:292. doi: 10.1186/1471-2105-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackenberg M, Matthiesen R. Annotation-Modules: a tool for finding significant combinations of multisource annotations for gene lists. Bioinformatics. 2008;24:1386–1393. doi: 10.1093/bioinformatics/btn178. [DOI] [PubMed] [Google Scholar]
- 19.Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 20.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie. 1994;125:167–188. [Google Scholar]
- 21.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2009;38:D453–D456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L, et al. The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 2008;36:D1009–D1014. doi: 10.1093/nar/gkm965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duvick J, Fu A, Muppirala U, Sabharwal M, Wilkerson MD, Lawrence CJ, Lushbough C, Brendel V. PlantGDB: a resource for comparative plant genomics. Nucleic Acids Res. 2008;36:D959–D965. doi: 10.1093/nar/gkm1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young ND, Cannon SB, Sato S, Kim D, Cook DR, Town CD, Roe BA, Tabata S. Sequencing the genespaces of Medicago truncatula and Lotus japonicus. Plant Physiol. 2005;137:1174–1181. doi: 10.1104/pp.104.057034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 29.Vaz C, Ahmad HM, Sharma P, Gupta R, Kumar L, Kulshreshtha R, Bhattacharya A. Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood. BMC Genomics. 2010;11:288. doi: 10.1186/1471-2164-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su RW, Lei W, Liu JL, Zhang ZR, Jia B, Feng XH, Ren G, Hu SJ, Yang ZM. The integrative analysis of microRNA and mRNA expression in mouse uterus under delayed implantation and activation. PLoS ONE. 2010;5:e15513. doi: 10.1371/journal.pone.0015513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghildiyal M, Xu J, Seitz H, Weng Z, Zamore PD. Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA. 2010;16:43–56. doi: 10.1261/rna.1972910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ. MicroRNAs both promote and antagonize longevity in C. elegans. Curr. Biol. 2010;20:2159–2168. doi: 10.1016/j.cub.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato M, de Lencastre A, Pincus Z, Slack FJ. Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol. 2009;10:R54. doi: 10.1186/gb-2009-10-5-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell. 2010;38:789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S, Li D, Li Q, Zhao P, Xiang Z, Xia Q. MicroRNAs of Bombyx mori identified by Solexa sequencing. BMC Genomics. 2010;11:148. doi: 10.1186/1471-2164-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Zhang Q, Zhang J, Wu L, Qi Y, Zhou JM. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2010;152:2222–2231. doi: 10.1104/pp.109.151803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moldovan D, Spriggs A, Yang J, Pogson BJ, Dennis ES, Wilson IW. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010;61:165–177. doi: 10.1093/jxb/erp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li T, Li H, Zhang YX, Liu JY. Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica) Nucleic Acids Res. 2010;39:2821–2833. doi: 10.1093/nar/gkq1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA methylation mediated by a microRNA pathway. Mol. Cell. 2010;38:465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 42.Wei F, Stein JC, Liang C, Zhang J, Fulton RS, Baucom RS, De Paoli E, Zhou S, Yang L, Han Y, et al. Detailed analysis of a contiguous 22-Mb region of the maize genome. PLoS Genet. 2009;5:e1000728. doi: 10.1371/journal.pgen.1000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62:960–976. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]