


Abstract
Peptides, either as protein fragments or as naturally occurring entities are characterized by their sequence and function features. Many times the researchers need to massively manage peptide lists concerning protein identification, biomarker discovery, bioactivity, immune response or other functionalities. We present a web server that manages peptide lists in terms of feature analysis as well as interactive clustering and visualization of the given peptides. PepServe is a useful tool in the understanding of the peptide feature distribution among a group of peptides. The PepServe web application is freely available at http://bioserver-1.bioacademy.gr/Bioserver/PepServe/.
INTRODUCTION
There is a plethora of tools that focus in proteomics sequence analysis, alignment and visualization but there are few examples for web servers and other software tools dedicated to peptide analysis. There are three major classes where the tools for peptide analysis can be classified: (i) the peptide/protein identification tools like RAId_DbS (1), Peptizer (2), DeNovoID (3) and PeptideFinder (4); (ii) tools for the mapping of post-translational peptide modifications like MODi (5), for peptide phosphorylation like PhosCalc (6) and proteolysis (7); (iii) tools for the investigation of specific characteristics of the peptide fragments like Remus (8) and UniMaP (9) for the property of uniqueness, like SignalP (10) for the signaling property, of the peptide–MHC binding affinity like BiodMHC (11) and MHCPred (12). However, to our knowledge, there is not any web server dedicated to the broad analysis of peptide characteristics performing peptide clustering and visualization in the peptide feature space.
Here, we present a web application which can provide information regarding Human peptide sequence analysis and clustering based on their related characteristics (annotated as ‘Features’ in the UniProt Database) along with information regarding peptide uniqueness and peptide similarity. PepServe is an application for analyzing and clustering peptide sequences according to a set of selected peptide features. The clusters of peptide sequences can be visualized as a graph where all the peptide sequences that share the same characteristics can be viewed.
MATERIALS AND METHODS
Web server
User input
The user may already have or may produce a peptide sequence list. In the second case, a peptide list can be retrieved in the following ways: (i) from a selected enzymatic digestion of a group of proteins; (ii) from the exhaustive peptide combinations around a specified position or within a position range of a protein sequence; and (iii) from the mapping on a given list of molecular masses, filtered or not with regular expression constraints as shown in Figure 1a.
Figure 1.

(a) The information flow of the system—the input can be protein(s) which is (i) digested or (ii) filtered by position and peptides, either sequences or molecular masses. The output is a list of peptide sequences which is subjected to feature retrieval and visualization. (b) A characteristic example of the feature-driven peptide visualization: the distribution of the peptides produced after tryptic digestion of three proteins from three different families (P14921, P22676 and P62166) according to the selected features (calcium binding and DNA binding).
In the first searching mode, the user can enter one or more protein UniProt accession numbers (AC) or protein names and select a set of enzymes that will computationally digest the specified proteins. Usually, tryptic digestion is preferred because it has high specificity and also 90% of the peptide fragments have less than 20 residues. Generally, the more enzymes are selected, the more peptides are produced with short length. This mode is useful when known protein sets are available and the researcher expects to find peptide characteristics and subsequently protein characteristics.
In the second searching mode, the protein accession number or the protein name is specified, along with the amino acid position of interest in the protein sequence. This position will define the part of a protein sequence which can be either around a selected position or within selected position boundaries in the protein sequence. The system searches for sequence characteristics within the specified positions and therefore this mode is preferred when a known protein is prone to mutagenesis or other protein states where the location of the sequence under investigation is important.
The third searching mode performs directly the analysis if a list of peptide sequences is at hand, whereas the final searching mode expects a list of peptide molecular masses in order to translate them to possible peptide sequences and then analyze them. The final search mode facilitates researchers using tandem mass spectrometry (MS/MS) analysis and for this reason there are more input parameters such as measured mass tolerance, protein molecular mass, isoelectric point range and pattern matching to refine the results.
Processing
The system uses the file repositories from our previous works (4,9) which contain peptide fragments classified according to their molecular mass, derived from extensive digestion of human proteins in the UniProt Database. The current repositories contain UniProt’s release 6/2010 and it has been scheduled for an update once a year. In addition, the system exploits the information from the UniProt flat files regarding the sequence positional features (Feature Table—FT lines) according to the position of the peptide fragment inside the corresponding protein. Complementary to the annotated features, peptide sequence uniqueness as well as sequence similarity are calculated and included in the collected peptide characteristics.
We define a peptide feature space with N dimensions defined from the collected features mentioned before. The number of dimensions is related with the number of peptide features which is selected by the user. The total number of features is 41, where the 38 features are those included in the ‘Sequence Annotation’ tag of UniProt and the three more features are described below. Each sequence has three possible states for each feature corresponding to (‘yes’, ‘partially’, ‘no’). When a sequence falls within the UniProt position boundaries for the given feature, the sequence state is characterized as ‘yes’, when it is outside the boundaries it is characterized as ‘no’ and the ‘partially’ state is when a part of the sequence is lying within the feature boundaries. For each two sequences Si and Sj, the system calculates their Euclidean distance as:
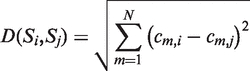 |
where cm,i and cm,j are the arithmetic state values (+1, 0, −1) of (‘yes’, ‘partially’, ‘no’) for the m-th characteristic of sequences Si and Sj, respectively. The peptides are clustered according to this Euclidian distance in the peptide feature space, and subsequently they are visualized within a graph. The graph contains nodes and edges where the nodes represent the peptides and the edges have a weight which corresponds to the Euclidian distance between the two nodes.
The performed clustering can be considered as a supervised one, since the classes are predefined from the combinations of the sequence features (i.e. all features included in the UniProt annotation scheme plus some extra features regarding uniqueness, sequence similarity and protein commonality). The clustering metric is the Euclidean distance in this feature space that controls the length of the edges in the graph topology.
Each peptide from the list is annotated according to the UniProt sequence positional features (Regions: Topological Domain, Transmembrane, Domain, Calcium Binding, Zinc Finger, DNA Binding, Nucleotide Binding, Coiled Coil, Motif, Compositional Bias; Molecule Processing: Signal, Transit Peptide, Propeptide, Peptide; Amino Acid Modifications: Lipidation, Disulfide Bond, Non-standard Residue, Modified Residue, Glycosylation, Cross-link; Experimental Information: Mutagenesis, Sequence Uncertainty, Sequence Conflict, Non-adjacent Residue, Non-terminal Residue; Sites: Active Site, Metal Binding, Binding Site, Site; Secondary Structure: Helix, Turn, Beta Strand; Natural Variations: Alternative Sequence, Natural Variant) enriched with the feature of sequence uniqueness. When a peptide is characterized as unique, it means that it exists only in one protein in the set of human proteins registered in the UniProt. This feature is provided for all other species in UniProt Database in order to compare with human peptide sequence uniqueness.
Output
The system produces a peptide sequence list accompanied by the corresponding list of peptide features (Table 1). All possible features are available to be selected for the clustering procedure. In the feature list, there are also two new characteristics related to the peptide pair-wise similarity: the sequence similarity and the number of common proteins the two peptide sequences can be found in. The sequence similarity is an index which shows the smallest number of edits to change one sequence into the other. This index has a value [0,1] and is produced by a Perl module.
Table 1.
An example of PepServe output
| Molecular mass | Peptide | Information | Unique peptide | Protein and features | ||
|---|---|---|---|---|---|---|
| 632.34 | STELLA | Peptide atlas | Yes | Q4KMX7 | No features found | – |
| Check across species | ||||||
| Check enymatic digestion. | ||||||
| 1016.46 | TYACFVSNL | Peptide atlas | Yes | P06731 | Domain | Ig-like 7 |
| check across species | ||||||
| Check enymatic digestion. | ||||||
| 587.34 | AVATAR | Peptide atlas | No | Q04609 | Topological domain | Cytoplasmic (Probable) |
| Check across species | Q8IWK6 | Topological Domain | Extracellular (Potential) | |||
| Check for other unique sequences | Q8N122 | No features found | – | |||
When peptide sequence ‘STELLA’, ‘AVATAR’ and ‘TYACFVSNL’ are served as an input, the system responds with the corresponding monoisotopic molecular mass and the peptide characteristics. As shown, ‘AVATAR’ is not unique in human proteome because it can be found in three proteins, whereas ‘STELLA’ and ‘TYACFVSNL’ are unique sequences and their corresponding features are displayed.
When the desired features are selected, the system calculates the Euclidian distance between the peptides. This way, the peptide relations in the feature space can be dynamically visualized in a graph by using properly created graphML files handled by Prefuse, a java-based visualization toolkit (13). The nodes of the graph are displayed as red circles representing the peptides and when they are double clicked they display the peptide sequences that belong to the same cluster along with the cluster’s features. The edge color varies according to the edge weight where a red edge represents the smallest weight. At last, the graph can be saved as an image.
RESULTS AND DISCUSSION
Feature-driven peptide visualization and clustering demonstrates the common peptide features of the various protein fragments. For example, in Figure 1b we show the distribution of the peptides produced after tryptic digestion of three proteins from three different families (P14921: ETS family; P22676: Calbindin family; and P62166: Recoverin family) according to the selected features (calcium binding and DNA binding).
Also, protein P27487-Dipeptidyl peptidase 4, which is a protein with 766 amino acids, has 12 positions in its sequence where eight of them can affect tryptic peptides. This protein is digested computationally with trypsin, producing 59 peptide sequences. Table 2 shows the number of peptide clusters along with their characteristics. There are 39 peptides out of 59 which are unique in the specified protein and they are not affected by experimental mutation of amino acid(s) on the biological properties of the protein.
Table 2.
Peptide clustering using the features of sequence uniqueness and mutagenesis
| Feature properties for peptide clustering | Number of peptides |
|---|---|
| Unique and mutagenesis | 4 |
| Unique and not mutagenesis | 39 |
| not Unique and mutagenesis | 4 |
| not Unique and not mutagenesis | 12 |
Tryptic digestion of P27487 dipeptidyl peptidase 4 produces 59 peptides which are grouped into 4 clusters.
In a third example, a group of immune peptide sequences were served as an input to PepServe (14). These peptides are differentiation antigens from four cases of cancer disease. There were 13 out of 14 peptides from gut carcinoma that were found to be unique in the human proteome. In the melanoma case, 54 out of 59 peptide sequences were unique, in prostate cancer all 5 peptides were unique and finally in breast cancer both peptides were also unique. The majority of the peptides from each tumor state share the same features, mainly the peptides from the melanoma are placed in the lumenal, melanosome subcellular compartment where there is a non-membrane region of a membrane-spanning protein. These 59 peptides are located in 7 proteins. Respectively, the prostate peptides are found in two proteins with Peptidase S1 protein domain, disulfide bonds and beta strand regions as peptide features.
Peptide clustering using the two features, disulfide bond and beta strand shows us the distinction between the two sets of peptides (Figure 2). The peptides related to gut carcinoma were found in one protein and those related to breast cancer in two proteins.
Figure 2.

Immune peptide sequences as differentiation antigens from prostate cancer disease and melanoma case are clustered according to the two selected features, disulfide bond and beta strand.
The immune peptides and especially the differentiation antigens are considered very useful in the design of monoclonal antibodies since targeted monoclonal antibody therapy is employed to treat diseases. The finding of unique differentiation antigens is considered very crucial and promising in this direction of targeted therapy.
When using the property of uniqueness, a biologist will be able to find the functions of specific peptides with high selectivity. Thus, one may perform studies on the opposite direction from the studies based on sequence and function similarity. That is, the property of sequence uniqueness may imply peptide function uniqueness and perhaps may give insights in the de novo design of differentiation antigens or antimicrobial peptides.
Peptide clustering and visualization offers useful information for the research community, since common peptide characteristics for a set of sequence annotations can emerge from a list of peptide fragments. The biologists can view groups of peptides that share the same characteristics, from a protein enzymatic digestion or mapped on a list of molecular masses from mass spectrometry. Furthermore, peptide feature clustering can be useful in the analysis of cell lines, and specifically cancer cell lines, and also to contribute in providing supplementary information concerning normal and disease states.
PepServe is hosted by an Apache server on a Linux platform and incorporates a script-based curation protocol of the repository files and the UniProt database. The system responds using reporting procedures based on dynamically generated html files (using CGI PERL scripts). The web application is developed in PHP language.
The updating procedure has been scheduled to run twice a year using UniProt’s Swiss-Prot database. PepServe is part of a group of tools, databases and web services developed in the Biomedical Research Foundation, Academy of Athens, called ‘BioServer’.
FUNDING
Funding for open access charge: Biomedical Research Foundation, Academy of Athens, 4 Soranou Ephessiou, 115 27 Athens.
Conflict of interest statement. None declared.
REFERENCES
- 1.Alves G, Ogurtsov AY, Yu YK. RAId_DbS: mass-spectrometry based peptide identification web server with knowledge integration. BMC Genomics. 2008;9:505. doi: 10.1186/1471-2164-9-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helsens K, Timmerman E, Vandekerckhove J, Gevaert K, Martens L. Peptizer, a tool for assessing false positive peptide identifications and manually validating selected results. Mol. Cell Proteomics. 2008;7:2364–2372. doi: 10.1074/mcp.M800082-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Halligan BD, Ruotti V, Twigger SN, Greene AS. DeNovoID: a web-based tool for identifying peptides from sequence and mass tags deduced from de novo peptide sequencing by mass spectroscopy. Nucleic Acids Res. 2005;33:W376–W381. doi: 10.1093/nar/gki461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexandridou A, Tsangaris GT, Vougas K, Nikita K, Spyrou G. Peptide Finder: mapping measured molecular masses to peptides and proteins. Bioinformatics. 2008;24:2267–2269. doi: 10.1093/bioinformatics/btn413. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Na S, Sim JW, Park H, Jeong J, Kim H, Seo Y, Seo J, Lee KJ, Paek E. MODi: a powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res. 2006;34:W258–W263. doi: 10.1093/nar/gkl245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacLean D, Burrell MA, Studholme DJ, Jones AM. PhosCalc: a tool for evaluating the sites of peptide phosphorylation from mass spectrometer data. BMC Res. Notes. 2008;1:30. doi: 10.1186/1756-0500-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beynon RJ. A simple tool for drawing proteolytic peptide maps. Bioinformatics. 2005;21:674–675. doi: 10.1093/bioinformatics/bti077. [DOI] [PubMed] [Google Scholar]
- 8.Pai TW, Chang MD, Tzou WS, Su BH, Wu PC, Chang HT, Chou WI. REMUS: a tool for identification of unique peptide segments as epitopes. Nucleic Acids Res. 2006;34:W198–W201. doi: 10.1093/nar/gkl188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexandridou A, Tsangaris GT, Vougas K, Nikita K, Spyrou G. UniMaP: finding unique mass and peptide signatures in the human proteome. Bioinformatics. 2009;25:3035–3037. doi: 10.1093/bioinformatics/btp516. [DOI] [PubMed] [Google Scholar]
- 10.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Pan D, Hu X, Xiao J, Gao Y, Zhang H, Zhang Y, Liu J, Zhu S. BiodMHC: an online server for the prediction of MHC class II-peptide binding affinity. J. Genet. Genomics. 2009;36:289–296. doi: 10.1016/S1673-8527(08)60117-4. [DOI] [PubMed] [Google Scholar]
- 12.Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003;31:3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heer J, Card SK, Landay JA. In Proceedings of the SIGCHI conference on Human factors in computing systems. USA: ACM, Portland, OR; 2005. Prefuse: a toolkit for interactive information visualization; pp. 421–430. [Google Scholar]
- 14.Jongeneel V. Towards a cancer immunome database. Cancer Immun. 2001;1:3. [PubMed] [Google Scholar]