


Abstract
MetalDetector identifies CYS and HIS involved in transition metal protein binding sites, starting from sequence alone. A major new feature of release 2.0 is the ability to predict which residues are jointly involved in the coordination of the same metal ion. The server is available at http://metaldetector.dsi.unifi.it/v2.0/.
INTRODUCTION
Metalloproteins are a large and diverse class of proteins which bind one or more metal ions in their native conformation (1). Metal atoms play a wide range of structural, regulatory or catalytic roles which are critical to protein function (2). Zinc ions contribute, for instance, to stabilize the structure of a huge number of transcription factors such as zinc fingers. Enzymes often employ metal ions as cofactors in their catalytic sites (3). Metal binding proteins are implicated in heavy metal toxicity, in processes such as apoptosis (4), aging (5) and carcinogenesis (6). Identifying metal binding sites in novel proteins can significantly contribute to their functional characterization, as well as help in understanding metal-related malfunctions.
X-ray absorption spectroscopy (HT-XAS) has recently proved capable of identifying metalloproteins with high reliability (7,8). However, the specific ligands involved in binding the metal ion(s) cannot be identified by these techniques. Bioinformatics tools can significantly contribute to a detailed annotation of metal binding sites, as well as in scaling-up to proteome-wide analyses. Motif-based approaches, relying on regular expression patterns or Pfam probabilistic models, have been employed (9) for sequence-based predictions on entire proteomes. The drawback of these methods is that they cannot identify novel sites: regular expression patterns tend to be quite specific but with low coverage (many false negatives), and Pfam models are limited to known metal-binding domains. In order to overcome these limitations, a number of supervised learning techniques [e.g. (10,11,12)] have been recently developed for predicting the metal bonding state of all residues in a sequence. The task consists of discriminating between free and metal-bonded residues (or disulfide bonded for cysteines).
MetalDetector (13) predicts metal-bonding state of CYS and HIS residues, focusing on transition metals, heme and Fe/S groups as candidate heterogens. The system has been active since April 2008 and has served roughly 10 000 queries so far. It was recently (8) employed in combination with HT-XAS in order to identify putative metal binding sites in a large set of protein targets generated within the Protein Structure Initiative (http://www.structuralgenomics.org).
Identification of binding sites geometry is the main new feature of release 2.0 presented in this article. The task consists in predicting the number of ions binding the protein together to their respective sets of ligands in the sequence. Figure 1 shows an example of a protein kinase C cystein-rich domain (PDB entry 1tbn). It highlights the 3D structure of the binding sites (top) and a graph-based representation of the input sequence together to the desired output (bottom). These predictions can have a significant impact in a number of tasks, including: detailed functional annotation of experimentally unsolved proteins, e.g. characterization of active sites in enzymes, many of which employ metal ions as cofactors (3); experimental determination of new metalloproteins, as the prediction of metal binding sites can guide the preparation of samples for in vitro studies (7).
Figure 1.

Metal binding prediction subtasks. (a): given sequence; (b) candidate ligands (CYS and HIS) are assigned bonding state (boldface for metal binding); (c) metal-binding residues are grouped to form binding site configurations.
There exist several web servers for metal-binding sites prediction. DiANNA (10) predicts cysteine-bonding states only, while it is not able to reconstruct metal-binding site geometry; MetSite (14) identifies sites using sequence profile information in combination with approximate structural data coming from low-resolution (or predicted) models; FINDSITE-metal (15) predicts metal-binding sites from evolutionarily related templates detected by threading; Feature (16) identifies zinc-binding sites for proteins whose 3D structure is given. The applicability of these web servers is thus limited to structurally determined proteins, or proteins for which a reasonable 3D model can be derived. SeqCHED (17) is a recently developed server predicting metal binding geometry from protein sequence, which relies on remote homology detection to create a structural model of the target protein, over which the original CHED (18) structure-based algorithm is applied. It thus cannot predict metal binding sites for proteins having novel folds. Similar limitations hold for the up-mentioned pattern-based or domain-based approaches. MetalDetector2 is the first server capable of predicting metal binding geometry for novel folds starting from sequence information alone.
MATERIALS AND METHODS
Overview
There are two crucial aspects concerning prediction of metal binding geometry. First, the number of admissible configurations can be extremely large. For a protein chain with n CYS and HIS (candidate ligands), m ions and ki ligands for the i-th ion, the number of configurations is the multinomial coefficient 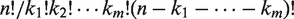 . In practice, each ion is coordinated by a variable number of ligands (typically ranging from 1 to 4, but occasionally more), and each protein chain binds a variable number of ions (typically ranging from 1 to 4). Assuming n = 12, m = 2 and ki = 4 (like in the small example shown in Figure 1), we obtain 831 600 alternative configurations. We are not considering the rare exceptions in which a CYS or HIS residue can bind multiple ions (in the December 2009 release of PDB, only 0.9% HIS and 1.6% CYS are found to be within 3 Å of two different ions). This assumption allows us to develop an efficient polynomial-time algorithm (19) for geometry prediction. To reduce the output search space and improve accuracy, we limit the maximum number of ions to 4 (covering 97% of known transition metal sites in current PDB). The second key aspect of the task is that the participation of a residue to a metal binding site should not be predicted independently from the other residues: interdependencies between candidates should be taken into account to form a collective prediction. These aspects strongly suggest solutions based on structured-output learning (20). This recent research field aims at generalizing learning algorithms, traditionally developed for classification or regression tasks, to predict outputs consisting of complex structures [like the one shown in Figure 1c].
. In practice, each ion is coordinated by a variable number of ligands (typically ranging from 1 to 4, but occasionally more), and each protein chain binds a variable number of ions (typically ranging from 1 to 4). Assuming n = 12, m = 2 and ki = 4 (like in the small example shown in Figure 1), we obtain 831 600 alternative configurations. We are not considering the rare exceptions in which a CYS or HIS residue can bind multiple ions (in the December 2009 release of PDB, only 0.9% HIS and 1.6% CYS are found to be within 3 Å of two different ions). This assumption allows us to develop an efficient polynomial-time algorithm (19) for geometry prediction. To reduce the output search space and improve accuracy, we limit the maximum number of ions to 4 (covering 97% of known transition metal sites in current PDB). The second key aspect of the task is that the participation of a residue to a metal binding site should not be predicted independently from the other residues: interdependencies between candidates should be taken into account to form a collective prediction. These aspects strongly suggest solutions based on structured-output learning (20). This recent research field aims at generalizing learning algorithms, traditionally developed for classification or regression tasks, to predict outputs consisting of complex structures [like the one shown in Figure 1c].
In MetalDetector2, identification of binding geometry is decomposed into two cascaded subtasks. The initial task consists of assigning bonding state to every CYS and HIS in two states (positive cases are metal-binding residues, negative cases are the rest, including half-cystines, i.e. cysteines forming disulfide bridges). The second task consists of grouping together metal-binding CYS and HIS, assigning them a conventional metal-ion identifier. This process is illustrated in Figure 1. Identification of the involved chemical element is not attempted.
The server uses a combination of different machine learning algorithms. The overall operation flow is shown in Figure 2.
Figure 2.

Schematic diagram of methods in MetalDetector v2.0.
Bonding state identification
This was the only functionality of MetalDetector1 (13) and the first stage of prediction in MetalDetector2. In Refs (11,13), we used a bidirectional recurrent neural network and Viterbi decoding with a simple probabilistic automaton to refine local predictions and obtain a collective assignment. In MetalDetector1, it was important to train the predictor including examples of non-metalloproteins and chains rich in disulfide bridges (since otherwise metal-binding CYS and half-cystines could be easily confused). When the input chain is not known to be a metalloprotein, we still rely on MetalDetector1 for prediction (Figure 2). On the other hand, if the input chain is known to be a metalloprotein (users can select a checkbox in the web interface to indicate this knowledge), then half-cystines are rare <3% and better accuracy can be obtained by training on metalloproteins only. In this case, half-cystines are not predicted and we solve the supervised sequence labeling task using SVM-HMM (20), a model that can be essentially interpreted as a hidden Markov model with discriminatively learned parameters, and that collectively assigns bonding state to all CYS and HIS in the sequence. The SVM-HMM sequence is the subsequence containing CYS and HIS only and observations (emissions) for each position include vectors of multiple alignment profiles among other features. Preliminary experiments showed that performance difference between MetalDetector1 and SVM-HMM is negligible under the same experimental conditions, while the latter is much simpler to train and engineer. Notably, knowing that a protein binds metal simplifies the prediction task by reducing the space of candidate outputs, resulting in better prediction accuracy on average.
Binding geometry identification
The core and novel feature in MetalDetector2 takes as input a protein chain and a (predicted) bonding state assignment and predicts binding geometry. This task is formalized as a link prediction in a bipartite graph, where a ligand node is connected to an ion node if and only if the residue coordinates that ion. In order to solve the structured-output learning problem, we introduce a function F(x, y) measuring the ‘compatibility’ between the input information x (sequence and bonding state assignment) and every admissible binding geometry y. The function is a linear combination of features of both x and y. The difficulty in this learning task is the inference step where F must be maximized with respect to y (in general, this is a hard combinatorial optimization problem). It turns out that under relatively mild assumptions, namely that every CYS or HIS coordinates at most one metal ion, there exists an optimal greedy algorithm that can identify very efficiently the binding configuration y that maximizes F—see Ref. (19) for details. Features of x and y required to construct F are defined by means of a kernel function that defines the similarity between two chains. The kernel takes into account several sources of information, including the coordination pattern of each (predicted) site and multiple alignment profiles.
THE WEB SERVER INTERFACE
Input
The input sequence can be entered either as a plain aminoacid string or in FASTA format. The web interface allows to choose between three different settings, corresponding to the three different paths in Figure 2: (i) no prior knowlegde (default operation mode); (ii) the chain is known to belong to a metalloprotein; (iii) the chain is known to belong to a metalloprotein, and the user can also provide (a guess for) the bonding state of each CYS and HIS. Note that checking in the web interface that a chain is known to bind metal is a form of positive evidence (i.e. not checking it means ignorance, not negative evidence). This knowledge can be obtained, for example, if the protein was annotated as a metalloprotein via HT-XAS (7,8).
Output
Output is either presented on a separate web page or delivered by via e-mail. It consists of a table having an entry for each CYS and HIS, with the indication of its position within the sequence, its predicted bonding state and, if the residue was predicted as metal bonded, the assigned metal ion identifier. Residues predicted to coordinate the same ion will share the same identifier. Every identifier is an integer ranging from 1 to 4 (maximum number of binding sites that can be predicted). Its value has no special biochemical semantics but lower values corresponds to a higher level of confidence for the predictor, as the greedy algorithm first builds sites where it is more confident. Figure 3 shows a web browser output for PDB entry 1t3qA.
Figure 3.

Output of the predictor for PDB entry 1t3qA.
RESULTS AND DISCUSSION
We evaluated performance according to several measures:
precision (PB) and recall (RB) of residue bonding state; precision is the ratio of true positives by the total number of residues predicted in metal-bonding state; recall or sensitivity is the ratio of true positives by the total number of metal-binding residues;
precision (PE) and recall (RE) of (ligand prediction, i.e. assignment of a residue to a metal ion. As we are not trying to predict ions of the chemical elements but to correctly group together ligands of the same ion, equivalence classes due to arbitrary reordering of ion identifiers are taken into account. In Figure 1, for instance, the correct labeling is {(3,33,36,52), (16,19,41,44)}. A prediction like {(16,19,41,52), (33,35,36)} would contain five out of seven correct assignments, while the true overall number of ligands is eight, giving PE = 5/7 and RE = 5/8. Note that the measure also accounts for residues predicted as non-metal-binding, like 3 or 44, and non-ligands predicted as metal binding, like 35. The former negatively affect recall, the latter precision.
true-positive hit rate (HT) and false-positive hit rate (HF) where a hit is counted whenever the intersection between a predicted and a true site is non-empty: HT is, therefore, the fraction of sites having at least one correctly identified ligand, and HF is the fraction of predicted sites having no correctly identified residues.
The server was tested on three distinct data sets, according to the different criteria for redundancy elimination. (All the data sets are available online at in the server website Supplementary Data). The first data set was obtained starting from the one in Ref. (11), where redundacy between sequences was removed using UniqueProt (21). the 199 metal-binding chains were collected from that data set, after removing sites containing residues different from CYS/HIS, or with a coordination number greater than four. Results in the first row of Table 1 are averages of 30 different train/test random splits, always in a ratio of 80/20. When starting from known bonding state, the predictor achieves on this data PE = RE = 90±3. We finally measured accuracy in the metalloprotein prediction task (i.e. classifying the whole sequence as metalloprotein or not), on the whole data set in Ref. (11): MetalDetector v2.0 correctly predicted as metalloproteins 65% of the ones in this data set, and as non-metalloproteins 96% of the 2362 chains having no metal-bonded CYS/HIS.
Table 1.
Evaluation of MetalDetector2
| Data set | Size | PB | RB | PE | RE | HT | HF |
|---|---|---|---|---|---|---|---|
| UniqueProt | 199 | 79 ± 4 | 88 ± 4 | 68 ± 4 | 74 ± 4 | 93 ± 4 | 10 ± 3 |
| SCOP-folds | 1824 | 62 ± 5 | 71 ± 10 | 61 ± 6 | 57 ± 7 | 70 ± 9 | 19 ± 4 |
| SCOP-superfamilies | 1466 | 60 ± 4 | 74 ± 10 | 56 ± 6 | 60 ± 10 | 74 ± 10 | 22 ± 5 |
| PDB 2010 | 549 | 60 | 75 | 50 | 62 | 77 | 20 |
The second data set was built according to the Structural Classification of Proteins (SCOP) hierarchy (22): the goal here was to test the predictor on new (i.e. not seen during the training phase) SCOP folds/superfamilies. We started from the December 2009 release of PDB, extracting 17 783 protein chains with at least a CYS or HIS bonded to a metal ion, and we retained only those chains which were mapped in SCOP 1.75 release (June 2009). After removing very few cases of chains bonded to more than five ions, we finally obtained a sequence-unique data set of 1 824 protein chains by running CD-HIT v4.0 (23) with sequence identity threshold set to 0.9 (default value).
Using this second data set, we partitioned the chains in 10 different subsets, maintaining the same average percentage of ligands in each subset, and allowing no pair of chains in different subsets to belong to the same SCOP superfamily. In a second version of this data set, we considered SCOP folds instead of superfamilies, and we therefore had to discard multi domain chains, as building the partition would have been otherwise unfeasible: this version of the data set was therefore reduced to 1466 chains. We trained 10 different models, using 9 of the subsets as the training set and the remaining subset as the test set. Results are summarized in the second and the third row in Table 1. Performance measures are averaged on the 10 splits.
The predictor available on the web server was trained on the whole SCOP-based data set. As a final test, we extracted 549 metal-bonded chains from PDB entries deposited in 2010 (after removing duplicates). Performance of the web server on this data set is reported in the fourth row of Table 1. Results in this setting are comparable to those obtained on the SCOP-based data sets.
In the Supplementary Data, we show the breakdown of prediction performance according to the number of coordinating ligands per ion. These results indicate that in the majority of cases MetalDetector2 is capable of identifying most of the binding site: in PDB 2010 data set, for example, among the 268 sites having 2 coordinating residues, MetalDetector2 correctly identifies both residues in 41.6% of the cases and one of the two 42.0% of the times. In 65 and 62% of the cases, the server misses at most one ligand in the sites with three and four coordinating residues, respectively. Concerning precision, at least half of the returned candidates actually belong to the site on average.
CONCLUSION
This release of MetalDetector adds an important feature to metalloproteins prediction, namely the ability to identify the number of binding sites and the involved CYS and HIS ligands. Unlike existing servers that can perform this task, MetalDetector does not rely on 3D structure similarity and can predict binding sites of proteins in novel folds.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: LION lab, DISI, Unitn.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bertini I, Sigel A, Sigel H. Handbook on Metalloproteins. 1st edn. New York: Marcel Dekker; 2001. [Google Scholar]
- 2.Degtyarenko K. Bioinorganic motifs: towards functional classification of metalloproteins. Bioinformatics. 2000;16:851–864. doi: 10.1093/bioinformatics/16.10.851. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002;324:105–121. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 4.Formigari A, Irato P, Santon A. Zinc, antioxidant systems and metallothionein in metal mediated-apoptosis: biochemical and cytochemical aspects. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007;146:443–459. doi: 10.1016/j.cbpc.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Mocchegiani E, Costarelli L, Giacconi R, Cipriano C, Muti E, Rink L, Malavolta M. Zinc homeostasis in aging: two elusive faces of the same “metal”. Rejuvenation Res. 2006;9:351–354. doi: 10.1089/rej.2006.9.351. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Bijur G, Jope RS. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4:137–144. doi: 10.1034/j.1399-5618.2002.40201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chance MR, Shi W. Metallomics and metalloproteomics. Cell Mol. Life Sci. 2008;65:3040–3048. doi: 10.1007/s00018-008-8189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi W, Punta M, Bohon J, Sauder JM, D'Mello R, Sullivan M, Toomey J, Abel D, Lippi M, Passerini A, et al. Characterization of metalloproteins by high-throughput x-ray absorption spectroscopy. Genome Res. 2011 doi: 10.1101/gr.115097.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertini I, Cavallaro G. Bioinformatics in bioinorganic chemistry. Metallomics. 2010;2:39–51. doi: 10.1039/b912156k. [DOI] [PubMed] [Google Scholar]
- 10.Ferrè F, Clote P. DiANNA 1.1: an extension of the DiANNA web server for ternary cysteine classification. Nucleic Acids Res. 2006;34:W182–W185. doi: 10.1093/nar/gkl189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Passerini A, Punta M, Ceroni A, Rost B, Frasconi P. Identifying cysteines and histidines in transition-metal-binding sites using support vector machines and neural networks. Proteins. 2006;65:305–316. doi: 10.1002/prot.21135. [DOI] [PubMed] [Google Scholar]
- 12.Shu N, Zhou T, Hovmoller S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics. 2008;24:775–782. doi: 10.1093/bioinformatics/btm618. [DOI] [PubMed] [Google Scholar]
- 13.Lippi M, Passerini A, Punta M, Rost B, Frasconi P. MetalDetector: a web server for predicting metal-binding sites and disulfide bridges in proteins from sequence. Bioinformatics. 2008;24:2094–2095. doi: 10.1093/bioinformatics/btn371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sodhi JS, Bryson K, McGuffin LJ, Ward JJ, Wernisch L, Jones DT. Predicting metal-binding site residues in low-resolution structural models. J. Mol. Biol. 2004;342:307–320. doi: 10.1016/j.jmb.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Brylinski M, Skolnick J. Findsite-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins. 2011;79:735–751. doi: 10.1002/prot.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebert JC, Altman RB. Robust recognition of zinc binding sites in proteins. Protein Sci. 2008;17:54–65. doi: 10.1110/ps.073138508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy R, Edelman M, Sobolev V. Prediction of 3D metal binding sites from translated gene sequences based on remote-homology templates. Proteins. 2009;76:365–374. doi: 10.1002/prot.22352. [DOI] [PubMed] [Google Scholar]
- 18.Babor M, Gerzon S, Raveh B, Sobolev V, Edelman M. Prediction of transition metal-binding sites from apo protein structures. Proteins. 2007;70:208–217. doi: 10.1002/prot.21587. [DOI] [PubMed] [Google Scholar]
- 19.Frasconi P, Passerini A. Predicting the geometry of metal binding sites from protein sequence. Adv. Neural Inform. Proces. Syst. 2009;21:465–472. [Google Scholar]
- 20.Bakir G, Hofmann T, Schölkopf B, Smola A, Taskar B, Vishwanathan SVN. Predicting Structured Data. Cambridge, MA: The MIT Press; 2007. [Google Scholar]
- 21.Mika S, Rost B. Uniqueprot: creating representative protein sequence sets. Nucleic Acids Res. 2003;31:3789–3791. doi: 10.1093/nar/gkg620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murzin AG, Brenner SE, Hubbard T, Chothia C. Scop: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]