


Abstract
The detection of functional motifs is an important step for the determination of protein functions. We present here a new web server SA-Mot (Structural Alphabet Motif) for the extraction and location of structural motifs of interest from protein loops. Contrary to other methods, SA-Mot does not focus only on functional motifs, but it extracts recurrent and conserved structural motifs involved in structural redundancy of loops. SA-Mot uses the structural word notion to extract all structural motifs from uni-dimensional sequences corresponding to loop structures. Then, SA-Mot provides a description of these structural motifs using statistics computed in the loop data set and in SCOP superfamily, sequence and structural parameters. SA-Mot results correspond to an interactive table listing all structural motifs extracted from a target structure and their associated descriptors. Using this information, the users can easily locate loop regions that are important for the protein folding and function. The SA-Mot web server is available at http://sa-mot.mti.univ-paris-diderot.fr.
INTRODUCTION
The identification of functional motifs in proteins is an effective method to infer protein functions. Different methods have been developed for extracting structural motifs (SMs) from proteins. For example MegaMotifBase (1) is a web server allowing the extraction from protein cores of SMs important for the three-dimensional (3D) structure in a given superfamily using sequence and structural feature conservation. Other methods focus on functional SMs. For example Webfeature (2) and SitePredict (3) extract SMs specific to a functional site and consist in learning the SMs of known functional sites. Other methods such as GASPS (4), for which no server is available and FunClust (5) look for conserved SMs in proteins with the same function. In contrast to the other methods, these two methods do not start from a known functional site and are able to discover new functional sites within superfamilies. Moreover, for all these methods, the extraction of SMs is based on structural alignment of proteins or the pairwise comparison of all fragments composing proteins by computing a root mean square deviation (RMSD) or distance between all fragments. Thus, using these geometrical parameters, many functional motifs are extracted from secondary structures. In spite of their large variability, loops are often involved in active or binding sites (6).
The structural alphanet HMM-SA is a collection of 27 structural prototypes of four residues called structural letters, permitting the simplification of all 3D protein structures into 1D sequences of structural letters (7). It has been shown that HMM-SA is an effective and relevant tool for the study of protein structures (8), to study protein contacts (9), or protein deformations (10,11), to search for 3D similarity across proteins (12), to predict the conformation of peptides in aqueous solutions (13,14), and to extract structural motifs from protein loops (15,16).
Here, we present a new web server, named SA-Mot (Structural Alphabet - Motif), allowing the extraction of SMs, which are important both for structure and function of protein loops. The first step, based on HMM-SA and the structural word notion, extracts SMs without pairwise comparisons of fragments (16). Then, SA-Mot provides a description of each SM (16,17) and identifies SMs of interest: recurrent and non random SMs with strong structural or amino acid sequence conservation and SMs likely to be involved in protein folding or function.
CONCEPTS AND METHODS
Protein dataset
A list of 4911 nonredundant (<50% sequence identity) protein structures listed in the SCOP classification (18) was extracted from the PDB of May 2008.
Extraction of SMs of interest
To extract SMs of interest we used a two step protocol: (i) extraction of all SMs from protein loops [for more details, see (16)], and (ii) selection of motifs of interest among all extracted SMs (16,17).
Step 1: Extraction of SMs from protein loops
This extraction is based on the notion of structural word (SA-W) derived from the structural alphabet HMM-SA (7). HMM-SA is a library of 27 structural prototypes, called structural letters, established after a geometric classification of protein fragments. Using HMM-SA, a 3D protein structure of n residues is simplified into a 1D sequence of n − 3 structural letters, in which each structural letter describes a four-residue geometry (7). Each 3D structure of our data set was encoded into structural-letter sequences, from which structural-letter sequences of loops were extracted using the HMM-SA definition (15). Each simplified loop was split into overlapping words (four consecutive structural letters, named SA-Ws), which correspond to SMs of seven residues (16). From the 90 811 loops, named loop data set, 25 304 different SA-Ws were extracted, describing the conformation of 238 158 seven-residue fragments.
Step 2: Extraction of SMs of interest
We defined different types of SA-Ws of interest using several parameters: (i) SA-W occurrence; (ii) structural and sequence conservation; and (iii) statistical overrepresentation (17)
Rare and recurrent SA-W
These two SA-W types were defined using SA-W occurrence that is the number of times a word is seen in the loop data set. SA-Ws with an occurrence <5 are qualified as rare. It has been shown that rare SA-Ws are linked to structural flexibility and regions with uncertain coordinates (16). SA-Ws observed more than 30 times are qualified as recurrent. They are found in a lot of proteins, which suggests they can be SMs with a key role in proteins.
SA-Ws presenting weak structural diversity and/or strong amino acid conservation
We can suppose that the structural and sequence conservation of these SA-Ws result from an evolutionary pressure since they are located in important protein sites. The structural variability and the amino acid conservation of a SA-W were quantified using the RMSD and Zmax parameters (16). The RMSD is the α-carbon RMSD between seven-residue fragments encoded by the same SA-W. The Zmax derives from the Zscore parameter that gives information about the conservation of each amino acid for the seven positions of a SA-W. A Zscore for amino acid a at position l in the SA-W w is defined by Equation (1) and corresponds to the comparison of its occurrence na,l,w in this position l to its expected occurrence Na,l,w
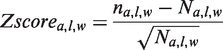 |
(1) |
| (2) |
where na,l is the occurrence of a at position l in the loop data set, nall is the total number of SA-Ws in the loop data set. A positive (resp. negative) Z-score corresponds to an overrepresentation (resp. underrepresentation) of a. Among the 140 (20 × 7) Zscores for a SA-W, we retain the maximal Zscore, named Zmax, which quantifies the amino acid conservation of the most significant position among the seven positions. Thus, the higher the Zmax of a SA-W is, the stronger the amino acid specificity of the SA-W is.
In order to have enough data for the computation of these structural and sequence parameters, they were computed only for recurrent SA-Ws.
Overrepresented SA-Ws
A statistically overrepresented SA-W has an unusual frequency in the data set (observed more than expected). As in DNA analyses methods (19), we suppose that this unusual frequency of a SA-W reflects a selective pressure on this SA-W, suggesting a functional role.
In this study, the overrepresentation was computed using the software SPatt (20) because it calculates exact statistics for sets of short sequences. The SPatt approach compares the occurrence of a SA-Ww in the data set (nw) and its expected one (Nw) computed under a background reference model (an 1-order Markov model) using the PMC (Pattern Markov Chain) notion (21). The overrepresentation score of w is named ORscore and is given by:
| (3) |
| (4) |
where P denotes the probability of the event. For more information, see (20). The ORscore threshold for statistical significance is set to 5.94, using the Bonferroni adjustment to take into account multiple tests. Thus, a SA-W with a ORscore above 5.94 is defined as significantly overrepresented.
In this study, we computed two types of overrepresentation of SA-Ws (i) in the loop data set, and (ii) in sets of proteins with similar function.
SA-W overrepresentation computed on the loop data set
It has been shown that overrepresented SA-Ws in the loop data set present particular properties such as a weak structural variability and strong amino acid specificities (16). Thus these SA-Ws seem correspond to nonrandom SMs that could be crucial for proteins.
SA-W overrepresentation computed on set of proteins with similar function
Sets of proteins sharing the same function are provided by superfamilies of the SCOP classification (18) that groups proteins according to their structure and function. The computation of SA-W overrepresentation, seen more than five times in SCOP superfamilies allowed us to distinguish two types of overrepresented SA-Ws (17). Ubiquitous SA-Ws are overrepresented in several superfamilies with different folds and functions that suggests they are important for protein structures. Functional candidate words correspond to SA-Ws highly overrepresented in one or few superfamilies with similar functions that suggests they are likely to be involved in functional sites. It has been shown that some functional candidate words correspond to SMs involved in binding or active site (17).
WEB SERVER IMPLEMENTATION
The purpose of SA-Mot web server is to allow researchers to easily identify SMs of interest in protein loops. The SA-Mot web server has been designed with a user-centered approach. Thus we tried to simplify the interface for new users that are not aware of all SA-W descriptors or statistics. To improve the user-friendliness of the server, we emphasized direct help through contextual menus or relevant links in the documentation. Results are concentrated on a single page, and are accessible by javascript hooks displaying layouts on-demand. In order to guarantee the stability of the appearance and the functionality of the server, we used standard libraries. This stability was checked on various operating systems and web browsers.
The duration of the process depends on the number and length of the chains of the target protein. For example the extraction of SA-Ws of interest in protein 105M (PDB code) containing one chain of 153 residues lasts about 13 s. The same extraction from protein 1BBR (PDB code) containing 10 chains with 11–250 residues lasts about 75 s.
SA-Mot inputs
Input data is a protein 3D structure. SA-Mot accepts as input either the PDB formatted coordinate file (22) or the PDB code of the protein target. Input PDB files can contain several chains.
SA-Mot outputs
Each chain of an uploaded protein structure is processed by SA-Mot separately and the results are presented accordingly, see Figure 1. First, SA-Mot presents the protein chain (see Figure 1A) using the different sequences corresponding to the primary sequence (AA), secondary structures (SS) and 3D structure through the structural-letter sequence (SL). These sequences allow the users to easily identify the loop regions of the studied chain.
Figure 1.

Output of SA-Mot run with protein 2RHM. (A) Information about studied chains with different sequences: amino acid (AA), structural-letter (SL) and secondary-structure (SS) sequences. (B) Counts of SA-Ws of interest extracted from the studied chain. (C) SA-W description. For each SA-W (column SW), the table provides its position and amino acid sequence in the studied chain (columns Pos and AA), its occurrence and over-representation in the loop dataset (columns Occ and OR), its structural and sequence conservation (columns RMSd and AACons) and its over-representation in SCOP superfamilies (column ORsf). By clicking on icons, users can access to a contextual window displaying parameter informations: the list of proteins (PDB ID) containing a given SA-W (D), the superimposition of all SA-W-fragments computed using the ProFit software (http://www.bioinf.org.uk/software/profit/) and represented using the Pymol software (http://www.pymol.org) (E), the logo (28) of the amino acid sequences of all SA-W-fragments (F), list of SCOP superfamilies where a SA-W is over-represented (G and H).
Then, SA-Mot provides a table containing the counts of extracted SA-Ws of interest, see Figure 1B. This table gives an overview of isolated SA-Ws of interest.
Finally, SA-Mot provides a second interactive table allowing the identification of SA-Ws of interest (Figure 1C). This interactive table contains, for each SA-W (column SW in Figure 1C) its positions and amino acid sequence (columns Pos and AA in Figure 1C). Other columns contain the values of parameters used for the identification of SMs of interest presented in ‘Concepts and Methods’ section. Thus the columns Occ and OR allow users to identify recurrent and nonrandom SA-Ws corresponding to SMs involved in the structural redundancy of loops. To illustrate the occurrence of a SA-W, users can access the list of proteins (and positions) containing the SA-W (see Figure 1D) by clicking on the related icon. The columns RMSd and AACons allow users to identify SA-Ws with a relevant structural or sequence conservation. These conservations are illustrated by figures obtained by clicking on the corresponding values. The first figure corresponds to the superimposition of all fragments encoded into the SA-W (Figure 1E) and the second corresponds to the logo of the amino acid sequences of all fragments encoded into a SA-W (Figure 1F). Finally, the column ORsf corresponding to the result of the computation of the SA-W overrepresentation in SCOP superfamilies allows the user to locate SMs which are likely to be involved in protein function (functional candidate words) or in protein structures (ubiquitous words). To help users to identify the role of these SMs, they have access to the SCOP id of each superfamily where the SA-W is overrepresented (Figure 1 H,G).
In the table, SA-Ws are first ranked according to their positions in the studied chain but can be sorted according to the different columns in order to facilitate the identification of SA-Ws of interest (Figure 1B).
USING SA-MOT
In this section, we will try to illustrate a concrete search case in which SA-Mot could be of interest. Our target is an uncharacterized protein: its structure is known and referenced in the PDB (2RHM) but its function has not been clearly established (putative kinase from Chloroflexus aurantiacus). Using DALI database (23), the structural closest protein (first hit) is the chain A of L-seryl-tRNA(sec) kinase of Methanocaldococcus jannaschii, (PDB code 3A4L). This hit presents a RMSD of 2.8 Å and a sequence identity of 25% with chain B of the protein target (see Supplementary Data), suggesting a kinase activity of protein target. This kinase activity is also suggested by CD-search web server (24,25) that identifies different conserved domains in the region 1–130, see Supplementary Data.
SA-Mot server is then used to locate motifs of interest in protein loops of the protein target. A part of the results for chain B is presented in Figure 1. First, chain B contains rare SA-Ws at positions 130–141 suggesting it may be a flexible region. We can observe that this region is located at the end of the conserved domain ‘Chloramphenicol phosphotransferase-like protein (CPT)’ detected by CD-search. Then, we located 19 SA-Ws that are recurrent and overrepresented in the loop data set and presenting a weak structural variability and some amino acid specificities. This suggests they correspond to SMs involved in crucial regions in the protein target. One of these motifs of interest, ZCDS, located at positions 25–31, corresponds to an ubiquitous word clearly associated with beta-turns (17). Chain B of the protein target contains also four functional candidate words: YUOD, UODO, CDSK, OZGB. For two SA-Ws, CDSK and OZGB, no available annotation was found to confirm their functionality that should be tested by experimental methods.
OZGB is located at positions 165–171 and is overrepresented in the ‘Trypsin-like serine proteases’ superfamily (SCOP id = 50494), suggesting that this SM is important for these proteases. It is surprising that a putative kinase shares a structural motif specific to trypsin-like serine proteases. However, this motif is not located in conserved kinase domains extracted from CD-search and it corresponds to a variable region in DALI alignment (see Supplementary Data). Moreover, using SA-Mot to analyze the closest (kinase) proteins provided by DALI, we found that these kinases do not contain OZGB. These results suggest this SA-W may identify a SM important for the C-terminal region of the target protein but not be involved in kinase activity.
CDSK is located at positions 26–32 and is overrepresented in the superfamily ‘YVTN repeat-like/Quinoprotein amine dehydrogenase’ (SCOP id = 50969). This suggests this SA-W is important for these dehydrogenase proteins. It is located in kinase conserved domains detected by CD-search and in a bit conserved region in the DALI alignment. Using DALI output, we observe that SA-W CDSK is found in uridylmonophosphate/cytidylmonophosphate kinases and adenylate kinases (for example PDB codes: 1QF9_A, 3UKD_A and 2C95_B), but not in L-Seryl-tRNA(sec) kinases (PDB code: 3A4L_A the first hit in DALI), putative gluconate kinases (PDB code: 2BDT_A the second hit in DALI), 6-Phosphofructo-2 kinases (PDB code: 1K6M_A). This suggests that this SA-W is important for a putative function common to some protein kinases (uridylmonophosphate/cytidylmonophosphate kinases and adenylate kinases).
Overlapping SA-Ws YUOD and UODO, located at positions 11–18, are strongly overrepresented in the superfamily ‘P-loop containing nucleotide triphosphate hydrolases’ (SCOPid = 52540). These SA-Ws have been identified as SMs with residues involved in nucleotide-binding sites (17). Moreover, the functionality of these two SA-Ws are confirmed by the fact they are located in the conserved domains with kinase activity detected by CD-seach and in a highly conserved region on DALI alignment (see Supplementary Data). Thus, SA-Mot detects an ATP/GTP-binding site.
The extraction of the four SA-Ws of interest in DALI hits suggests the protein target is closer to uridylmonophosphate/cytidylmonophosphate kinases or adenylate kinases, proteins containing CDSK word like the target, rather than a L-Seryl-tRNA(sec) kinases, as suggested by DALI results, see Supplementary Data. Thus, SA-Mot could be used in complementarity with DALI to order of DALI hits.
These four motifs of interest extracted by SA-Mot were not detected by other methods dedicated to the extraction of functional sites. For example the extraction of functional sites from the protein target is impossible using the following web servers: MegaMotifBase (1), Webfeature (2), FunClust (5), because the protein target is not in their current databases. SitePredict (3) predicts a calcium-binding site in chain B at position 137 and a copper, iron, manganese, zinc-binding sites at position 132, but with a weak prediction probability (i.e. <60%). PROSITE (26) leads to the identification of no functional pattern. The IBIS web server (27) annotates the protein target as putative kinase and extracts a binding site to a benzoic acid. In Swiss-Prot, the protein target (corresponding to the A9WDG5 id) does not contain any functional sites despite its annotation by the Gene Ontology term ‘ATP binding’ (molecular function). We can observe that for this uncharacterized protein, there is no consensus between methods that extract functional motifs.
In addition to the knowledge of the putative kinase activity suggested by IBIS, Swiss-Prot, CD-search and DALI methods, SA-Mot results allow the location of ATP-binding site, that is the functional site needed for this activity. SA-Mot also locates three other SMs of putative interest for target structure and function.
CONCLUSION
SA-Mot is a new web server for the identification of SMs of interest extracted from protein loop structures. It is based on two steps: first the extraction of all SMs from loops of protein structures, and secondly the description of all extracted SMs. This description is based on statistics of SMs computed either on the loop data set or on SCOP superfamilies, on geometric and sequence parameters and is summed up in an interactive table.
SA-Mot allows the users to easily identify SMs of interest such as those involved in the structural and sequence redundancy and those with a putative role in protein structure or function. Thus, in contrast to classical methods, SA-Mot does not focus only on the detection of a binding site associated with a ligand, but explores all crucial SMs for proteins such as motifs of interest for protein folding or structural motifs involved in active site or REPEAT regions (17). Another interest of SA-Mot is that the learning of motifs of interest is not based on the knowledge of functional sites. Thus, SA-Mot is able to propose novel SMs putatively important for the protein function.
In the current SA-Mot release, the overrepresentation of SA-Ws is computed in each SCOP superfamily. As our method is effective for any protein classification, it may be extended to CATH superfamilies.
Moreover, SA-Mot only runs on proteins for which the 3D structure has been resolved but it could be extended to circumstances where only the sequence is known. For this last case, we are currently developing a method to predict the SA-Ws of interest directly from amino acid sequences. Then it will be possible to integrate this new method into the SA-Mot server to detect SMs of interest from protein loops using either the protein structure or the protein sequence.
Currently the only piece of information about the nature of the potential functional role of some SMs extracted by SA-Mot is provided by the id of the SCOP superfamily where a SM is overrepresented. Thus, the next step of this work is the identification of the role of the functional candidate words.
It is clear that methods dedicated to functional motifs extraction are complementary between both them and methods allowing protein global structural comparisons such as DALI. Thus, the next release of SA-Mot will be integrated in a meta-server including both local and global approaches in a high-throughput functional annotation pipeline.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: INSERM UMR-S 973 recurrent funding, University Paris Diderot.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Dr Christelle Reynès and Dr Jennifer Becq for their critical reading of the manuscript, Dr Juliette Martin for her useful comments and suggestions for the improving the web site. The authors acknowledge preliminary work done by Sébastien Lementec. The authors thank Dr Gilles Labesse for his discussions about protein kinases, Dr Henri Xhaard for his discussions about serine proteases and Dr Lukasz Jaroszewski and Mark Knuth for their discussions about the protein corresponding to the PDB code 2RHM.
REFERENCES
- 1.Pugalenthi G, Suganthan PN, Sowdhamini R, Chakrabarti S. MegaMotifBase: a database of structural motifs in protein families and superfamilies. Nucl Acids Res. 2008;36:D218–D221. doi: 10.1093/nar/gkm794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halperin I, Glazer D, Wu S, Altman R. The FEATURE framework for protein function annotation: modeling new functions, improving performance, and extending to novel applications. BMC Genomics. 2008;9:S2. doi: 10.1186/1471-2164-9-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bordner A. Predicting small ligand binding sites in proteins using backbone structure. Bioinformatics. 2008;24:2865–2871. doi: 10.1093/bioinformatics/btn543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polacco BJ, Babbitt PC. Automated discovery of 3D motifs for protein function annotation. Bioinformatics. 2006;22:723–730. doi: 10.1093/bioinformatics/btk038. [DOI] [PubMed] [Google Scholar]
- 5.Ausiello G, Gherardini P, Marcatili P, Tramontano A, Via A, Helmer-Citterich M. FunClust: a web server for the identification of structural motifs in a set of non-homologous protein structures. BMC Bioinformatics. 2008;9:S2. doi: 10.1186/1471-2105-9-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Via A, Ferre F, Brannetti B, Valencia A, Helmer-Citterich M. Three-dimensional view of the surface motif associated with the P-loop structure: cis and trans cases of convergent evolution. J. Mol. Biol. 2000;303:455–465. doi: 10.1006/jmbi.2000.4151. [DOI] [PubMed] [Google Scholar]
- 7.Camproux AC, Gautier R, Tufféry P. A hidden Markov model derivated structural alphabet for proteins. J. Mol. Biol. 2004;339:561–605. doi: 10.1016/j.jmb.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Regad L, Guyon F, Maupetit J, Tufféry P, Camproux AC. A hidden Markov model applied to the protein 3D structure analysis. CSDA. 2008;52:3198–3207. [Google Scholar]
- 9.Martin J, Regad L, Etchebest C, Camproux AC. Taking advantage of local structure descriptors to analyze interresidue contacts in protein structures and protein complexes. Proteins. 2008;73:672–689. doi: 10.1002/prot.22091. [DOI] [PubMed] [Google Scholar]
- 10.Martin J, Regad L, Lecornet H, Camproux AC. Structural deformation upon protein-protein interaction: a structural alphabet approach. BMC Struct. Biol. 2008;8:12. doi: 10.1186/1472-6807-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baussand J, Camproux AC. Deciphering the shape and deformation of secondary structures through local conformation analysis. BMC Struct. Biol. 2011;11:9. doi: 10.1186/1472-6807-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyon F, Camproux AC, Hochez J, Tufféry P. SA-Search: a web tool for protein structure mining based on a structural alphabet. Nucleic Acids Res. 2004;32:W545–W548. doi: 10.1093/nar/gkh467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maupetit J, Derreumaux P, Tufféry P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maupetit J, Derreumaux P, Tufféry P. A fast method for large-scale de novo peptide and miniprotein structure prediction. J. Comput. Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- 15.Regad L, Martin J, Camproux AC. In Proceedings of IEEE Symposium on Computational Intelligence in Bioinformatics and Computational. Toronto: 2006. Identification of non random motifs in loops using a structural alphabet; pp. 92–100. September. [Google Scholar]
- 16.Regad L, Martin J, Nuel G, Camproux AC. Mining protein loops using a structural alphabet and statistical exceptionality. BMC Bioinformatics. 2010;11:75. doi: 10.1186/1471-2105-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regad L, Martin J, Camproux AC. Dissecting protein loops with a statistical scalpel suggests a functional implication of some structural motifs. BMC Bioinfo. 2011 doi: 10.1186/1471-2105-12-247. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 19.Karlin S, Burge C, Campbell AM. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 1992;20:1363–1370. doi: 10.1093/nar/20.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuel G, Regad L, Martin J, Camproux AC. Exact distribution of pattern in a set of random sequences generated by a Markov source: application to biological data. Algo. Mol. Biol. 2010;5:15. doi: 10.1186/1748-7188-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuel G. Numerical solutions for patterns statistics on Markov chains. Stat. Appl. Genet. Mol. Biol. 2006;5:26. doi: 10.2202/1544-6115.1219. [DOI] [PubMed] [Google Scholar]
- 22.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 24.Marchler-Bauer A, Bryant S. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchler-Bauer A, Lu S, Anderson J, Chitsaz F, Derbyshire M, DeWeese-Scott C, Fong J, Geer L, Geer R, Gonzales N, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche B, De Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJA. The 20 years of PROSITE. Nucleic Acids Res. 2007;36:245–249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoemaker B, Zhang D, Thangudu R, Tyagi M, Fong J, Marchler-Bauer A, Bryant S, Madej T, Panchenko A. Inferred biomolecular interaction server–a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 2010;38:D518–D524. doi: 10.1093/nar/gkp842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]