


Abstract
The products of many bacterial non-ribosomal peptide synthetases (NRPS) are highly important secondary metabolites, including vancomycin and other antibiotics. The ability to predict substrate specificity of newly detected NRPS Adenylation (A-) domains by genome sequencing efforts is of great importance to identify and annotate new gene clusters that produce secondary metabolites. Prediction of A-domain specificity based on the sequence alone can be achieved through sequence signatures or, more accurately, through machine learning methods. We present an improved predictor, based on previous work (NRPSpredictor), that predicts A-domain specificity using Support Vector Machines on four hierarchical levels, ranging from gross physicochemical properties of an A-domain’s substrates down to single amino acid substrates. The three more general levels are predicted with an F-measure better than 0.89 and the most detailed level with an average F-measure of 0.80. We also modeled the applicability domain of our predictor to estimate for new A-domains whether they lie in the applicability domain. Finally, since there are also NRPS that play an important role in natural products chemistry of fungi, such as peptaibols and cephalosporins, we added a predictor for fungal A-domains, which predicts gross physicochemical properties with an F-measure of 0.84. The service is available at http://nrps.informatik.uni-tuebingen.de/.
INTRODUCTION
Non-ribosomally synthesized peptides are a class of highly important metabolites in the secondary metabolisms of bacteria and fungi (1,2). Important representatives of this family are mostly antibiotics like penicillin or vancomycin but also the immunosuppressant cyclosporin. The precursor peptides of these compounds are synthesized by non-ribosomal peptide synthetases (NRPSs), which are multi-modular megasynthetases with molecular weights up to 2.3 MDa (tex1 NRPS from Trichoderma virens). NRPSs act as an assembly line that produces the final peptide by a chain of reactions occuring along that line. The primary sequence of the peptide product is determined by the sequential arrangement of minimal repetitive modules of an NRPS. The minimal module consists of three domains termed adenylation domain (A-domain), peptidyl carrier domain (PCP-domain) and condensation domain (C-domain). The A-domain is responsible for the recruitment of the amino acid monomers that are to be incorporated into the final product. Several hundred different A-domain substrate specificities have been biochemically characterized and each A-domain recruits a specific amino acid as monomer. Accordingly, the sequential order of A-domains along the assembly line determines (in the majority of cases) the primary sequence of the final peptide product. A comprehensive source of NRPS peptides and monomers is the NORINE database assembled by Caboche et al. which currently features over 1000 peptide products and over 500 monomers (3) . The cross linking between each adjacent monomer is carried out by the help of the other two domains that synthesize the peptide bond between these monomers. The minimal module is often equipped with additional domains that allow for modifications of the recruited amino acid monomers like epimerization, methylation or formylation.
The structure–function relationship for monomer recruitment by A-domains has been further elucidated by Stachelhaus et al. and Challis et al. by examining the crystal structure of the peptide synthetase gramicidin S synthetase 1 (GrsA, PDB-ID: 1AMU) (4–6). The structure of the GrsA adenylation domain was determined with a co-crystallized phenylalanine monomer and thus delivers additional structural information about the binding pocket of the A-domain, which enabled Stachelhaus et al. to propose a specificity conferring-code of A-domains by relating the active site configuration of A-domains to the corresponding substrates.
The specificity-conferring code was based on 10 active site residues and it could be used to predict the putative substrates of A-domains for which only the sequence was known. Many NRPS services like the NRPS-PKS knowledgebase, the NP.searcher or the system devised by Bachmann et al. make use of this specificity-conferring code to predict putative A-domain substrates (7–9). The specificity-conferring code was further refined by Rausch et al. (10) by not only considering these 10 residues but by using all active site residues within 8 Å of the amino acid substrate. A predictor, NRPSpredictor, based on Transductive Support Vector Machines (TSVMs) was built on these 34 active site residues to predict A-domain specificity. In the following part of this article we will present details about the new version of this predictor, termed NRPSpredictor2, namely the improved prediction performance, simplified descriptor set used for signature encoding and estimation of the applicability domain of the predictor.
MATERIALS AND METHODS
Method outline
The predictions of substrate specificity are based on the configuration of the residues in the active site of an A-domain. We therefore made use of an A-domain crystal structure (PDB-ID: 1AMU) as a template to determine these active site residues. The positions of these residues were then located in the A-domain sequences of our training data set, and for each domain we extracted those positions. Having labeled sequence data, we applied machine learning methods, namely SVMs, to train predictors of substrate specificity. The predictions are based on numerical representations of the extracted signatures. The predictors were trained as detectors for each known substrate specificity in a one-versus-rest scheme, so every predictor that gives a positive prediction signals that the query A-domain might activate the corresponding substrate. Using this scheme, a query A-domain might yield positive signals from more than one predictor and thereby giving the user additional information about possible substrate promiscuity of the A-domain or ambiguity of the prediction.
Training data
The starting point for this work were the 397 labeled A-domains collected by Rausch et al. for which the specificity had been harvested from scientific literature describing their experimental characterization (10). We added 79 labeled bacterial A-domains and 100 labeled fungal A-domains to the database of NRPSpredictor. Furthermore, we added 4282 unlabeled bacterial and 814 unlabeled fungal A-domains to the data set (see Supplementary Material S1). These A-domains were retrieved from the UniProt database by an automated BLAST search for A-domains that are embedded within a minimal NRPS module, which requires the existence of an A-domain (Pfam-ID: PF00501), C-domain (Pfam-ID: PF00668) and PCP-domain (Pfam-ID: PF00550) (11,12).
Signature extraction
The set of all active site amino acids, called the signature, was identified by extracting all residues within 8 Å of the substrate phenylalanine in the crystal structure of GrsA (PDB-ID: 1AMU). These 34 positions were then extracted from the set of training sequences using an A-domain profile HMM and selecting relevant positions from the alignment. The specificity conferring code proposed by Stachelhaus et al. is a subset of these 34 residues and is also reported by the web server (6). Handling of protein structures, extraction of signatures and further processing was carried out using the Active Site Classification (ASC) software (13).
Encoding
NRPSpredictor2 makes use of two feature encodings for amino acids: one is the original encoding proposed by Rausch et al. based on 12 AAindex (14) descriptors and the other is a reduced encoding based on three z-scales descriptors devised by Wold et al. (15). The z-scales descriptors represent the following physicochemical properties: hydrophobicity (WOLS870101), size (WOLS870102) and electronic properties (WOLS870103). Each signature can be embedded in 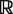 n by encoding each residue into a descriptor tuple and concatenating these tuples. The predictive models are then trained on the transformed data.
n by encoding each residue into a descriptor tuple and concatenating these tuples. The predictive models are then trained on the transformed data.
SVMs
SVMs are classifiers based on the maximum margin principle (16,17). During SVM training a hyperplane in feature space is determined that gives the largest possible margin between the positive and negative class, thereby yielding an intuitively robust classifier. The hyperplane gives a decision surface defined by f(x) = Σi yiαi k(x, xi) whose functional value is zero for data points directly on the hyperplane, +1 or more for data points in the positive half-space and −1 or less for points in the negative half-space. The margin is determined by the geometric distance of points with functional value of +1 or −1 (support vectors) to the hyperplane. NRPSpredictor2 uses the RBF kernel k(x, y) = exp(−γ||x−y||2) and the linear kernel k(x, y) = xty on the physico-chemical feature vectors. For the training of SVMs a set of labeled data points (xi, yi) is needed where xi is from 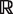 n and the labels yi are in (+1, −1) for two-class problems.
n and the labels yi are in (+1, −1) for two-class problems.
TSVMs
TSVMs extend classical SVMs by the property of making use of unlabeled data to train more robust classifiers, especially in the case of scarce labeled training data (18). TSVMs try to determine a separating hyperplane that does not cut clusters of data by forcing the hyperplane to go through low data density regions. This is enforced by keeping the margin clear of unlabeled data points. However, the objective function of TSVMs is not that easily optimized as the classical SVM objective, hence heuristics have to be used to optimize the objective. For NRPSpredictor2 we make use of the SVMlight package that offers such an heuristic to train TSVM classifiers (18).
Prediction levels and predictor quality
NRPSpredictor2 was designed to predict the putative substrate specificity on four different hierarchical levels for bacterial A-domains and on one level for fungal A-domains. The bacterial levels are: gross physico-chemical properties of the substrate (hydrophobic–aromatic, hydrophobic–aliphatic and hydrophilic), large clusters, small clusters and on a single amino acid level (Table 1). The fungal predictor predicts only on the gross physico-chemical properties level (hydrophobic-aromatic, hydrophobic-aliphatic and hydrophilic) due to the lack of sufficient fungal training data to allow further subdivision of substrate clusters. However, within the web server we trigger the bacterial models to give also more fine grained predictions for fungal signatures. An overview of the set of bacterial prediction levels is given in Table 1. For many substrates there are only very few labeled A-domains, like the 2-amino-butyric acid (Abu) specificity with less than five known A-domain sequences. For these specificities no SVM-model was built. Instead, we make use of the Nearest-Neighbor Rule to get a specificity prediction, by reporting for each query the substrate specificity of the most similar active-site signature (based on the Stachelhaus code) in our database, along with the sequence identity.
Table 1.
Prediction levels and predictor quality (bacterial)
| Classname | Members | Type | NRPSpredictor2 |
NRPSpredictor1 | ||
|---|---|---|---|---|---|---|
| F | Prec. | Rec. | F | |||
| Three class | ||||||
| Hydrophobic aliphatic | Ala, Gly, Val, Leu, Ile, Abu, Iva Ser, Thr, Hpg, Dhpg, Cys, Pro, Pip | W,R,T | 0.974 | 0.974 | 0.974 | – |
| Hydrophilic | Arg, Asp, Glu, His, Asn, Lys, Gln, Orn, Aad | W,R,T | 0.940 | 0.940 | 0.940 | – |
| Hydrophobic aromatic | Phe, Tyr, Trp, Dhb, Phg, Bht | W,R,T | 0.890 | 0.889 | 0.892 | – |
| Large clusters | ||||||
| Hydroxy-benzoic acid derivates | Dhb, Sal | W,R,T | 0.982 | 1.000 | 0.967 | 0.982 |
| Polar, uncharged (aliphatic with -SH) | Cys | R,R,T | 0.976 | 0.975 | 0.975 | 0.954 |
| Aliphatic chain or phenyl group with -OH | Ser, Thr, Dhpg, Hpg | R,R,T | 0.968 | 0.967 | 0.969 | 0.963 |
| Aliphatic chain with H-bond donor | Asp, Asn, Glu, Gln, Aad | W,R,C | 0.958 | 0.969 | 0.950 | 0.942 |
| Apolar, aliphatic | Gly, Ala, Val, Leu, Ile, Abu, Iva | W,R,T | 0.940 | 0.947 | 0.934 | 0.940 |
| Aromatic side chain | Phe, Trp, Phg, Tyr, Bht | W,R,T | 0.881 | 0.881 | 0.881 | 0.881 |
| Cyclic aliphatic chain (polar NH2 group) | Pro, Pip | R,R,T | 0.867 | 0.867 | 0.867 | 0.811 |
| Long positively charged side chain | Orn, Lys, Arg | W,R,T | 0.864 | 0.898 | 0.833 | 0.861 |
| Ø | 0.930 | – | – | 0.917 | ||
| Small clusters | ||||||
| 2-amino-adipic acid | Aad | W,L,C | 1.000 | 1.000 | 1.000 | 1.000 |
| Dhb, Sal | Dhb, Sal | W,L,C | 1.000 | 1.000 | 1.000 | 0.940 |
| Polar, uncharged (hydroxy-phenyl) | Dhpg, Hpg | R,L,T | 1.000 | 1.000 | 1.000 | 0.981 |
| Cys | Cys | R,L,T | 0.983 | 0.983 | 0.983 | 0.950 |
| Serine-specific | Ser | W,R,T | 0.972 | 1.000 | 0.947 | 0.936 |
| Threonine-specific | Thr | W,L,C | 0.969 | 0.978 | 0.961 | 0.942 |
| Asp-Asn | Asp, Asn | W,L,C | 0.948 | 0.969 | 0.931 | 0.942 |
| Orn and hydroxy- Orn specific | Orn | R,L,T | 0.900 | 0.900 | 0.900 | 0.800 |
| Aliphatic, branched hydrophobic | Val, Leu, Ile, Abu, Iva | W,R,T | 0.893 | 0.892 | 0.895 | 0.887 |
| Tiny, hydrophilic, transition to aliphatic | Gly, Ala | W,L,C | 0.886 | 0.938 | 0.843 | 0.859 |
| Pro-specific | Pro | R,L,T | 0.882 | 0.938 | 0.833 | 0.900 |
| Polar aromatic ring | Tyr, Bht | W,R,T | 0.857 | 0.892 | 0.825 | 0.793 |
| Glu-Gln | Glu, Gln | W,L,C | 0.813 | 0.850 | 0.791 | 0.860 |
| Arg-specific | Arg | W,L,C | 0.740 | 1.000 | 0.600 | 0.800 |
| Unpolar aromatic ring | Phe, Trp | W,L,C | 0.538 | 0.608 | 0.500 | 0.671 |
| Ø | 0.892 | – | – | 0.884 | ||
| Single substrates | ||||||
| Aad | Aad | W,R,T | 1.000 | 1.000 | 1.000 | – |
| Cys | Cys | R,R,T | 1.000 | 1.000 | 1.000 | – |
| Hpg | Hpg | R,R,T | 0.974 | 1.000 | 0.950 | – |
| Ser | Ser | W,R,T | 0.962 | 0.993 | 0.933 | – |
| Thr | Thr | W,R,T | 0.949 | 0.976 | 0.922 | – |
| Dhb | Dhb | W,R,T | 0.947 | 1.000 | 0.900 | – |
| Dhpg | Dhpg | W,R,T | 0.943 | 0.967 | 0.925 | – |
| Asn | Asn | R,R,T | 0.939 | 0.934 | 0.944 | – |
| Orn | Orn | R,R,T | 0.933 | 0.933 | 0.933 | – |
| Ile | Ile | R,R,T | 0.918 | 1.000 | 0.850 | – |
| Gly | Gly | R,R,T | 0.906 | 0.902 | 0.910 | – |
| Ala | Ala | W,R,T | 0.878 | 0.901 | 0.856 | – |
| Arg | Arg | W,R,T | 0.833 | 0.833 | 0.833 | – |
| Iva | Iva | W,R,T | 0.814 | 0.933 | 0.725 | – |
| Val | Val | W,R,T | 0.801 | 0.828 | 0.777 | – |
| Leu | Leu | W,R,T | 0.784 | 0.782 | 0.787 | – |
| Pro | Pro | W,R,T | 0.755 | 0.792 | 0.722 | – |
| Bht | Bht | W,R,T | 0.717 | 0.782 | 0.675 | – |
| Glu | Glu | R,R,T | 0.704 | 0.760 | 0.657 | – |
| Pip | Pip | W,R,T | 0.700 | 0.800 | 0.625 | – |
| Asp | Asp | R,R,T | 0.700 | 0.700 | 0.700 | – |
| Tyr | Tyr | W,R,T | 0.696 | 0.671 | 0.725 | – |
| Gln | Gln | W,R,T | 0.689 | 0.775 | 0.620 | – |
| Phe | Phe | W,R,T | 0.688 | 0.740 | 0.643 | – |
| Lys | Lys | R,R,T | 0.400 | 0.500 | 0.333 | – |
| Trp | Trp | W,R,T | 0.320 | 0.400 | 0.267 | – |
The column type gives the best performing predictor encoded by three letters: the first letter represents the used encoding (W: Wold, R: Rausch), the second letter the used kernel (L: linear, R: RBF) and the third letter the used SVM type (C: classical SVM T: transductive SVM). The columns F, Prec. and Rec. give the F-measure, Precision and Recall of the best predictor, respectively. Aad: 2-amino-adipic-acid; Bht: beta-hydroxy-tyrosine; Hpg: 4-hydoxy-phenyl-glycine; Dhb: 2,3-dihydroxy-benzoic acid; Dhpg: 3,5-dihydroxy-phenyl-glycin; Iva: isovaline; Orn: ornitine; Pip: pipecolic acid; Sal: salicylic acid.
Predictor validation
To quantify the performance of the NRPSpredictor2 we used the F-measure as quality criterion, which is defined as the harmonic mean of precision and recall. The precision is defined by prec = tp/(tp + fp) and the recall (or sensitivity) is defined by rec = tp/(tp + fn), where tp, fp and fn are the number of true positives, false positives and false negatives, respectively. The precision (or positive predictive value) measures how reliable a positive prediction of a substrate specificity detector is and the recall measures how good the detector is in finding the true positives. To determine the performance on new test data we applied a repeated external validation scheme. We split the whole data set into half, selected and trained a SVM model on one half of the data and evaluated the predictor performance on the other half, the independent test set. This procedure was repeated on 10 shuffled versions of the whole data set to get a more robust average of the predictor performance on new test data.
Applicability domain
The applicability domain of a predictor is a concept that helps to give for each predictor query a feedback whether that query is too far away from the data used during training or whether that instance lies within the, say, 95% support volume of the training data. Predictions for queries that do not lie within the applicability domain of the model should be handled with more care. To model the applicability domain of our model we made use of the 1-Class SVM concept as described by Schölkopf et al. (19). Therefore, we modelled the 95% support of our data using the 1-Class SVM functionality of LIBSVM. We selected values for γ and ν in such a way as to achieve a recall of ∼95% on left out data and then trained a 1-class SVM for the whole data set using these parameters to describe the 95% support volume in feature space of our data.
RESULTS
Predictor quality
The quality of each bacterial predictor as determined by our model validation is given in Table 1. It can be observed that the predictors at the highest hierarchical level are the best-performing ones. At the level of gross physico-chemical properties we have an average F-measure of F = 0.94, whereas the average F-measure at the most fine-grained level (single substrates) is F = 0.80. Generally, the average performance as quantified by the F-measure is F = 0.94 for the three class level, F = 0.93 for the large clusters level, F = 0.89 for the small clusters level and F = 0.80 for the single substrate level. The fungal predictor has an average F-measure of F = 0.84 at the three class level. Table 1 also gives for each prediction task the best performing kernel, feature encoding and SVM type (classic or TSVM).
A general trend is that, except from the more exotic aromatic substrates, like the hydroxy-benzoic derivatives that can be predicted very well, the other more common aromatic substrates are predicted less reliably. One reason might be the observed promiscuity of the A-domains utilizing these substrates (10). When compared with the original version of the NRPSpredictor (Table 1) the new version could improve the performance (F-measure) on the large cluster level and on the small clusters level by roughly one percentage point. While the original NRPSpredictor was able to predict the membership to clusters of amino acids only, NRPSpredictor2 also can predict single amino acid specificities. The newly introduced applicability domain gives further information on the quality of the specificity prediction. Upon request of many colleagues working on fungal NRPSs, a predictor specific for fungal NRPS sequences was included in NRPSpredictor2.
Web server
Users of the NRPSpredictor2 web server can submit their data as full NRPS sequences in multi-FASTA format and the signatures will be extracted automatically. Another option is to directly supply the extracted signatures and request a prediction from the predictor, thus users are not required to disclose the full NRPS sequence. After short extraction and prediction phases the user receives a list of detected A-domains along with the predictions of NRPSpredictor2 at each hierarchical level. For user convenience we report the predictions of the original version of the NRPSpredictor. A typical report for one particular extracted A-domain is given in Figure 1. For each extracted A-domain the ID of the parent sequence is given with the number of the A-domain added as suffix. The exact location of the A-domain within the parent sequence is also reported, along with the bit score of the Pfam HMM that extracted this domain. The result of the applicability check is given by either a green checkmark (as shown in Figure 1) if the query signatures lies within the applicability domain of our predictor or as red X if the signature is most likely outside the applicability domain of the model. In this case the prediction should be taken with caution. Finally, the specificity predictors that give positive predictions for this signature are listed for each hierarchical level. The scores of the SVMs along with the precision of the SVM predictors, determined during model validation, are given in the last two columns. The last row gives the nearest neighbor to the query signature found in our database of annotated A-domain signatures (based on Stachelhaus code) along with the sequence identity. Using this rule NRPSpredictor2 can even detect specificities for which no SVM model could be learned, due to scarcity of labeled training data.
Figure 1.

NRPSpredictor2 prediction report for one extracted A-domain. On top, the ID of the parent sequence, location of the A-domain within the sequence and the bit score of the PFAM-HMM are given. The green checkmark signals that the signature sequence lies within the applicability domain of the model. The extracted 8 Å signature and Stachelhaus code are given directly below. Subsequently, the list of predictions is given along with the score of the respective SVM predictors. For each predictor we also report the reliability of that predictor as determined during model validation. The last row gives the nearest sequence neighbor in the NRPSpredictor2 database (based on Stachelhaus code) and the respective sequence identity.
DISCUSSION
We have presented the NRPSpredictor2 that predicts A-domain substrate specificity based on sequence and structural information about the active site of the domain. The new predictor comes with an improved prediction performance over the previous version and also with two new prediction levels, namely the gross physico-chemical properties level and the detailed prediction level, which predicts the single amino acid likely to be activated by the given A-domain. The performance improvement was mainly due to the additional labeled training data as well as the use of an additional encoding of A-domain signatures (Wold encoding). The transductive SVM method, which makes use of unlabeled data, is very important in the settings with scarce training data per class, as can be seen in the most detailed prediction tasks (single amino acid level) where the transductive SVM is the best performing type of SVM. In the upper prediction levels classical SVMs quite often suffice to build a well-performing predictive model. In some of these cases the use of a transductive SVM might even hurt performance due to the heuristic training procedure that may yield suboptimal models, when compared to the classical SVM models, which use only labeled training data. We also created a new web interface for the predictor, allowing prediction of either bacterial or fungal sequences based on full NRPS sequences or already extracted signatures. For comparison purposes the web server also reports the predictions of the original NRPSpredictor. Finally, NRPSpredictor2 has also been incorporated into antiSMASH, a new comprehensive pipeline for secondary metabolite gene cluster detection and annotation, which allows users to rapidly analyze complete NRPS gene clusters or even whole genomes containing multiple NRPS gene clusters (M. H. Medema et al., submitted for publication).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Ministry for Education and Research (BMBF) [0315585A (GenBioCom) to T.W.]. The work of MHM was supported by the Dutch Technology Foundation (STW), which is the applied-science division of The Netherlands Organisation for Scientific Research (NWO) and the Technology Programme of the Ministry of Economic Affairs (grant STW 10463). Funding for open access charge: University of Tübingen.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Rainer Breitling for constructive comments and suggestions.
REFERENCES
- 1.Marahiel MA, Stachelhaus T, Mootz HD. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem. Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 2.Schwarzer D, Finking R, Marahiel MA. Nonribosomal peptides: from genes to products. Nat. Prod. Rep. 2003;20:275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 3.Caboche S, Pupin M, Leclere V, Fontaine A, Jacques P, Kucherov G. NORINE: a database of nonribosomal peptides. Nucleic Acids Res. 2008;36:D326–D331. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 5.Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 7.Ansari MZ, Yadav G, Gokhale RS, Mohanty D. NRPS-PKS: a knowledge-based resource for analysis of NRPS/PKS megasynthases. Nucleic Acids Res. 2004;32:W405–413. doi: 10.1093/nar/gkh359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S, Sherman DH. Automated genome mining for natural products. BMC Bioinformatics. 2009;10:185. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachmann BO, Ravel J. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 10.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Röttig M, Rausch C, Kohlbacher O. Combining structure and sequence information allows automated prediction of substrate specificities within enzyme families. PLoS Comput Biol. 2010;6:e1000636. doi: 10.1371/journal.pcbi.1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wold S, Eriksson L, Hellberg S, Jonsson J, Sjöström M, Skagerberg B, Wikström C. Principal property-values for 6 nonnatural amino-acids and their application to a structure activity relationship for oxytocin peptide analogs. Can. J. Chem. 1987;65:1814–1820. [Google Scholar]
- 16.Boser BE, Guyon IM, Vapnik VN. Proceedings of the Fifth Annual Workshop on Computational Learning Theory. Pittsburgh, Pennsylvania, United States: ACM; 1992. pp. 144–152. [Google Scholar]
- 17.Cortes C, Vapnik V. Support-vector networks. Mach. Lear. 1995;20:273–297. [Google Scholar]
- 18.Joachims T. Proceedings of the Sixteenth International Conference on Machine Learning. Morgan Kaufmann Publishers Inc.; 1999. pp. 200–209. [Google Scholar]
- 19.Schölkopf B, Platt JC, Shawe-Taylor JC, Smola AJ, Williamson RC. Estimating the Support of a High-Dimensional Distribution. Neural Comput. 2001;13:1443–1471. doi: 10.1162/089976601750264965. [DOI] [PubMed] [Google Scholar]