


Abstract
With the immense growth in the number of available protein structures, fast and accurate structure comparison has been essential. We propose an efficient method for structure comparison, based on a structural alphabet. Protein Blocks (PBs) is a widely used structural alphabet with 16 pentapeptide conformations that can fairly approximate a complete protein chain. Thus a 3D structure can be translated into a 1D sequence of PBs. With a simple Needleman–Wunsch approach and a raw PB substitution matrix, PB-based structural alignments were better than many popular methods. iPBA web server presents an improved alignment approach using (i) specialized PB Substitution Matrices (SM) and (ii) anchor-based alignment methodology. With these developments, the quality of ∼88% of alignments was improved. iPBA alignments were also better than DALI, MUSTANG and GANGSTA+ in >80% of the cases. The webserver is designed to for both pairwise comparisons and database searches. Outputs are given as sequence alignment and superposed 3D structures displayed using PyMol and Jmol. A local alignment option for detecting subs-structural similarity is also embedded. As a fast and efficient ‘sequence-based’ structure comparison tool, we believe that it will be quite useful to the scientific community. iPBA can be accessed at http://www.dsimb.inserm.fr/dsimb_tools/ipba/.
INTRODUCTION
Continuous increase in number of 3D structures of proteins necessitates development of efficient tools for structure comparison. Such developments facilitate characterization of function of a protein of known structure (1) or aid in evolutionary studies (2–4). Considering the complexity involved in obtaining an optimal superposition solely by global structural searches, a large majority of the structural alignment approaches focus on optimizing a combination of local segments of similarity to derive the global alignment (5–7). Many of the very recent approaches consider the match between secondary structural elements (8–10) while others are fragment based (11–16). This idea is extended further to investigate flexibility of protein structures (17,18).
Local backbone conformations such as α-helices, β-strands, β-turns and PPII helices characterize a large part tertiary structure of a protein chain. A complete protein backbone can be approximated with a limited set of local conformations. Such a collection of local structural prototypes is called Structural Alphabets (SA). Protein Blocks (PBs) (19–21) is one such SA involving 16 pentapeptide conformations (represented by alphabets a to p), characterized by backbone dihedral angles. Several biological questions could be addressed based on PB-based abstraction.
The main chain 3D information can be represented as a sequence in 1D, using PBs. This reduces the problem of protein structural comparison to a classical sequence alignment. Dynamic programming algorithms like Needleman Wunsch (22) and Smith Waterman (23) were used earlier for PB alignment and PB substitution matrix was generated for scoring the alignment (24–26). We propose an improved and novel version of PB alignment using (i) specialized substitution matrices for pairwise alignment and database search and (ii) an anchor-based dynamic programming algorithm. Most of the recent web tools for structure comparison are either dedicated to a database search (9–10,13,27,28) or for pairwise structural alignments (29–32). As an efficient tool for both pairwise alignments and database searches, this web-server serves as a good platform for such studies. A local alignment strategy for motif or sub-structure search is also available. The proposed development provides output such as: (i) different scoring schemes to indicate the quality of the alignment, (ii) user-friendly interface to view and analyze the 3D superposition and (iii) downloadable alignment files (both sequence and structural alignment).
MATERIALS AND METHODS
The server can be used to search for structural relatives of a query protein (Figure 1A) or to compare two protein structures (Figure 1B). In both cases, the user can decide whether to carry out alignments for the complete structure (global) or to look for the best local similarity (local).
Figure 1.

The framework of iPBA and underlying methods. User can either compare two structures or search for structural neighbors (mining) from a databank. The input and output web interfaces for pairwise structural alignment are highlighted with a blue background. The web interfaces for mining has a green background. The rest of the figure (white background) gives the outline of underlying methodological aspects. (A) Search for structural similar protein in 3D database. (B) Compare two protein structures. (C) Alignment approach. (D) Main outputs.
Input
For comparing two structures, the user can either provide the coordinates in the standard PDB format or enter the PDB code. The identifiers of chains to be compared should also be given. For searching related protein structure in database, only one PDB file or code is necessary (Figure 1A and B).
Pre-processing
Atomic coordinate sets are first translated into sequence of PBs (Figure 1C). PBs constitute 16 pentapeptide conformations (labeled from a to p) each described by a series of Φ, Ψ dihedral angles. A reasonable approximation of local structures (19) with a root mean square deviation (RMSD) of 0.42 Å could be obtained (33).
Computing pairwise alignment
The alignment method implemented in this server represents a significant improvement over our earlier work (24). In the previous work, the PB substitution matrix was generated from pairwise alignments in PALI database (3). This database was redundant in terms of the distribution of related proteins. We have so refined the databank. Hence the PB substitutions were calculated from a non-redundant subset sharing sequence identity <40% and a refined substitution matrix was generated. Also, in our previous approach, a simple Needleman–Wunsch (22) algorithm was used for alignment. Protein structural homologues are often characterized by conserved stretches separated by variable regions. Hence a combination of local and global alignment is expected to give a better performance.
A set of local alignments (anchors) associated with these two sequences is derived using a modified version of SIM algorithm (34). The remaining segments between anchors (linkers) are then aligned using the Needleman–Wunsch algorithm (Figure 1C). Affine gap penalties are used for the anchor and linker alignments. Distance constraints on the structures are included to identify false anchors. The different parameters were optimized as done in the previous work based on alignments of proteins in PALI data set (3). A total of 80% of the alignments were better when compared to that obtained with our previous work (24).
Different scores are used to quantify the quality of PB alignment:
A score similar to Global Distance Test Total score (GDT_TS) (35) for PB sequence alignment, derived using seven decreasing cut-offs of PB substitution scores (similar to distance cut-offs for GDT_TS).
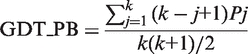 |
where k corresponds to the total number of thresholds used, i.e. 7. Pj is the percentage of PB substitutions that are within the cut-off level j. The residue equivalences from the PB alignment then guides the 3D fitting of the structures by ProFit (36) (http://www.bioinf.org.uk/software/profit/) which reports the RMSD and number of aligned residues (within 5 Å) (Figure 2). The GDT_TS score for the alignment is also provided along with the Aln_Score and GDT_PB. Note that the GDT_TS score used for comparison of iPBA with other web-tools (Table 1) was computed with a maximum distance threshold of 5 Å. The percentage of equivalent residues was calculated from only one of the protein lengths. These variations were included to avoid bias in the score due to the different distance thresholds used by different methods and also due to incomplete alignment outputs provided by the servers.
Figure 2.

Comparison of iPBA with other Rigid Body alignment methods. The 3D superposition of Nucleotide Kinases (PDB IDs: 1AKY and 1GKY) by different methods is shown. The RMSD (in bold) and the number of aligned residues (as reported by the tool) are also given.
Table 1.
Comparison of iPBA with different structural alignment tools (web services)
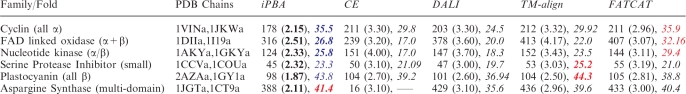 |
Each protein pair is chosen in random from different structural classes (in parentheses), from the HOMSTRAD database (4). The number of aligned residues (as defined by different methods) and their RMSD is given within parentheses. The GDT_TS score calculated for increasing distances of 0.5 Å in the range 0.5–5 Å, is also shown in italics. The best and second best scores are highlighted in red and blue. (–––) reflects the incomplete output of the program which limits GDT_TS calculation. Rigid-body approaches have been tested with CE, DALI and TM-Align. Best RMSD and GDT_TS of the rigid-body approaches have been highlighted in bold.
Database search
A sequence of PBs can also be used to search for structurally related proteins from a data set of structures (Figure 1A). SCOP version 1.75 SCOP (37) is used as the structure data set and the user can also search refined subsets derived at different sequence identity cut-offs. The top 100 hits are reported based on the PB alignment score which is scaled to values between −13 and 17. Values >1.5 are generally associated with high confidence. GDT_PB scores are also provided for the hits obtained. To account for the speed, structure based refinements are not included. User can carry out further alignments of the hits obtained (Figure 1A and B).
Output for pairwise alignments
With the help of Jmol applet, users can have a 3D analysis of superposed structures and also choose different visual representations of structure (Figure 1D). Images of aligned structures rendered in PyMol are also provided. The residue equivalences in the 3D alignment are given as a complete sequence alignment. The corresponding PBs are also shown in the alignment. PB stretches of high similarity, identified as anchors, are also highlighted (Figure 1D). The user can download coordinates of aligned structures in PDB format and PyMol scripts for local analysis of the superposition. Raw output file with sequence alignment and quality scores is also downloadable in text format.
Implementation
Implementation of this tool is mainly done in C, Python, HTML and also using Jmol and PyMol programs. The front-end use is based on html and php. Perl/cgi programs control the input while python and C based programs carry out the processing behind the database search and pairwise comparisons. Direct visualization and manipulation of aligned structured is enabled with a Jmol applet and static images of superposed structures are rendered in PyMol using internal ‘raytracer’ option. Supplementary Data S1 shows the schematic representation of series of steps involved in iPBA webserver.
DISCUSSION
As shown in Figure 1, it is quite simple to use the web-based iPBA alignment tool. User only needs to give the coordinates to mine SCOP (Figure 1A) or for pairwise superimposition (Figure 1B). Outputs are mainly given visually as sequence alignments and 3D structure superimpositions (Figure 1D). Output alignment files can be also downloaded for local use. The local alignment strategy also provides a route to detect specific structural motifs in proteins.
The improvement in the alignment methodology and the use of specialized PB substitution matrices has greatly enhanced the quality of alignments and the mining efficiency. The PB-based alignment approach had shown an impressive performance as a structure comparison tool (24). Supplementary Figure 2 highlights the gain in alignment quality with respect to the earlier approach [PBALIGN, (24)]. One hundred randomly chosen SCOP domain pairs sharing <40% sequence identity were used for comparison. 89% of the alignments have a better RMSD when compared to PBALIGN (Supplementary Data S2). Comparison performed on a bigger benchmark data set also suggested that a significant gain of 82% in alignment quality could be achieved. The mining efficiency also improved by 6.8% and the gain was largely uniform across different structural classes.
To present a picture on the performance, the quality of alignments generated by iPBA was compared with the output alignments of some of the other well-established tools like CE, DALI, FATCAT and TMalign (7,18,38,39) (Table 1). For the full-length chains (‘global’ alignment option), the alignments generated using iPBA has the least RMSD. However, the number of aligned residues is also lower in many cases. GDT_TS scores are more appropriate in such cases to give a better idea of the alignment quality. As highlighted in Table 1, iPBA generates alignments of very high quality. Among the non-flexible aligners (CE, DALI and TMalign), iPBA alignments have the best quality scores in the majority of cases. FATCAT produces flexible alignments and it is expected to give the best performance when flexible movements are involved. This is true for the first three cases in Table 1 where iPBA scores next to FATCAT. Thus the quality of iPBA alignments is largely comparable. In a systematic comparison using the standalone version of iPBA, the alignments were found to be better than DALI and MUSTANG in >80% of the cases. To demonstrate this, we chose the data set of 100 domain pairs from SCOP database, sharing <40% sequence identity. On this set of domain pairs, the alignments generated by iPBA were compared to those obtained with DALI (38), MUSTANG (40), GANGSTA+ (41) and TMalign (39). A total o 93.2 and 95.1% of the alignments had a better GDT_TS score compared to DALI and MUSTANG alignments respectively (Supplementary Data 3A and B). The quality of ∼81.6% of alignments were better than GANGSTA+ while the difference was less striking when compared to TMalign. About 45% of the alignments had a GDT_TS score lower than TMalign (Supplementary Data 3D), however the difference in scores for 80% of these cases was <3, reflecting a similar alignment.
Figure 2 presents a view of the 3D alignments of two Nucleotide Kinase structures with similar folds, using different non-flexible alignment approaches like DALI, CE, TM-Align, GANGSTA+ and ALADYN. As highlighted (also see Table 1), the alignment quality is better with iPBA. A closer look on the figure can show that iPBA gives a more refined alignment with the equivalent secondary structural elements well fitted onto each other.
CONCLUSION
The ability to represent complete backbone conformation of the protein chain as a series of alphabets followed by the use of sequence alignment techniques mainly distinguishes iPBA from other structure comparison tools. In terms of alignment quality and the efficiency in detecting structural relatives, iPBA has been quite successful among the wide range of methods available (42). The local alignment option further adds to the utility of this approach. The web tool also provides an interface for the visualization and analysis of the alignments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French Ministry of Research; University of Paris Diderot – Paris 7; French National Institute for Blood Transfusion (INTS); French Institute for Health and Medical Research (INSERM) (to A.P.J., J.-C.G. and A.G.d.B.); Department of Biotechnology, India (to N.S.); CEFIPRA number 3903-E (to A.P.J.); CEFIPRA for collaborative grant (number 3903-E) (to N.S. and A.G.d.B.). Funding for open access charge: INSERM (NAR membership).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would also like to thank the anonymous reviewers for their help in improving the manuscript.
REFERENCES
- 1.Skolnick J, Fetrow JS, Kolinski A. Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 2000;18:283–287. doi: 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal G, Rajavel M, Gopal B, Srinivasan N. Structure-based phylogeny as a diagnostic for functional characterization of proteins with a cupin fold. PLoS ONE. 2009;4:e5736. doi: 10.1371/journal.pone.0005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaji S, Sujatha S, Kumar SS, Srinivasan N. PALI-a database of Phylogeny and ALIgnment of homologous protein structures. Nucleic Acids Res. 2001;29:61–65. doi: 10.1093/nar/29.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizuguchi K, Deane CM, Blundell TL, Overington JP. HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci. 1998;7:2469–2471. doi: 10.1002/pro.5560071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 6.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 7.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 8.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 9.Shi S, Chitturi B, Grishin NV. ProSMoS server: a pattern-based search using interaction matrix representation of protein structures. Nucleic Acids Res. 2009;37:W526–W531. doi: 10.1093/nar/gkp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang ZH, Bharatham K, Sherman WA, Mihalek I. deconSTRUCT: general purpose protein database search on the substructure level. Nucleic Acids Res. 38:W590–W594. doi: 10.1093/nar/gkq489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedberg I, Harder T, Kolodny R, Sitbon E, Li Z, Godzik A. Using an alignment of fragment strings for comparing protein structures. Bioinformatics. 2007;23:e219–e224. doi: 10.1093/bioinformatics/btl310. [DOI] [PubMed] [Google Scholar]
- 12.Madhusudhan MS, Webb BM, Marti-Renom MA, Eswar N, Sali A. Alignment of multiple protein structures based on sequence and structure features. Protein Eng. Des. Sel. 2009;22:569–574. doi: 10.1093/protein/gzp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Margraf T, Schenk G, Torda AE. The SALAMI protein structure search server. Nucleic Acids Res. 2009;37:W480–W484. doi: 10.1093/nar/gkp431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tung CH, Huang JW, Yang JM. Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for rapid search of protein structure database. Genome Biol. 2007;8:R31. doi: 10.1186/gb-2007-8-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Zheng WM. CLePAPS: fast pair alignment of protein structures based on conformational letters. J. Bioinform. Comput. Biol. 2008;6:347–366. doi: 10.1142/s0219720008003461. [DOI] [PubMed] [Google Scholar]
- 16.Yang J. Comprehensive description of protein structures using protein folding shape code. Proteins. 2008;71:1497–1518. doi: 10.1002/prot.21932. [DOI] [PubMed] [Google Scholar]
- 17.Shatsky M, Nussinov R, Wolfson HJ. Flexible protein alignment and hinge detection. Proteins. 2002;48:242–256. doi: 10.1002/prot.10100. [DOI] [PubMed] [Google Scholar]
- 18.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19(Suppl. 2):ii246–ii255. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 19.de Brevern AG, Etchebest C, Hazout S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins. 2000;41:271–287. doi: 10.1002/1097-0134(20001115)41:3<271::aid-prot10>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Etchebest C, Benros C, Hazout S, de Brevern AG. A structural alphabet for local protein structures: improved prediction methods. Proteins. 2005;59:810–827. doi: 10.1002/prot.20458. [DOI] [PubMed] [Google Scholar]
- 21.Joseph AP, Agarwal G, Mahajan S, Gelly J-C, Swapna LS, Offmann B, Cadet F, Bornot A, Tyagi M, Valadié H, et al. A short survey on protein blocks. Biophys. Rev. 2010;2:137–145. doi: 10.1007/s12551-010-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 23.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 24.Tyagi M, de Brevern AG, Srinivasan N, Offmann B. Protein structure mining using a structural alphabet. Proteins. 2008;71:920–937. doi: 10.1002/prot.21776. [DOI] [PubMed] [Google Scholar]
- 25.Tyagi M, Gowri VS, Srinivasan N, de Brevern AG, Offmann B. A substitution matrix for structural alphabet based on structural alignment of homologous proteins and its applications. Proteins. 2006;65:32–39. doi: 10.1002/prot.21087. [DOI] [PubMed] [Google Scholar]
- 26.Tyagi M, Sharma P, Swamy CS, Cadet F, Srinivasan N, de Brevern AG, Offmann B. Protein Block Expert (PBE): a web-based protein structure analysis server using a structural alphabet. Nucleic Acids Res. 2006;34:W119–W123. doi: 10.1093/nar/gkl199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim BH, Cheng H, Grishin NV. HorA web server to infer homology between proteins using sequence and structural similarity. Nucleic Acids Res. 2009;37:W532–W538. doi: 10.1093/nar/gkp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konagurthu AS, Stuckey PJ, Lesk AM. Structural search and retrieval using a tableau representation of protein folding patterns. Bioinformatics. 2008;24:645–651. doi: 10.1093/bioinformatics/btm641. [DOI] [PubMed] [Google Scholar]
- 29.Potestio R, Aleksiev T, Pontiggia F, Cozzini S, Micheletti C. ALADYN: a web server for aligning proteins by matching their large-scale motion. Nucleic Acids Res. 2010;38:W41–W45. doi: 10.1093/nar/gkq293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teichert F, Bastolla U, Porto M. SABERTOOTH: protein structural alignment based on a vectorial structure representation. BMC Bioinformatics. 2007;8:425. doi: 10.1186/1471-2105-8-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosca R, Schneider TR. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res. 2008;36:W42–W46. doi: 10.1093/nar/gkn197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sippl MJ. On distance and similarity in fold space. Bioinformatics. 2008;24:W872–W873. doi: 10.1093/bioinformatics/btn040. [DOI] [PubMed] [Google Scholar]
- 33.de Brevern AG. New assessment of a structural alphabet. In Silico Biol. 2005;5:283–289. [PMC free article] [PubMed] [Google Scholar]
- 34.Huang X, Miller W. A time-efficient linear-space local similarity algorithm. Advances in Applied Mathematics. 1991;12:337–357. [Google Scholar]
- 35.Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLachlan A. Rapid comparison of protein structres. Acta Cryst A. 1982;38:871–873. [Google Scholar]
- 37.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 38.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: a multiple structural alignment algorithm. Proteins. 2006;64:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- 41.Guerler A, Knapp EW. Novel protein folds and their nonsequential structural analogs. Protein Sci. 2008;17:1374–1382. doi: 10.1110/ps.035469.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joseph AP, Srinivasan N, de Brevern AG. Improvement of protein structure comparison using a structural alphabet. Biochimie. 2011 doi: 10.1016/j.biochi.2011.04.010. in press. [DOI] [PubMed] [Google Scholar]