


Abstract
Sequence-derived structural and physicochemical features have been extensively used for analyzing and predicting structural, functional, expression and interaction profiles of proteins and peptides. PROFEAT has been developed as a web server for computing commonly used features of proteins and peptides from amino acid sequence. To facilitate more extensive studies of protein and peptides, numerous improvements and updates have been made to PROFEAT. We added new functions for computing descriptors of protein–protein and protein–small molecule interactions, segment descriptors for local properties of protein sequences, topological descriptors for peptide sequences and small molecule structures. We also added new feature groups for proteins and peptides (pseudo-amino acid composition, amphiphilic pseudo-amino acid composition, total amino acid properties and atomic-level topological descriptors) as well as for small molecules (atomic-level topological descriptors). Overall, PROFEAT computes 11 feature groups of descriptors for proteins and peptides, and a feature group of more than 400 descriptors for small molecules plus the derived features for protein–protein and protein–small molecule interactions. Our computational algorithms have been extensively tested and used in a number of published works for predicting proteins of specific structural or functional classes, protein–protein interactions, peptides of specific functions and quantitative structure activity relationships of small molecules. PROFEAT is accessible free of charge at http://bidd.cz3.nus.edu.sg/cgi-bin/prof/protein/profnew.cgi.
INTRODUCTION
Sequence-derived structural and physicochemical features are highly useful for representing and distinguishing proteins or peptides of different structural, functional and interaction properties, and have been widely used in developing methods and software for predicting protein structural and functional classes (1–7), protein–protein interactions (8–10), protein–ligand interactions (11,12), protein substrates (13,14), molecular binding sites on proteins (15–20), subcellular locations (21), protein crystallization propensity (22–24) and peptides of specific properties (25–30). Web servers, such as PROFEAT (31) and PseAAC (http://www.csbio.sjtu.edu.cn/bioinf/PseAA/) (32), have been built to facilitate the computation of protein and peptide features.
Nonetheless, some features important for studying proteins, peptides and molecular interactions have not been provided in these web servers. Examples of these features include atomic-level topological descriptors that are useful for structure–property correlations (33) and descriptors of total amino acid properties (TAAPs) that have been used for modeling protein conformational stability (34), ligand binding site structural features (35) and interaction with small molecules (36). Moreover, the descriptors provided in those available web servers are not suitable for analyzing local properties of sequence subsections, and additional works are needed to use descriptors to study protein–protein and protein–ligand interactions. Therefore, it is desirable to provide segment descriptors for local properties of subsections of protein sequences, and descriptors that can be straightforwardly used for exploring protein–protein and protein–small molecule interactions.
We updated PROFEAT by adding new functions for computing descriptors of protein–protein and protein–small molecule interactions, segment descriptors for local properties of subsections of protein sequences, atomic-level topological descriptors for peptide sequences and small molecule structures, and topological polar surface areas of small molecules. Moreover, we added new feature groups such as pseudo-amino acid composition (PAAC), amphiphilic PAAC (APAAC), TAAPs, and atomic-level topological descriptors. The computational algorithms of these newly added feature groups have been extensively tested and used in a number of published works for predicting proteins and peptides of specific properties, protein–protein interactions, and quantitative structure activity relationships of small molecules. A list of publications using features covered by PROFEAT is provided in Supplementary Table S1 and in PROFEAT online server which can be accessed at http://bidd.cz3.nus.edu.sg/prof/part_of_publications.htm. PROFEAT homepage is shown in Figure 1. A list of features for proteins and peptides covered by this version of PROFEAT is summarized in Table 1 and a list of the topological descriptors for peptides and small molecules computed by PROFEAT is summarized in Supplementary Table S2.
Figure 1.

PROFEAT new web page.
Table 1.
List of PROFEAT computed features for proteins, peptides and protein–protein interactions
| Feature group | Features | No. of descriptors | No. of descriptor values |
|---|---|---|---|
| Composition-1 | Amino acid composition | 1 | 20 |
| Composition-2 | Dipeptide composition | 1 | 400 |
| Autocorrelation 1 | Normalized Moreau–Broto autocorrelation | a | a |
| Autocorrelation 2 | Moran autocorrelation | a | a |
| Autocorrelation 3 | Geary autocorrelation | a | a |
| Composition, Transition, Distribution | Composition | 7 | 21 |
| Transition | 7 | 21 | |
| Distribution | 7 | 105 | |
| Quasi-sequence order descriptors | Sequence order coupling number | 2 | 90 |
| Quasi-sequence order descriptors | 2 | 150 | |
| PAAC | PAAC | b | b |
| APAAC | APAAC | c | c |
| Topological descriptors | Topological descriptors | 405 | |
| TAAPs | TAP | d | d |
aThe number depends on the choice of the number of properties of amino acid and the choice of the maximum values of the lag.
bThe number depends on the choice of the number of the set of amino acid properties and the choice of the λ value.
cThe number depends on the choice of the λ value.
dThe numbers depend on the choice of the number of properties of amino acid.
METHODS FOR NEWLY ADDED FEATURES AND FUNCTIONS
PAAC descriptors
First, three variables are derived from the original hydrophobicity values 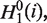 hydrophilicity values
hydrophilicity values 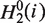 and side chain masses
and side chain masses 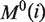 of 20 amino acids (i = 1, 2, … , 20) (32):
of 20 amino acids (i = 1, 2, … , 20) (32):
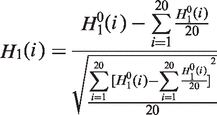 |
(1) |
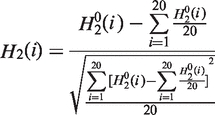 |
(2) |
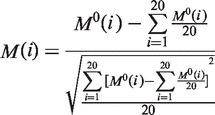 |
(3) |
Then, a correlation function can be computed as:
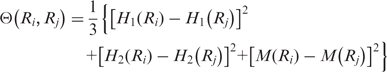 |
(4) |
from which, sequence order-correlated factors are defined as:
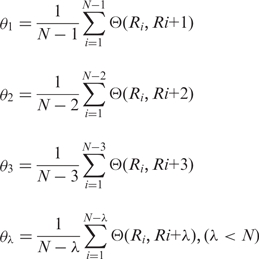 |
(5) |
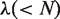 is a parameter. Let fi be the normalized occurrence frequency of 20 amino acids in the protein sequence, a set of
is a parameter. Let fi be the normalized occurrence frequency of 20 amino acids in the protein sequence, a set of 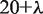 descriptors called the PAAC are defined as:
descriptors called the PAAC are defined as:
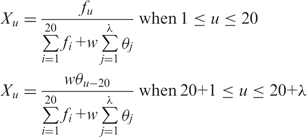 |
(6) |
where w is the weighting factor for the sequence-order effect and is set to be w = 0.05 as suggested by Shen (32).
APAAC
From  defined in Equation (1) and (2), the hydrophobicity and hydrophilicity correlation functions are defined (32), respectively, as:
defined in Equation (1) and (2), the hydrophobicity and hydrophilicity correlation functions are defined (32), respectively, as:
from which sequence order factors can be defined as:
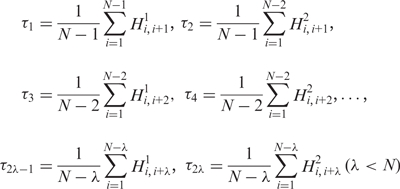 |
(7) |
and APAAC are defined as:
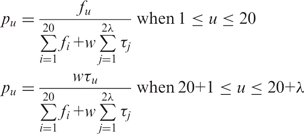 |
(8) |
where w is the weighting factor and is taken as  .
.
Topological descriptors at atomic level
Topological descriptors are based on graph theory and encode information about the types of atoms and bonds in a molecule and the nature of their connections. Examples of topological descriptors include counts of atom and bond types and indexes that encode the size, shape and types of branching in a molecule (37). These descriptors can be calculated from the 2D structure of a peptide automatically generated from its sequence based on the molecular structures of the amino acid residues in the sequence. Supplementary Table S2 gives a list of the topological descriptors computed by PROFEAT.
TAAP
TAAP descriptor for a specific physicochemical property i is defined as: 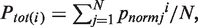 where
where 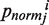 represents the property i of amino acid Rj that is normalized between 0 and 1 using the following expression,
represents the property i of amino acid Rj that is normalized between 0 and 1 using the following expression, 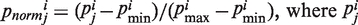 is the original amino acid property i for residue j.
is the original amino acid property i for residue j. 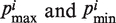 are, respectively, the minimum and maximum values of the original amino acid property i, and N is the length of the sequence (38–40).
are, respectively, the minimum and maximum values of the original amino acid property i, and N is the length of the sequence (38–40).
Protein–protein interaction descriptors
Protein–protein interaction descriptors can be computed from the descriptors Va = {Va(i), i = 1, 2, … , n} and Vb = {Vb(i), i = 1, 2, … , n} of individual proteins A and B by three methods. In the first method, two protein-pair vectors Vab and Vba with dimension of 2n are constructed with Vab = (Va, Vb) for interaction between proteins A and B and Vba = (Vb, Va) for interaction between proteins B and A (8,9). In the second method, one vector V with dimension of 2n is constructed: V = {Va(i) + Vb(i), Va(i) × Vb(i), i = 1, 2, … , n} which has the property that V is unchanged when a and b are exchanged. In the third method, one vector V with dimension of n2 is constructed by the tensor product: V = {V(k) = Va(i) × Vb(j), i = 1, 2, … , n, j = 1, 2, … , n, k = (i − 1) × n + j}.
Protein–ligand interaction descriptors
Protein–ligand interaction descriptor vector V can be constructed from the protein descriptor vector Vp (Vp(i), i = 1, … , np) and ligand descriptor vector Vl (Vl(i), i = 1, … , nl) by two methods similar to the first and third method for constructing protein pair descriptors. In the first method, one vector V with dimension of np + nl are constructed V = (Vp,Vl) for interaction between protein and ligand. In the second method, one vector V with dimension of np × nl is constructed by the tensor product: V = {v(k) = Vp(i) × Vl(j), i = 1, 2 , … , np, j = 1, 2, … , nl, k=(i − 1) × np + j}.
Segmented sequence descriptors
To characterize the local feature of a protein sequence, a protein sequence can be divided into several segments and descriptors are calculated for each segment.
Topological descriptors for small molecules
For small molecules, topological descriptors are calculated from the input 2D structures of small molecules in mol or sdf format. Names of these descriptors are the same as those for protein segments which are listed in Supplementary Table S2.
REMARKS
Compared with its earlier version, the updated PROFEAT is significantly enhanced in both the number of newly added features useful for representing various protein properties, and newly added functions for computing features for local properties of protein segments, protein–protein interactions, protein–small molecule interactions and small molecules. These enhancements are intended to provide more comprehensive features for facilitating the analysis and prediction of proteins, peptides, small molecules of different properties and molecular interactions involving proteins, peptides and small molecules. With continued interest in using molecular and interaction features and developing new algorithms for representing these features, new descriptors and functions such as those involving DNA, RNA and other nucleotides can be integrated into PROFEAT in the near future to better facilitate the study of molecular and bio-molecular functions and interactions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Natural Science Foundation of China (grant number 20973118).
Conflict of interest statement. None declared.
REFERENCES
- 1.Karchin R, Karplus K, Haussler D. Classifying G-protein coupled receptors with support vector machines. Bioinformatics. 2002;18:147–159. doi: 10.1093/bioinformatics/18.1.147. [DOI] [PubMed] [Google Scholar]
- 2.Cai CZ, Han LY, Ji ZL, Chen X, Chen YZ. SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003;31:3692–3697. doi: 10.1093/nar/gkg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubchak I, Muchnik I, Mayor C, Dralyuk I, Kim SH. Recognition of a protein fold in the context of the Structural Classification of Proteins (SCOP) classification. Proteins. 1999;35:401–407. [PubMed] [Google Scholar]
- 4.Han L, Cui J, Lin H, Ji Z, Cao Z, Li Y, Chen Y. Recent progresses in the application of machine learning approach for predicting protein functional class independent of sequence similarity. Proteomics. 2006;6:4023–4037. doi: 10.1002/pmic.200500938. [DOI] [PubMed] [Google Scholar]
- 5.Langlois RE, Lu H. Boosting the prediction and understanding of DNA-binding domains from sequence. Nucleic Acids Res. 2010;38:3149–3158. doi: 10.1093/nar/gkq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan RX, Si JN, Wang C, Zhang Z. DescFold: a web server for protein fold recognition. BMC Bioinformatics. 2009;10:416. doi: 10.1186/1471-2105-10-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu F, Han L, Zheng C, Xie B, Tammi MT, Yang S, Wei Y, Chen Y. What are next generation innovative therapeutic targets? Clues from genetic, structural, physicochemical, and systems profiles of successful targets. J. Pharmacol. Exp. Ther. 2009;330:304–315. doi: 10.1124/jpet.108.149955. [DOI] [PubMed] [Google Scholar]
- 8.Bock JR, Gough DA. Predicting protein–protein interactions from primary structure. Bioinformatics. 2001;17:455–460. doi: 10.1093/bioinformatics/17.5.455. [DOI] [PubMed] [Google Scholar]
- 9.Lo SL, Cai CZ, Chen YZ, Chung MC. Effect of training datasets on support vector machine prediction of protein-protein interactions. Proteomics. 2005;5:876–884. doi: 10.1002/pmic.200401118. [DOI] [PubMed] [Google Scholar]
- 10.Qiu J, Noble WS. Predicting co-complexed protein pairs from heterogeneous data. PLoS Comput. Biol. 2008;4:e1000054. doi: 10.1371/journal.pcbi.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24:i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia Z, Wu LY, Zhou X, Wong ST. Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. BMC Syst. Biol. 2010;4(Suppl. 2):S6. doi: 10.1186/1752-0509-4-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barkan DT, Hostetter DR, Mahrus S, Pieper U, Wells JA, Craik CS, Sali A. Prediction of protease substrates using sequence and structure features. Bioinformatics. 2010;26:1714–1722. doi: 10.1093/bioinformatics/btq267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rottig M, Rausch C, Kohlbacher O. Combining structure and sequence information allows automated prediction of substrate specificities within enzyme families. PLoS Comput. Biol. 2010;6:e1000636. doi: 10.1371/journal.pcbi.1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006;34:W243–W248. doi: 10.1093/nar/gkl298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terribilini M, Lee JH, Yan C, Jernigan RL, Honavar V, Dobbs D. Prediction of RNA binding sites in proteins from amino acid sequence. RNA. 2006;12:1450–1462. doi: 10.1261/rna.2197306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L. Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics. 2010;26:1616–1622. doi: 10.1093/bioinformatics/btq253. [DOI] [PubMed] [Google Scholar]
- 18.Carson MB, Langlois R, Lu H. NAPS: a residue-level nucleic acid-binding prediction server. Nucleic Acids Res. 2010;38:W431–W435. doi: 10.1093/nar/gkq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami Y, Spriggs RV, Nakamura H, Jones S. PiRaNhA: a server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res. 2010;38:W412–W416. doi: 10.1093/nar/gkq474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CT, Yang EW, Hsu HJ, Sun YK, Hsu WL, Yang AS. Protease substrate site predictors derived from machine learning on multilevel substrate phage display data. Bioinformatics. 2008;24:2691–2697. doi: 10.1093/bioinformatics/btn538. [DOI] [PubMed] [Google Scholar]
- 21.Rastogi S, Rost B. Bioinformatics predictions of localization and targeting. Methods Mol. Biol. 2010;619:285–305. doi: 10.1007/978-1-60327-412-8_17. [DOI] [PubMed] [Google Scholar]
- 22.Overton IM, Padovani G, Girolami MA, Barton GJ. ParCrys: a Parzen window density estimation approach to protein crystallization propensity prediction. Bioinformatics. 2008;24:901–907. doi: 10.1093/bioinformatics/btn055. [DOI] [PubMed] [Google Scholar]
- 23.Kurgan L, Razib AA, Aghakhani S, Dick S, Mizianty M, Jahandideh S. CRYSTALP2: sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 2009;9:50. doi: 10.1186/1472-6807-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kandaswamy KK, Pugalenthi G, Suganthan PN, Gangal R. SVMCRYS: an SVM approach for the prediction of protein crystallization propensity from protein sequence. Protein Pept. Lett. 2010;17:423–430. doi: 10.2174/092986610790963726. [DOI] [PubMed] [Google Scholar]
- 25.Schneider G, Wrede P. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys J. 1994;66:335–344. doi: 10.1016/s0006-3495(94)80782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui J, Han LY, Lin HH, Zhang HL, Tang ZQ, Zheng CJ, Cao ZW, Chen YZ. Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. Mol. Immunol. 2007;44:866–877. doi: 10.1016/j.molimm.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, Cherkasov A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009;52:2006–2015. doi: 10.1021/jm8015365. [DOI] [PubMed] [Google Scholar]
- 28.Khatun J, Hamlett E, Giddings MC. Incorporating sequence information into the scoring function: a hidden Markov model for improved peptide identification. Bioinformatics. 2008;24:674–681. doi: 10.1093/bioinformatics/btn011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah AR, Agarwal K, Baker ES, Singhal M, Mayampurath AM, Ibrahim YM, Kangas LJ, Monroe ME, Zhao R, Belov ME, et al. Machine learning based prediction for peptide drift times in ion mobility spectrometry. Bioinformatics. 2010;26:1601–1607. doi: 10.1093/bioinformatics/btq245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacob L, Vert JP. Efficient peptide-MHC-I binding prediction for alleles with few known binders. Bioinformatics. 2008;24:358–366. doi: 10.1093/bioinformatics/btm611. [DOI] [PubMed] [Google Scholar]
- 31.Li ZR, Lin HH, Han LY, Jiang L, Chen X, Chen YZ. PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006;34:W32–W37. doi: 10.1093/nar/gkl305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen HB, Chou KC. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008;373:386–388. doi: 10.1016/j.ab.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Ren B. Atomic-level-based AI topological descriptors for structure-property correlations. J. Chem. Inf. Comput. Sci. 2003;43:161–169. doi: 10.1021/ci020382n. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez L, Caballero J, Abreu JI, Fernandez M. Amino acid sequence autocorrelation vectors and Bayesian-regularized genetic neural networks for modeling protein conformational stability: gene V protein mutants. Proteins. 2007;67:834–852. doi: 10.1002/prot.21349. [DOI] [PubMed] [Google Scholar]
- 35.Niwa T. Elucidation of characteristic structural features of ligand binding sites of protein kinases: a neural network approach. J. Chem. Inf. Model. 2006;46:2158–2166. doi: 10.1021/ci050528t. [DOI] [PubMed] [Google Scholar]
- 36.Niu B, Jin Y, Lu L, Fen K, Gu L, He Z, Lu W, Li Y, Cai Y. Prediction of interaction between small molecule and enzyme using AdaBoost. Mol. Divers. 2009;13:313–320. doi: 10.1007/s11030-009-9116-1. [DOI] [PubMed] [Google Scholar]
- 37.Todeschini R, Consonni V. Handbook of Molecular Descriptors. Weinheim: Wiley-VCH; 2000. [Google Scholar]
- 38.Gromiha MM, Suwa M. Influence of amino acid properties for discriminating outer membrane proteins at better accuracy. Biochim. Biophys. Acta. 2006;1764:1493–1497. doi: 10.1016/j.bbapap.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Huang LT, Gromiha MM. Analysis and prediction of protein folding rates using quadratic response surface models. J. Comput. Chem. 2008;29:1675–1683. doi: 10.1002/jcc.20925. [DOI] [PubMed] [Google Scholar]
- 40.Gromiha MM. Importance of native-state topology for determining the folding rate of two-state proteins. J. Chem. Inf. Comput. Sci. 2003;43:1481–1485. doi: 10.1021/ci0340308. [DOI] [PubMed] [Google Scholar]