

Abstract
Fibronectin-binding protein A (FnBPA) and FnBPB are important adhesins for Staphylococcus aureus infection. We constructed fnbA and/or fnbB mutant strains from S. aureus SH1000, which possesses intact rsbU, and studied the role of these adhesins in in vitro and in vivo infections. In intravenous infection, all fnb mutants caused a remarkable reduction in the colonization rate in kidneys and the mortality rate of mice. fnbB mutant caused a more severe decrease in body weight than that caused by fnbA mutant. Serum levels of interleukin-6 and nuclear factor κB (NF-κB) activation in spleen cells were remarkably reduced in fnbA or fnbA fnbB mutant infections; however, there was no significant reduction in fnbB mutant infections. In in vitro cellular infection, FnBPA was shown to be indispensable for adhesion to and internalization by nonprofessional phagocytic cells upon ingestion by inflammatory macrophages and NF-κB activation. However, both FnBPs were required for efficient cellular responses. The results showed that FnBPA is more important for in vitro and in vivo infections; however, cooperation between FnBPA and FnBPB is indispensable for the induction of severe infection resulting in septic death.
INTRODUCTION
Staphylococcus aureus is a commensal bacterium in humans and a major cause of community- and hospital-acquired infectious diseases (3). This pathogen causes a variety of diseases in humans, ranging from superficial infections of the skin to severe systemic infections such as endocarditis, osteomyelitis, pneumonia, and sepsis.
The nasal cavities of 30% of the human population are consistently colonized by S. aureus (59). Nasal colonization by S. aureus is a cause of sepsis because most S. aureus isolates from patient blood with S. aureus sepsis are identical to those isolated from nasal cavities (14, 55). Furthermore, intracellular S. aureus was detected in the nasal epithelium of patients with recurrent rhinosinusitis. This shows that intracellularly invasive S. aureus may also be a source of chronic infections and sepsis. Of late, methicillin-resistant S. aureus (MRSA) has spread not only in hospitals but also in the community (31). This increase in MRSA infections is alarming.
S. aureus possesses a large number of virulence factors, including toxins and enzymes. Furthermore, S. aureus expresses a repertoire of cell wall-anchored proteins containing the LPXTG sequence (36). Some of these proteins interact with host serum or extracellular matrix proteins, so they are thought to be involved in the colonization and invasion of host tissues (17). Of these proteins, two fibronectin (Fn)-binding protein (FnBP) homologues, FnBPA and FnBPB, have been demonstrated to be involved in not only adhesion to cells but also internalization by cells (14, 17). Binding of S. aureus to α5β1-integrin on cells via Fn induces the formation of an adherent apparatus-like structure beneath the cell membrane so that the activation signal is transduced (1, 13, 17, 49). FnBPs have also been demonstrated to be important in in vivo infection by S. aureus (26, 44). However, many studies have been carried out with fnbA alone (44, 46) or performed with mutants constructed from S. aureus strain 8325-4, which contains a small deletion in rsbU, a positive regulator of σB (22, 27, 51), resulting in the extraordinarily large expression of exoproteins due to high activation of agr. Subsequently, clfA and fnbA are positively regulated either directly or indirectly by σB (4, 7, 11, 24, 30, 38, 60). For years, the NCTC8325 strain and its phage-cured derivative 8325-4 have been widely used as model strains for basic research on the genetics and virulence of S. aureus. In addition to the high level of characterization, this strain lineage has seemed representative of the S. aureus species for studying the mechanism of infection because NCTC8325 was clinically isolated from a sepsis patient (35). However, the 8325 lineage contains mutational defects in two regulatory genes, rsbU and tcrA. It has been demonstrated that tcrA has little effect on the physiology of bacteria such as extracellular protein production, production of staphyloxanthin, hemolytic activity, and biofilm formation, whereas rsbU profoundly alters these properties (18, 19). In in vitro infection, restoration of rsbU induces high internalization and intracellular growth in human umbilical vein endothelial cells and macrophages (38). In vivo, intravenous infection with SH1000, a strain of 8325-4 in which rsbU has been repaired, resulted in more significant severe arthritis and sepsis than that with the parental strain (24). These studies strongly demonstrate that pathogenicity can be remarkably altered by rsbU dysfunction.
In this study, using fnbA and/or fnbB mutants constructed from SH1000, we investigated the roles of FnBPA and FnBPB in a mouse sepsis model and infection of L929 nonprofessional phagocytes and inflammatory macrophages.
MATERIALS AND METHODS
Plasmids and bacteria.
The plasmids and bacteria used in this study are listed in Table 1. fnbA mutant strain JS1078 was constructed as follows. A 3,075-bp fragment containing the fnbA gene from SH1000 was amplified by PCR with ExTaq polymerase (Takara Bio Inc., Tokyo, Japan) and primers fnbA-F3 (5′-AAACAATCTTCGGTACGGCA-3′) and fnbA-R3 (5′-TAAACCCGTCAATTTTTGTT-3′). The fragment was inserted into the SmaI site of plasmid PKF3 (Takara Bio Inc.), and the resulting plasmid was designated pHS1. The DNA fragment lacking the 410-bp central part of the fnbA gene was amplified from pHS1 using primers fnbA-XF (5′-AGCGAACGGAAATGAGAAAA-3′) and fnbA-XR (5′-AAATGCAACATGCGAAAATC-3′). The fragment containing the 410-bp deletion was ligated to a 1,701-bp Tcr gene fragment that was amplified from plasmid pHY300 (Takara Bio Inc.) using primers Tet-F1 (5′-CCCCGGGSAATTCCTGTTAT-3′) and Tet-R1 (5′-CTTGTTGGTTTTATGCGTGC-3′) to create pHS2. The DNA fragment containing the fnbA::Tcr gene was cut out of the plasmid with HindIII and SacI and then ligated into plasmid p159, which had been digested with the same enzymes to create pHS3. pHS3 was electroporated into restriction-negative S. aureus 1039 (61). Because plasmid pHS3 is not able to replicate at high temperatures, allele replacement was achieved after growth at 42°C for 42 h without antibiotics. The cells were spread onto LB agar plates containing 5 μg/ml of tetracycline and incubated at 42°C. PCR analysis was used to verify the fnbA::Tcr allele on the chromosome. Subsequently, fnbA::Tcr was transduced into SH1000 using phage 80α.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant marker(s) | Property(ies) | Source or reference |
|---|---|---|---|
| S. aureus strains | |||
| 8325-4 | NCTC8325 cured of prophages | 35 | |
| RN4220 | Restriction-negative derivative of 8325-4 | 28 | |
| 1039 | Restriction-negative strain | 61 | |
| SH1000 | Functional rsbU derivative of 8325-4 | 19 | |
| DU5882 | fnbB::Emr | Mutant of 8325-4 defective in expression of FnBPB | 15 |
| JS1078 | fnbA::Tcr | Mutant of SH1000 defective in expression of FnBPA | This study |
| JS2072 | fnbB::Emr | Mutant of SH1000 defective in expression of FnBPB | This study |
| JS3094 | fnbA::Tcr, fnbB::Emr | Mutant of SH1000 defective in expression of FnBPA and FnBPB | This study |
| Plasmids | |||
| pSK265 | CmrS. aureus replicon | 23 | |
| pFNBA4 | Shuttle plasmid derived from pGEM-7Zf(Apr) and pSK265(Cmr) carrying full-length fnbA gene | 15 | |
| pFNBB4 | Shuttle plasmid derived from pGEM-7Zf(Apr) and pSK265(Cmr) carrying full-length fnbB gene | 15 | |
| p159 | Shuttle plasmid derived from pUC119 and pLE194Ts | Gift from K. Hiramatsu | |
| pHS1 | 3,095-bp insert of fnbA in pKF3 | This study | |
| pHS2 | fnbA::Tcr | 3,095-bp insert of fnbA lacking 410 bp of central part and 1,071-bp insert of Tcr gene | This study |
| pHS3 | fnbA::Tcr, Ts | Insert of 4,366-bp fnbA::Tcr fragment from pHS2 in p159 | This study |
fnbB::Emr was transduced from DU5882 (a generous gift from T. Foster) into SH1000 or JS1078 using phage 80α to create JS2072 and JS3094, respectively. Plasmids pFNBA4 and pFNBB4 (a generous gift from T. Foster) were electroporated into JS1078 and JS2072 to create the corresponding complemented strains.
Bacteria were grown overnight in brain heart infusion medium at 37°C with shaking to steady state, and then 1/20 volume of each culture was inoculated into heart infusion and cultivated at 37°C with shaking for 1.5 h into the early exponential growth phase. The bacteria were washed three times with phosphate-buffered saline (PBS) and used in in vivo and in vitro experiments.
Analysis of transcription levels of fnbA and fnbB.
RNA samples were prepared from SH1000 and its fnb mutants in the early exponential growth phase by using TRI-REAGENT (Molecular Research Center, Inc., Cincinnati, OH). For analysis of fnbA or fnbB transcription levels, cDNA was prepared from the isolated RNA with random primers and SuperScript II reverse transcriptase (TaKaRa Bio Inc.) according to the instructions of the manufacturer. cDNA (50 ng) was used as the template for PCR with ExTaq polymerase (TaKaRa Bio Inc.) for detection of fnbA and/or fnbB. Primers A-F1 (5′-GACAAAGAAGCTGCAGCATC-3′) and A-R3 (5′-TAAACCCGTCAATTTTTGTT-3′) were used to amplify a 3.0-kbp fragment of fnbA that did not contain the signal sequence. Primers A-F1 and B-R3 (5′-GAACGCCTTCATAGTGTCATT-3′) were used to amplify a 2.8-kbp fragment of fnbB that did not contain the signal sequence.
West-Western blot analysis of Fn-binding components in cell wall fraction.
For detection of expressed FnBPs, bacteria in the early exponential growth or stationary phase were lysed with 40 μg/ml lysostaphin in the presence of 0.1 M sucrose and 10 μg/ml DNase I for 30 min at 37°C with gentle shaking. After centrifugation at 14,000 × g for 10 min, the supernatant was collected and used as cell wall fractions. SDS and 2-mercaptoethanol were added to the supernatant to final concentrations of 0.1% and 0.1 M, respectively, and the supernatant was boiled. Samples were separated by SDS-PAGE and transferred to an Immobilon-P membrane (Millipore Corp., Bedford, MA), where a volume of supernatant corresponding to 5 × 108 CFU of bacteria was loaded in each lane. The membrane was blocked and treated with purified bovine serum Fn (50), which was biotinylated with ImmunoPure Sulfo-NHS-Biotin (Thermo Fisher Scientific Inc., Rockford, IL). The membrane was washed with PBS containing 0.1% Tween 20 and treated with horseradish peroxidase (HRP)-conjugated streptavidin (Sigma-Aldrich Co.). HRP signals were detected with ECL Western blotting detection reagents (GE Healthcare).
Mouse sepsis model.
The Animal Care Committee of the Jikei University School of Medicine granted permission for experimentation with live animals in the present work. Female BALB/c mice (5 weeks old) were raised for a week in a constant atmosphere in our animal laboratory. Mice (10 to 12 per group) were then infected intravenously with 0.1 ml of saline containing 5 × 107CFU of S. aureus. Survival and body weight were monitored for more than 2 weeks. In another experiment, kidneys were obtained at 30 min or 24 h after infection and mashed into homogeneous suspensions in nutrient broth (NB). After appropriate dilutions with NB, 0.1 ml of each suspension was plated onto an NB agar plate and then cultivated overnight at 37°C before the colonies were counted. Simultaneously, mouse sera were obtained; interleukin-6 (IL-6) was measured by enzyme-linked immunosorbent assay (Pierce Biotechnology Inc., Rockford, IL). For histological examination, kidneys of infected mice were fixed with 4% paraformaldehyde and embedded in paraffin. Organ sections 4 μm thick were then stained with hematoxylin and eosin (H&E).
In vitro infection assays.
Peritoneal macrophages were obtained from female BALB/c mice (5 weeks of age; Charles River Laboratories Japan Inc.) as previously described (48). L929 fibroblasts were maintained in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS).
In the infection assay, cells were cultured in 8-well slide chambers (Nalgene Nunc International) without FBS (105 cells/well). Fn-treated or untreated bacteria were added to the macrophage cultures at a multiplicity of infection (MOI) of 50. After 30 min of incubation, the cultures were washed with RPMI and treated with 20 μg/ml lysostaphin and antibiotics (100 units/ml penicillin G and 100 ng/ml streptomycin) for 30 min at 37°C. For the experiment with fibroblasts, cells were cocultured with Fn-untreated bacteria at an MOI of 50 for 60 min and then treated with lysostaphin and antibiotic as described above. Cells were then fixed with methanol and stained with Giemsa's solution (Merck KGaA, Darmstadt, Germany). The stained cells were observed with a Nikon Optiphoto microscope (Nikon Co. Ltd., Tokyo, Japan).
Nuclear factor κB (NF-κB) activation.
L929 cells or mouse inflammatory macrophages were cultured in 35-mm petri dishes (Nalgene Nunc International) at a density of 2 × 105/cm2 without FBS. Bacteria were added to the cultures (MOI = 50) and left standing for 1 h at 37°C. After washing, cells were cultured for 1 h with 20 μg/ml lysostaphin and antibiotics. Spleens were obtained from S. aureus-infected mice, and spleen cells were harvested in RPMI medium containing 100 μg/ml penicillin and 200 U/ml streptomycin. After passage through a nylon mesh strainer, cells were washed with ice-cold PBS. Nuclear extracts from these cells were prepared with NE-PER Nuclear and Cytoplasmic Extraction Reagent (Pierce Biotechnology). 3′-biotinylated double-stranded DNA (dsDNA) oligonucleotides with a consensus binding sequence of NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) were synthesized by Sigma Aldrich Japan (Hokkaido, Japan). In each reaction mixture, 4 μg of nuclear protein and 4 pmol/ml of biotinylated probe were incubated with 5 mM MgCl2, 2.5 mM EDTA, 5 mM dithiothreitol, 50 mM Tris-HCl (pH 7.5), 50 μg/ml poly(dI-dC), and 5% glycerol at room temperature for 30 min. DNA-protein complexes were separated from unbound probes on 4% polyacrylamide gel and transferred onto Hybond-N+ membrane (Amersham Bioscience Corp., Piscataway, NJ); this was followed by cross-linking by UV irradiation. Biotin signals on the membrane were detected with a Chemiluminescent Nucleic Acid Detection Module (Pierce Biotechnology). To confirm that the observed signal was specific, a 200-fold molar excess of unlabeled dsDNA oligonucleotide was added to the reaction mixture as a competitor.
Reverse transcription (RT)-PCR of tumor necrosis factor alpha (TNF-α) and IL-6.
Total cellular RNA was isolated from mouse inflammatory macrophages using TRI-REAGENT (Molecular Research Center, Inc.). Total RNA (100 ng) was reverse transcribed and amplified with gene-specific primers using RT-PCR high-Plus- (TOYOBO). The primer sequences for the genes were 5′-TGAACTTCGGGGTGATCGGTC-3′ (forward) and 5′-AGCCTTGTCCCTTGAAGAGAAC-3′ (reverse) for TNF-α, 5′-GAAAATCTGCTCTGGTCTTCTGG-3′ (forward) and 5′-TTTTCTGACCACAGTGAGGAATG-3′ (reverse) for IL-6, and 5′-ATGTCAGATCCACAACGGATACAT-3′ (forward) and 5′-ACTCCCTCAAGATTGTCAGCAAT-3′ (reverse) for glyceraldehyde 3-phosphate dehydrogenase.
Statistical analysis.
Statistical significance was determined with a log-rank test for mortality rates and a Student t test for differences in body weight and for differences in the number of internalized bacteria in the cells.
RESULTS
Expression of FnBPA and FnBPB in SH1000 and its fnb mutants.
Expression of Fn-binding factors in cell wall fractions from wild-type strain SH1000 was investigated by West-Western blot analysis. In the early exponential growth phase, two specific Fn-binding components were detected whose molecular masses were determined to be approximately 200 kDa by SDS-PAGE, while in the stationary phase, these components were not detected (Fig. 1A). Considering their molecular masses on SDS-PAGE and the expression phases observed, the upper and lower signals were determined to be FnBPA and FnBPB, respectively (32). These two signals were either equivalent in strength or the lower FnBPB signal appeared to be slightly stronger than the upper FnBPA signal (Fig. 1A and B). A previous study has shown that FnBPA possesses more Fn-binding segments than FnBPB (6); Fig. 1 shows that SH1000 expresses FnBPA and FnBPB at comparable levels or the expression of FnBPB is slightly higher than that of FnBPA. Furthermore, transcription of fnbA and fnbB was studied by RT-PCR and equivalent expression was observed in the early exponential growth phase (Fig. 1C). This confirms that equivalent levels of both FnBPs are expressed in SH1000. Only the lower or upper signal was detected in JS1078 (fnbA mutant) or JS2072 (fnbB mutant), while none of the Fn binding signal was observed in JS3094 (Fig. 1B).
Fig. 1.

Expression of FnBPA and FnBPB on SH1000 and its fnb mutants. (A and B) Fn-binding factors in the cell wall preparations from 3 × 107 bacteria were subjected to West-Western blot analysis by using 100 μg/ml of purified bovine serum Fn. (A) Fn-binding factors in SH1000 in the early exponential growth phase and those in the stationary phase. (B) Fn-binding factors in SH1000 and its mutant strains in the early exponential growth phase. Arrows at the left side of each panel show the Fn binding signals. (C) mRNA expression of fnbA and fnbB in SH1000 and its mutant strains in the early exponential growth phase. RNA samples were prepared from SH1000 and its fnb mutants in the early exponential growth phase. cDNA was prepared from the isolated RNA with random primers and reverse transcriptase, and cDNA (50 ng) was used as a template for PCR for detection of fnbA and/or fnbB. Primers A-F1 and A-R3 were used to amplify a 3.0-kbp fragment of fnbA. Primers A-F1 and B-R3 were used to amplify a 2.8-kbp fragment of fnbB.
Role of FnBPs in death and weight change of mice during septic infection by S. aureus.
We used a mouse sepsis model to compare SH1000 and its fnb mutant strains JS1078, JS2072, and JS3094 for virulence. Mice were intravenously administered 5 × 107 CFU, and their survival (Fig. 2A) and changes in body weight (Fig. 2B) were observed for more than 2 weeks. In this experiment, all of the mice infected with SH1000 died within 6 days whereas no fnb mutants were so severely lethal, especially in infections with JS1078 and JS3094. Weight was checked daily, and mice infected with SH1000 displayed significantly more weight loss than the other 3 groups of mice; weight loss in SH1000-infected mice started at day 1 and continued until the mice died. The initial loss of body weight was also seen in mice infected with JS1078 or JS2072. In JS1078 infections, weight loss was seen only on the first day. In contrast, it continued for 5 days in JS2072 infections. In JS3094 infections, no weight loss was recorded at least for the first 5 days. We also observed the appearance of the mice following S. aureus infection. The mice infected with JS1078, as well as JS3094, had lustrous fur and remained active; in contrast, mice infected with JS2072 developed unhealthy coats and a remarkable decrease in movement. These results show that the contribution of FnBPA to septic symptoms is greater than that of FnBPB, although both FnBPA and FnBPB are required for septic death.
Fig. 2.

Effects of FnBPA and FnBPB on the mortality rate of BALB/c mice after intravenous infection with SH1000 or its fnb mutants. Female BALB/c mice were infected with 5 × 107 CFU of SH1000 or its mutant strains, and their survival (A) and weight changes (B) were monitored. Closed circles, SH1000; open circles, JS3094; closed triangles, JS1078; open triangles, JS2072. Statistical significance of mortality rate differences: P < 0.001 for SH1000 versus JS1078, SH1000 versus JS2072, and SH1000 versus JS3094. Statistical significance of body weight differences: asterisk, P < 0.01 for SH1000 versus each mutant; cross, P < 0.01 for JS3094 versus JS1078 or JS2072. P < 0.01 for JS1078 versus JS2072 from day 2 to day 8.
Colonization and proliferation of the wild-type strain and its fnb mutants in kidneys.
We observed bacterial colonization and multiplication in the kidneys at 30 min and 24 h after infection, respectively (Table 2). The number of SH1000 bacteria that colonized a kidney at 30 min was significantly greater than the number of mutant bacteria that did so. The number of bacteria at 24 h increased remarkably only in SH1000 infection (approximately 18-fold). This result shows that both FnBPA and FnBPB are required for efficient colonization and multiplication of S. aureus in kidneys.
Table 2.
Numbers of bacteria that colonized mouse kidneysa
| Strain | No. of CFU/kidney (104) at: |
|
|---|---|---|
| 30 min | 24 h | |
| SH1000 | 8.1 ± 3.5 (a) | 102 ± 66 (e) |
| JS1078 (A−) | 2.4 ± 1.0 (b) | 0.3 ± 0.2 (f) |
| JS2072 (B−) | 2.9 ± 1.1 (c) | 5.7 ± 7.5 (g) |
| JS3094 (A−/B−) | 1.2 ± 0.3 (d) | 0.4 ± 0.2 (h) |
BALB/c mice were intravenously infected with SH1000 or fnb mutant strains (5 × 107 CFU/head). After 30 min or 24 h, kidneys were removed and the number of bacteria in each kidney was analyzed. P < 0.05 for a versus d and c versus d, P < 0.01 for a versus c and e versus g, P < 0.005 for a versus b, and P < 0.0001 for e versus h.
To prove the pathological consequence of infection, another septic infection experiment was performed. Similar to the result shown in Fig. 2, only 30% of the SH1000-infected mice survived for 7 days, whereas all of the mice survived following JS1078 infection (Fig. 3A). At day 7, the kidneys of mice infected with SH1000 or its fnb mutants were removed, and inspection of kidneys and microscopic observation of H&E-stained thin sections were done (Fig. 3B). Infection with the wild-type strain generated multiple raised lesions in the renal cortex. H&E staining showed that the lesions contained bacterial foci surrounded by a large number of infiltrated cells. Such abscess formation was not observed in kidneys from mice infected with JS1078. This shows that S. aureus expressing only the FnBPB homologue exhibited attenuated virulence with a defect in the ability to multiply in the kidneys.
Fig. 3.

Abscess formation in kidneys of mice infected with the wild-type strain or the fnbA mutant strain. (A) Mice were monitored for death for 7 days upon intravenous infection with the wild-type strain or JS1078. (B) Kidneys from mice in panel A were obtained at 1 week after infection. The outside and inside appearance of kidneys is shown at the top. The white arrow at the right shows a piece of ureter and not an abscess. Tissue sections stained with H&E are shown at the bottom. Clumps of bacteria are indicated by black arrows.
NF-κB activation in the spleen.
NF-κB is an important transcription factor that regulates inflammatory and antimicrobial responses, including the expression of many inflammatory and immune factors (5, 43). To prove the contribution of FnBPs to the activation of this transcription factor in S. aureus infection, translocation of NF-κB into the nuclei in spleen cells was studied. Spleen tissue was used because it is a major secondary lymphoid tissue responsible for systemic inflammatory and immune responses. As shown in Fig. 4, NF-κB was obviously translocated into the nucleus in infection with the wild-type strain. However, translocation of NF-κB was scarcely detected in JS1078 infection. This shows that inflammatory and immune responses declined remarkably in FnBP mutant infection.
Fig. 4.
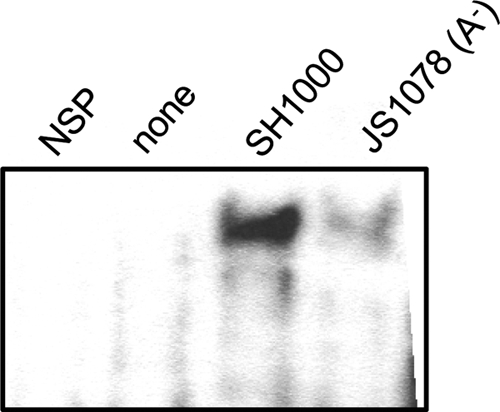
Activation of NF-κB in spleens from mice infected with the wild-type strain or the fnbA mutant strain. BALB/c mice were infected with SH1000 or JS1078. At 5 h after infection, spleens were removed and the spleen cells were harvested. Activation of NF-κB in the spleen cells was studied by electrophoretic mobility shift assay (EMSA). NSP, nuclear extract from spleen cells of SH1000-infected mice with a 200-fold molar excess of unlabeled dsDNA oligonucleotide.
Accumulation of IL-6 in serum.
The serum level of IL-6 was another parameter we examined for systemic inflammation; it is a pleiotropic cytokine that plays an important role in the regulation of a variety of inflammatory and immune responses, acute-phase responses, and hematopoiesis (2, 20, 21, 34, 52, 53) in both the mouse-infective wild-type strain and its fnb mutants. Serum from SH1000-infected mice contained high levels of IL-6 at 5 h postinfection; the level returned to the baseline at 24 h postinfection. The level at 5 h postinfection was significantly higher than that in JS1078- and JS3094-infected mice at 5 h after infection (Table 3). The level of serum IL-6 in JS2072-infected mice was lower than that in SH1000-infected mice, but not significantly so. This indicates that FnBPs contribute to IL-6 production in systemic infection and that FnBPA is a more potent inducer of IL-6.
Table 3.
Serum IL-6 levels in mice infected with SH1000 or fnb mutant strainsa
| Strain | IL-6 concn (pg/ml serum) at: |
|
|---|---|---|
| 5 h | 24 h | |
| SH1000 | 235 ± 28 (a) | 50 ± 7.5 (e) |
| JS1078 (A−) | 101 ± 47 (b) | 56 ± 8.5 (f) |
| JS2072 (B−) | 192 ± 132 (c) | 48 ± 6.0 (g) |
| JS3094 (A−/B−) | 129 ± 16 (d) | 50 ± 5.2 (h) |
BALB/c mice were intravenously infected with SH1000 or fnb mutant strains (5 × 107 CFU/head). After 5 or 24 h, serum was removed from the collected blood and the concentration of IL-6 was measured by ELISA. P < 0.01 for a versus b and a versus d.
Role of FnBPs in in vitro infection of nonprofessional phagocytes.
S. aureus is known to be internalized in nonprofessional phagocytic cells via FnBP-α5β1-integrin interaction (17). We corroborated the effect of FnBPs on cellular infection by S. aureus SH1000 and its fnb mutants in vitro. Figure 5A shows images of lysostaphin-treated and untreated cultures of L929 fibroblasts after 1 h of cocultivation with bacteria (MOI = 50), and the mean ± the standard deviation of internalized bacteria is shown in Fig. 5B. SH1000 adhered well and was internalized by the cells, whereas JS1078 and JS3094 were barely adherent and internalized. In the case of JS2072, adhesion and internalization of bacteria were not completely abolished. These results show that FnBPA and FnBPB cooperatively interact with L929 fibroblasts, although FnBPA works more efficiently than FnBPB. Similar results were obtained with other nonprofessional phagocytes, MDCK epithelial cells, and human umbilical vein endothelial cells (data not shown).
Fig. 5.

Effects of FnBPA and FnBPB on in vitro infection with S. aureus. Adhesion of S. aureus to and internalization in L929 fibroblasts were studied. L929 cells were cocultured with SH1000 or its fnb mutant strains for 60 min (MOI = 50) and stained by Giemsa's solution. Microscopic images with or without lysostaphin treatment are shown in panel A, and the mean numbers of incorporated bacteria per cell ± standard deviations are shown in panel B. P < 0.0001 for a versus b, a versus f, b versus c, b versus d, b versus e, c versus f, d versus e, d versus f, and e versus f. P < 0.01 for a versus d and a versus e.
Next, we examined the effect of FnBPs on the activation of NF-κB in L929 fibroblasts (Fig. 6). Cocultivation of SH1000 with L929 fibroblasts strongly induced the translocation of NF-κB. Translocation of NF-κB was also seen in JS2072-treated cells, but the amount was smaller than that in SH1000-treated cells. Both JS1078 and JS3094 did not induce translocation. Complementation with fnbA and fnbB completely restorered this response. These results indicate that the interaction of fibroblasts with S. aureus via FnBPs is associated with the induction of NF-κB activation; furthermore, FnBPA is more potent for induction of NF-κB activation than is FnBPB.
Fig. 6.

Activation of NF-κB in L929 fibroblasts after infection with the wild-type strain or the fnbA mutant strain. SH1000 or the fnb mutant in the early exponential growth phase was cocultured with L929 fibroblasts at an MOI of 50. After 60 min of cultivation, cells were washed with PBS and treated with 20 μg/ml lysostaphin plus antibiotics for 30 min. Binding of nuclear extracts with 3′-biotinylated oligonucleotides having the consensus binding sequence of NF-κB was analyzed by EMSA. NSP shows the nonspecific signal obtained with the nuclear extract from SH1000-treated cells with a 200-fold molar excess of unlabeled oligonucleotides.
Effect of FnBPs on activation of inflammatory macrophages.
Previously, we reported that Fn-treated S. aureus was well ingested by inflammatory macrophages through α5β1-integrin on the macrophages (49). To determine the effect of FnBPs on this type of ingestion, thioglycolate-elicited mouse peritoneal macrophages were treated with SH1000 and the mutants. The number of ingested bacteria decreased in all 3 mutants, namely, JS1078, JS2072, and JS3094 (38%, 54%, and 4% of the number of ingested SH1000 bacteria, respectively), and was restored by complementation with fnbA or fnbB (129% and 162% of the number of ingested SH1000 bacteria for JS1078/pFNBA4 and JS2072/pFNBB4, respectively).
Inflammatory macrophages are thought to be among the most important cells in inflammatory responses. Therefore, we studied the activation of NF-κB and the expression of the inflammatory cytokines TNF-α and IL-6 in inflammatory macrophages. As shown in Fig. 7A, SH1000 induced remarkable activation of NF-κB, and Fn pretreatment of SH1000 enhanced this activation. In JS1078, however, the amount of activated NF-κB was reduced compared to that in SH1000 infection, irrespective of Fn pretreatment. In accordance with this result, SH1000-ingesting macrophages expressed definite levels of mRNA or TNF-α and IL-6; however, JS1078 induced much lower expression of these cytokines (Fig. 7B). These results indicate that FnBPs are responsible for Fn-mediated ingestion and the induction of an inflammatory response in inflammatory macrophages.
Fig. 7.

Inflammatory responses of macrophages cocultivated with the wild-type strain or the fnbA mutant strain. (A) Activation of NF-κB was analyzed by EMSA. (B) mRNA expression of proinflammatory cytokines TNF-α and IL-6 in mouse inflammatory macrophages. SH1000 or its fnbA mutant in the early exponential growth phase was cocultured with mouse inflammatory macrophages at an MOI of 50. Bacteria left untreated or pretreated with 100 μg/ml Fn were used in the NF-κB activation analysis (A). Fn-pretreated bacteria were used in the proinflammatory cytokine expression analysis (B). After a 30-min cultivation, cells were washed with PBS and treated with 20 μg/ml lysostaphin plus antibiotics for 30 min, and then the nuclear extracts or RNAs were prepared.
DISCUSSION
Sepsis is a major life-threatening symptom with severe systemic inflammation attributed to infectious microorganisms. S. aureus, especially MRSA, is one of the typical causes of septicemia following trauma, surgery, and catheter-related bloodstream infection. Using a mouse model and administration of fnbA and/or fnbB mutant strains derived from the parental strain SH1000, which expressed similar amounts of FnBPA and FnBPB in the exponential growth phase, our present study clearly shows that both FnBPA and FnBPB are indispensable factors in the establishment of septicemia.
In a mouse model, the weight loss and lethal effect of intravenously infected S. aureus were dramatically abolished in fnbA or fnbB mutants. An initial reduction in weight was seen in fnbA and fnbB mutant infections; however, a prolonged reduction was only seen in fnbB mutant infections. These results demonstrate that FnBPA and FnBPB work together for the establishment of septic infection, although FnBPA contributes more to infection potency than FnBPB does. The intense effect of FnBPA is obvious in the appearance of infected mice. In fnbA fnbB mutant infections, no obvious change in weight was observed during the first 5 days; this was followed by transient weight loss. Similar transient changes were seen in fnbA mutant infections. The cause of this time profile of body weight changes is not clear, but one possibility is endogenous reinfection by bacteria, as discussed further on.
The cooperative function of FnBPA and FnBPB was also seen in the other parameters indicating the progress of systemic infection, i.e., bacterial colonization, multiplication, and abscess formation in the kidneys. These results clearly show that both FnBPA and FnBPB are indispensable for the initial adhesion of S. aureus, followed by further multiplication of bacteria in organs.
The parameters for systemic inflammation, activation of NF-κB, and serum IL-6 levels also indicate that both FnBPs are required for severe inflammatory responses, although FnBPA is necessary for the induction of these responses. IL-6 is known as one of the most important proinflammatory cytokines detected in serum and tissues in mouse models of S. aureus-induced septicemia (2, 20, 21, 34) and in patients with S. aureus septicemia (16, 39, 52). The time profile of serum IL-6 levels shown in Table 3 is similar to the profile described in another sepsis model of S. aureus intraperitoneal infection (2) and in sepsis caused by another Gram-positive bacterium, Streptococcus suis (9). Previous studies have demonstrated that IL-10, which appears later than proinflammatory cytokines, inhibits these cytokines (9, 12), resulting in initially high cytokine levels declining to basal levels at 24 h postinfection. Excess production of proinflammatory cytokines damages the endothelium and other organs (37). Therefore, high production of IL-6 in wild-type strain- or fnbB mutant-infected mice should be one of the major causes of significant alteration of body weight, unhealthy appearance, and drastic lethality. Palmqvist et al. also demonstrated the contribution of FnBPs to systemic infection (40). They compared the pathogenicity of wild-type S. aureus strain LS-1 with its mutant lacking both fnbA and fnbB and demonstrated that the fnbA fnbB mutant caused no change in the body weights of mice and lower production of serum IL-6 than the wild-type strain. Furthermore, Kerdudou et al. investigated the role of FnBPs in S. aureus adhesion to and invasion of the endothelium by using FnBPA- or FnBPB-complemented transformants in nonadhesive S. carnosus (26). These transformants displayed enhanced binding to the vascular endothelium after intra-arterial application compared to that of the wild-type strain. They also demonstrated that FnBPA contributed to bacterial adhesion to the endothelium more efficiently than FnBPB. These studies clearly show the importance of FnBPs in S. aureus infection; however, the contribution of the respective FnBPs to infection with S. aureus in terms of the physiological condition of bacteria has yet to be elucidated. Our present study clearly demonstrates that FnBPA is more crucial than FnBPB, whereas both FnBPA and FnBPB are indispensable for the establishment of sepsis.
The mechanism of in vivo infection is partly explained based on evidence of in vitro cellular studies. FnBPA was crucial for adhesion to and internalization by the cells in the interaction of S. aureus with nonprofessional phagocytic cells, while the mutation in fnbB did not show a significant reduction in this aspect. The adhesion and internalization abilities were in good accordance with the level of NF-κB activation. These results suggest that the inflammatory response in nonprofessional phagocytes is associated with systemic inflammation factors such as serum levels of IL-6. Sinha et al. previously suggested that heterologous overexpression of FnBPA or FnBPB in the Du5883 strain, the fnbA fnbB double mutant of strain 8325-4, or the coagulase-negative species S. carnosus was sufficient for staphylococcal invasion of epithelial cells (51). Under experimental conditions, either FnBPA or FnBPB definitely promotes cellular internalization; however, in physiological expression of FnBPs as in our present study, FnBPA is essential for S. aureus to adhere to and be internalized by host cells.
Macrophages are among the most important professional phagocytes following bacterial infection (54). We previously reported that S. aureus was ingested by macrophages via Fn and α5β1-integrin (49). This type of ingestion is seen only in inflammatory macrophages and not in resident macrophages because of the difference in their expression of integrin (48). As mentioned in Results, a mutation(s) in fnbA and/or fnbB resulted in the reduction of bacteria ingested by inflammatory macrophages, suggesting that FnBPs are responsible for α5β1-integrin-mediated ingestion. Recently, Wang et al. demonstrated that macrophages lacking β1-integrin exhibit reduced phagocytosis of bacteria, including S. aureus, and that β1-integrin works as a phagosome maturation regulator by inducing the expression of Rho family GTPases in macrophages (56). On the basis of their results and ours, the wild-type strain is expected to be well excluded by macrophages as a demonstration of its lower pathogenicity than its fnb mutants, even if the other phagocytic pathways are activated. However, as shown by our present results, the severity of the wild-type strain in in vivo infection was extremely high compared to that of the fnb mutants. In this regard, Kubica et al. reported that S. aureus SH1000, but not 8325-4, was well phagocytosed by macrophages; however, it persisted intracellularly for a few days without being killed and subsequently escaped into the cytoplasm to lyse the cells (29). Thus, S. aureus ingested by macrophages may survive in the cells and thereafter cause reinfection in vivo. This may be one of the reasons for the severity of SH1000 infection and for the secondary transient weight loss of fnbA or fnbA fnbB mutant-infected mice. Our study further proved that interaction of α5β1-integrin with FnBPs induced the activation of NF-κB and the following expression of proinflammatory cytokines in inflammatory macrophages. Since macrophages are a major source of proinflammatory cytokines (8, 57), these cells are responsible for the high levels of serum IL-6 observed in wild-type strain infections.
Our present study shows that FnBPA and FnBPB function cooperatively in in vivo and in vitro infections, primarily through initial adhesion to cells or tissues, although FnBPA plays a more crucial role than FnBPB. This cooperative function between FnBPA and FnBPB is thought to induce severe septic symptoms resulting in septic death from S. aureus infection. The understanding of the structures of FnBPA and FnBPB shows that both proteins have high sequence homology (68%), especially in Fn-binding repeats (FnBRs), which are responsible for binding of F1 modules in the N-terminal domain of Fn (25, 33). FnBPA possesses 11 potential FnBRs containing 6 high-affinity sites with dissociation constants (KDs) in the nanomolar range and 5 low-affinity sites. Similarly, FnBPB possesses 10 Fn-binding sites and 6 of them are suggested to show high-affinity binding to Fn (33, 42, 47). A recent study demonstrated that multiple FnBRs were required for virulence in a murine sepsis model, although only a single high-affinity FnBR facilitated the invasion of endothelial cells in vitro (10). N-terminal A domains in FnBPA and FnBPB are approximately 40% identical in amino acid sequence and have fibrinogen and elastin binding capacity (45, 58). These findings indicate that FnBPA and FnBPB are similar in organization and function in S. aureus infection. However, the two proteins work cooperatively in in vitro cellular and in vivo septic infections, and further, FnBPA is indispensable, as demonstrated above. The statistics show that the majority (>70%) of clinical isolates encode both FnBPA and FnBPB, while 20% encode only FnBPA. Isolates encoding only FnBPB are very few (1%) (41). This reflects the importance of both FnBPs, especially FnBPA, in S. aureus infection and is in accordance with our results. One possible mechanism of infection is the difference between the KDs of the high-affinity Fn-binding motifs in FnBPA and FnBPB; the high-affinity Fn-binding motif in FnBPB has 2 to 3 times lower affinity than that of FnBPA (42). This difference may not be so significant in each FnBR; however, it may be critical overall, allowing FnBPA to achieve a dominant role. Another possible explanation is the structural difference between the A domains of FnBPA and FnBPB. If the two domains show different tertiary structures on the bacterial surface, functions such as Fn binding would be affected. The third possible mechanism is the difference in the distribution of FnBPA and FnBPB on the bacterial surface. If FnBPA molecules were distributed in clusters on the bacterial surface and FnBPB molecules were distributed all over the surface, integrins would easily be activated by FnBPA because the cluster of FnBPAs could efficiently organize integrins. As a result, S. aureus would be well internalized or well ingested by nonprofessional phagocytes or professional phagocytes, resulting in strong inflammatory responses. Further investigation of the mechanism by which FnBPs contribute to infections caused by clinical isolates is necessary.
ACKNOWLEDGMENTS
This study was supported by Grant-in-Aid for Scientific Research (C) JSPS22590404, the Jikei University Research Fund, and the Jikei University Graduate Research Fund.
We thank T. J. Foster for kindly providing strains and plasmids. We are grateful to S. Masuda for his accurate advice on this study. We thank K. Hiramatsu for the generous gift of a plasmid.
Footnotes
Published ahead of print on 21 March 2011.
REFERENCES
- 1. Agerer F., et al. 2005. Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalization. J. Cell Sci. 118:2189–2200 [DOI] [PubMed] [Google Scholar]
- 2. Ahn J.-Y., Song J.-Y., Yun Y.-S., Jeong G., Choi I.-S. 2006. Protection of Staphylococcus aureus-infected septic mice by suppression of early acute inflammation and enhanced antimicrobial activity by ginsan. FEMS Immunol. Med. Microbiol. 46:187–197 [DOI] [PubMed] [Google Scholar]
- 3. Archer G. L., Climo M. W. 2001. Staphylococcus aureus bacteremia—consider the source. N. Engl. J. Med. 344:55–56 [DOI] [PubMed] [Google Scholar]
- 4. Bischoff M., et al. 2004. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 186:4085–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonizzi G., Karin M. 2004. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25:280–288 [DOI] [PubMed] [Google Scholar]
- 6. Burke F. M., McCormack N., Rind S., Speziale P., Foster T. J. 2010. Fibronectin-binding protein B variation in Staphylococcus aureus. BMC Microbiol. 10:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheung A. L., Yang S.-J., Bayer A. S., Xiong Y. Q. 2009. Disparity in the in vitro versus in vivo regulation of fibronectin-binding proteins by 2 global regulators, saeRS and sigB, in Staphylococcus aureus. J. Infect. Dis. 200:1371–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen J. 2002. The immunopathogenesis of sepsis. Nature 420:885–891 [DOI] [PubMed] [Google Scholar]
- 9. de la Cruz Domínguez-Punaro M., Segura M., Radzioch D., Rivest S., Gottschalk M. 2008. Comparison of the susceptibilities of C57BL/6 and A/J. mouse strains to Streptococcus suis serotype 2 infection. Infect. Immun. 76:3901–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edwards A. M., Potts J. R., Josefsson E., Massey R. C. 2010. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog. 6:e10009645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Entenza J.-M., et al. 2005. Role of σB in expression of Staphylococcus aureus cell wall adhesins ClfA and FnbA and contribution to infectivity in a rat model of experimental endocarditis. Infect. Immun. 73:990–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fiorentino D. F., Zlotnik A., Mosmann T. M., Howard H., O'Garra A. 1991. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 147:3815–3822 [PubMed] [Google Scholar]
- 13. Fowler T., Johansson S., Wary K. K., Höök M. 2003. Src kinase has a central role in in vivo cellular internalization of Staphylococcus aureus. Cell. Microbiol. 5:417–426 [DOI] [PubMed] [Google Scholar]
- 14. Garzoni C., Kelley W. L. 2009. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 17:59–65 [DOI] [PubMed] [Google Scholar]
- 15. Greene C., et al. 1995. Adhesion properties of mutants of Staphylococcus aureus defective in fibronectin-binding proteins and studies on the expression of fnb genes. Mol. Microbiol. 17:1143–1152 [DOI] [PubMed] [Google Scholar]
- 16. Groeneveld A. B. J., Tacx A. N., Bossink A. W. J., van Mierlo G. J., Hack C. E. 2003. Circulating inflammatory mediators predict shock and mortality in febrile patients with microbial infection. Clin. Immunol. 106:106–115 [DOI] [PubMed] [Google Scholar]
- 17. Hauck C. R., Ohlsen K. 2006. Sticky connection: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr. Opin. Microbiol. 9:5–11 [DOI] [PubMed] [Google Scholar]
- 18. Herbert S., et al. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78:2877–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horsburgh M. J., et al. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hultgren O. H., Stenson M., Tarkowski A. 2001. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 3:41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hultgren O. H., Svensson L., Tarkowski A. 2002. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J. Immunol. 168:5207–5212 [DOI] [PubMed] [Google Scholar]
- 22. Jett B. D., Gilmore M. S. 2002. Internalization of Staphylococcus aureus by human corneal epithelial cells: role of bacterial fibronectin-binding protein and host cell factors. Infect. Immun. 70:4697–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones C. L., Khan S. 1986. Nucleotide sequence of the enterotoxin B gene from Staphylococcus aureus. J. Bacteriol. 166:29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jonsson I.-M., Arvidson S., Foster S., Tarkowski A. 2004. Sigma factor B and RsbU are required for virulence in Staphylococcus aureus-induced arthritis and sepsis. Infect. Immun. 72:6106–6111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jönsson K., Signäs C., Müller H. P., Lindberg M. 1991. Two different genes encode fibronectin binding proteins in Staphylococcus aureus. The complete nucleotide sequence and characterization of the second gene. Eur. J. Biochem. 18:1041–1048 [DOI] [PubMed] [Google Scholar]
- 26. Kerdudou S., et al. 2006. Fibronectin binding proteins contribute to the adherence of Staphylococcus aureus to intact endothelium in vivo. Thromb. Haemost. 96:183–189 [PubMed] [Google Scholar]
- 27. Kintarak S., Whawell S. A., Speight P. M., Packer S., Nair S. P. 2004. Internalization of Staphylococcus aureus in human keratinocytes. Infect. Immun. 72:5668–5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kreiswirth B. N., et al. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:680–685 [DOI] [PubMed] [Google Scholar]
- 29. Kubica M., et al. 2008. A potential new pathway for Staphylococcus aureus dissemination. The silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 3:e1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li D., et al. 2005. Induction of fibronectin adhesins in quinolone-resistant Staphylococcus aureus by subinhibitory levels of ciprofloxacin or by sigma B transcription factor activity is mediated by two separate pathways. Antimicrob. Agents Chemother. 49:916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loughman J. A., Fritz S. A., Storch G. A., Hunstad D. A. 2009. Virulence gene expression in human community-acquired Staphylococcus aureus infection. J. Infect. Dis. 199:294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGavin M. J., Zahradka C., Rice K., Scott J. E. 1997. Modification of the Staphylococcus aureus fibronectin binding phenotype by V8 protease. Infect. Immun. 65:2621–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meenan N. A. G., et al. 2007. The tandem β-zipper model defines high affinity fibronectin-binding repeats within Staphylococcus aureus FnBPA. J. Biol. Chem. 282:25893–25902 [DOI] [PubMed] [Google Scholar]
- 34. Nakane A., Okamoto M., Asano M., Kohanawa M., Minagawa T. 1995. Endogenous gamma interferon, tumor necrosis factor, and interleukin-6 in Staphylococcus aureus infection in mice. Infect. Immun. 63:1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Novick R. P. 1967. Properties of a cryptic high frequency transducing phage in Staphylococcus aureus. Virology 33:155–166 [DOI] [PubMed] [Google Scholar]
- 36. Novick R. P. 2003. Autoinduction and signal transduction in the regulation of staphylococcus virulence. Mol. Microbiol. 48:1429–1449 [DOI] [PubMed] [Google Scholar]
- 37. Nyström P.-O. 1998. The systemic inflammatory response syndrome: definitions and aetiology. J. Antimicrob. Chemother. 41(Suppl. A):1–7 [DOI] [PubMed] [Google Scholar]
- 38. Olivier A. C., Lemaire S., Bambeke F. V., Tulkens P. M., Oldfield E. 2009. Role of rsbU and staphyloxanthin in phagocytosis and intracellular growth of Staphylococcus aureus in human macrophages and endothelial cells. J. Infect. Dis. 200:1367–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Osuchowski M. F., Welch K., Siddiqui J., Remick D. G. 2006. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 177:1967–1974 [DOI] [PubMed] [Google Scholar]
- 40. Palmqvist N., Foster T., Fitzgerald J. R., Josefsson E., Tarkowski A. 2005. Fibronectin-binding proteins and fibrinogen-binding clumping factors play distinct roles in staphylococcal arthritis and systemic inflammation. J. Infect. Dis. 191:791–798 [DOI] [PubMed] [Google Scholar]
- 41. Peacock S. J., Day N. P. J., Thomas M. G., Berendt A. R., Foster T. J. 2000. Clinical isolates of Staphylococcus aureus exhibit diversity in fnb genes and adhesion to human fibronectin. J. Infect. 41:23–31 [DOI] [PubMed] [Google Scholar]
- 42. Provenza G., et al. 2010. Functional analysis of a murine monoclonal antibody against the repetitive region of the fibronectin-binding adhesins fibronectin-binding protein A and fibronectin-binding protein B from Staphylococcus aureus. FEBS J. 277:4490–4505 [DOI] [PubMed] [Google Scholar]
- 43. Pulai J. I., et al. 2005. NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 174:5781–5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Que Y.-A., et al. 2005. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 201:1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roche F. M., et al. 2004. The N-terminal A domain of fibronectin-binding proteins A and B promotes adhesion of Staphylococcus aureus to elastin. J. Biol. Chem. 279:38433–38440 [DOI] [PubMed] [Google Scholar]
- 46. Schröder A., et al. 2006. Staphylococcus aureus fibronectin binding protein-A induces motile attachment sites and complex actin remodeling in living endothelial cells. Mol. Biol. Cell 17:5198–5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schwarz-Linek U., et al. 2003. Pathogenic bacteria attach to human fibronectin through a tandem β-zipper. Nature 423:177–181 [DOI] [PubMed] [Google Scholar]
- 48. Shinji H., et al. 2007. Expression and distribution of very late antigen-5 in mouse peritoneal macrophages upon ingestion of fibronectin-bound Staphylococcus aureus. Microbiol. Immunol. 51:63–71 [DOI] [PubMed] [Google Scholar]
- 49. Shinji H., Seki K., Tajima A., Uchida A., Masuda S. 2003. Fibronectin bound to the surface of Staphylococcus aureus induces association of very late antigen 5 and intracellular signaling factors with macrophages. Infect. Immun. 71:140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shinji H., Sakurada J., Seki K., Murai M., Masuda S. 1998. Different expression of Staphylococcus aureus and coagulase-negative staphylococci by murine peritoneal macrophages. Microbiol. Immunol. 42:851–861 [DOI] [PubMed] [Google Scholar]
- 51. Sinha B., et al. 2000. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect. Immun. 68:6871–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Söderquist B., Sundqvist K. G., Jones I., Holmberg H., Vikerfors T. 1995. Interleukin-6, C-reactive protein, lactoferrin and white blood cell count in patients with S. aureus septicemia. Scan. J. Infect. Dis. 27:375–380 [DOI] [PubMed] [Google Scholar]
- 53. Suárez-Santamaría M., et al. 2010. Prognostic value of inflammatory markers (notably cytokines and procalcitonin), nutritional assessment, and organ function in patients with sepsis. Eur. Cytokine Netw. 21:19–26 [DOI] [PubMed] [Google Scholar]
- 54. Verdrengh M., Tarkowski A. 2000. Role of macrophages in Staphylococcus aureus-induced arthritis and sepsis. Arthritis Rheum. 43:2276–2282 [DOI] [PubMed] [Google Scholar]
- 55. von Eiff C., Becker K., Machka K., Stammer H., Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. N. Engl. J. Med. 344:11–16 [DOI] [PubMed] [Google Scholar]
- 56. Wang Q.-Q., et al. 2008. Integrin β1 regulates phagosome maturation in macrophages through Rac expression. J. Immunol. 180:2419–2428 [DOI] [PubMed] [Google Scholar]
- 57. Wang Z.-M., Liu C., Dziarsk R. 2000. Chemokines are the main proinflammatory mediators in human monocytes activated by Staphylococcus aureus, peptidoglycan, and endotoxin. J. Biol. Chem. 275:20260–20267 [DOI] [PubMed] [Google Scholar]
- 58. Wann E. R., Gurusiddappa S., Höök M. 2000. The fibronectin-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. J. Biol. Chem. 275:13863–13871 [DOI] [PubMed] [Google Scholar]
- 59. Wertheim H. F. L., et al. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 5:751–762 [DOI] [PubMed] [Google Scholar]
- 60. Wolz C., et al. 2000. Agr-independent regulation of fibronectin-binding protein(s) by the regulatory locus sar in Staphylococcus aureus. Mol. Microbiol. 36:230–233 [DOI] [PubMed] [Google Scholar]
- 61. Yoshizawa Y. 1985. Isolation and characterization of restriction negative mutants of Staphylococcus aureus. Jikeikai Med. J. 32:413–421 [Google Scholar]