

Abstract
Clostridium perfringens causes clostridial myonecrosis or gas gangrene and produces several extracellular hydrolytic enzymes and toxins, many of which are regulated by the VirSR signal transduction system. The revR gene encodes a putative orphan response regulator that has similarity to the YycF (WalR), VicR, PhoB, and PhoP proteins from other Gram-positive bacteria. RevR appears to be a classical response regulator, with an N-terminal receiver domain and a C-terminal domain with a putative winged helix-turn-helix DNA binding region. To determine its functional role, a revR mutant was constructed by allelic exchange and compared to the wild type by microarray analysis. The results showed that more than 100 genes were differentially expressed in the mutant, including several genes involved in cell wall metabolism. The revR mutant had an altered cellular morphology; unlike the short rods observed with the wild type, the mutant cells formed long filaments. These changes were reversed upon complementation with a plasmid that carried the wild-type revR gene. Several genes encoding extracellular hydrolytic enzymes (sialidase, hyaluronidase, and α-clostripain) were differentially expressed in the revR mutant. Quantitative enzyme assays confirmed that these changes led to altered enzyme activity and that complementation restored the wild-type phenotype. Most importantly, the revR mutant was attenuated for virulence in the mouse myonecrosis model compared to the wild type and the complemented strains. These results provide evidence that RevR regulates virulence in C. perfringens; it is the first response regulator other than VirR to be shown to regulate virulence in this important pathogen.
INTRODUCTION
Clostridium perfringens is ubiquitous in soil and sewage and is a commensal of the gastrointestinal tracts of both humans and animals (51); therefore, it needs to be able to adapt to an ever-changing environment. Two-component signal transduction systems enable bacteria to sense changes in their environment and generally consist of a membrane-bound sensor histidine kinase and a cytoplasmic response regulator (27, 63, 69). Upon detection or binding of the stimulus, the sensor histidine kinase autophosphorylates at a conserved histidine residue present within the cytoplasmic C-terminal region of the protein (27, 48). The phosphoryl group is then transferred from the sensor histidine kinase to a conserved aspartate residue present within the N-terminal domain of the response regulator (24, 27, 63). The phosphorylated response regulator usually acts by binding to a specific DNA target and altering the transcription of the target genes (27, 48, 63). Signal transduction systems often can be directly linked to the expression of specific virulence factors involved in disease pathogenesis (10, 13).
C. perfringens causes human gas gangrene and food poisoning and several diseases of animals (61, 62). These diseases are mediated by a diverse range of extracellular toxins and extracellular enzymes (50), two of which, alpha-toxin and perfringolysin O, have been shown to be important for the development of gas gangrene or clostridial myonecrosis (2, 3). The genes encoding these toxins, as well as other genes, have been shown to be regulated by the global VirSR two-component signal transduction pathway (32, 44, 54). In this system the response regulator VirR directly regulates the expression of pfoA (encoding perfringolysin O or theta-toxin) and ccp (encoding the cysteine protease α-clostripain) and indirectly regulates the expression of plc (encoding alpha-toxin), colA (encoding collagenase or kappa-toxin), and other genes involved in cellular metabolism (7, 9, 57).
Although the VirSR network regulates several potential virulence genes in C. perfringens, there are other putative virulence factors whose expression is not understood. As part of a broader study, we initially focused on the use of bioinformatics to identify regulatory genes that may have a potential role in virulence. In this study we report the results of genetic studies, coupled with microarray analysis and virulence experiments, which have led to the identification and characterization of a novel orphan response regulator, RevR, which also regulates the ability of C. perfringens to cause clostridial myonecrosis.
MATERIALS AND METHODS
Plasmids, strains, and media.
All plasmids and bacterial strains used in this study are listed in Table 1. All C. perfringens strains were derived from the strain 13 derivative JIR325 (32). Culture medium was from Oxoid and chemicals were from Sigma or Amresco, unless otherwise stated. C. perfringens broth cultures were grown in either fluid thioglycolate broth (FTG) (Difco) or TPYG (5% [wt/vol] tryptone, 0.5% [wt/vol] proteose peptone, 0.3% [wt/vol] yeast extract, 0.1% [wt/vol] sodium thioglycolate) with glucose added to a final concentration of 0.38% (wt/vol) after sterilization. Agar cultures of C. perfringens were grown on nutrient agar (NA) (49) in an atmosphere of 10% (vol/vol) H2, 10% (vol/vol) CO2, and 80% (vol/vol) N2. Where appropriate, C. perfringens media were supplemented with rifampin (10 μg/ml), nalidixic acid (10 μg/ml), erythromycin (Amresco) (50 μg/ml), or chloramphenicol (30 μg/ml). To visualize cellular morphology, bacterial cells were Gram stained and examined at a magnification of ×1,000 using an Olympus BX51 microscope. Digital images were acquired using DPController software (Olympus).
Table 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Bacterial strains | ||
| E. coli DH5α | F− φ80dlacZΔM15Δ(lacZYA-argF)U169 endA1 recA1 hadR17 (rK− mK−) deoR thi-1 supE44 gyrA96 relA1 | Life Technologies |
| C. perfringens | ||
| JIR325 | Rifr Nalr derivative of strain 13 | 32 |
| JIR12233 | JIR325ΔrevRΩ erm(Q) | This study |
| Plasmids | ||
| pT7Blue-3 | Commercial cloning vector; Apr | Novagen |
| pJIR750 | E. coli-C. perfringens shuttle vector, carries pIP404 replication origin; Cmr | 5 |
| pJIR2715 | Suicide vector base for C. perfringens; Cmr Ermr | 6 |
| pJIR3374 | pJIR2715 (Asp718/BamHI)Ω JRP3520/JRP3521 (Asp718/BamHI, 1.8 kb) PCR product, Cmr Ermr | This study |
| pJIR3429 | pJIR3474 (XhoI/NotI)Ω JRP3718/JRP3719 (XhoI/NotI, 1.2 kb) PCR product, Cmr Ermr (revR suicide vector) | This study |
| pJIR3534 | pJIR750 (BamHI/Asp718)Ω JRP3954/JRP3966 (BamHI/Asp718, 1.2 kb, Cmr (revR+ complementation plasmid) | This study |
Escherichia coli strain DH5α (Life Technologies) was used for the cloning and propagation of plasmids. E. coli cells were incubated in 2YT medium (52) under aerobic conditions. Where applicable, the medium was supplemented with either erythromycin (150 μg/ml) or chloramphenicol (30 μg/ml). The turbidity of broth cultures was measured at 600 nm using a WPA Biowave CO8000 cell density meter.
Genetic and molecular manipulations.
Plasmids from E. coli cells were prepared as described previously (36) or by the use of a Qiagen miniprep purification system. When plasmid preparations were required for nucleotide sequencing, a modified polyethylene glycol (PEG) precipitation method was employed as described in the ABI BigDye manual (Applied Biosystems). All restriction endonucleases and enzymes were used according to the manufacturer's instructions (Roche Diagnostics; New England BioLabs). Transformation of C. perfringens cells was carried out by electroporation (53) with at least 5 μg of purified plasmid DNA per experiment, using a BTX ECM-630 Electro Cell Manipulator (BTX Laboratories) with a single electric pulse of 1.8 kV, a resistance of 200 Ω, and a capacitance of 25 μF. E. coli cells were made chemically competent and transformed as described previously (29). C. perfringens chromosomal DNA (40) and total RNA (17) were isolated as before. Repeated cycles of DNase I digestion of the RNA samples and subsequent Trizol extraction were carried out until the RNA was free of contaminating DNA. Standard methods were used for the modification, ligation, and analysis of plasmid and genomic DNA preparations and PCR products (52). DNA and RNA concentrations were determined using a NanoDrop Technologies spectrophotometer.
DNA sequencing reactions were performed using a PRISM BigDye Terminator Mix (Applied Biosystems). Signal detection was performed on an Applied Biosystems 3730S Genetic Analyser and sequences analyzed using ContigExpress software (Invitrogen). All oligonucleotide primers for PCR or sequencing were obtained from Sigma-Aldrich and are listed in Table S1 in the supplemental material.
Construction and complementation of a revR mutant.
The suicide plasmid pJIR3429 was constructed by generating an 1,867-bp PCR fragment that encompassed the region directly upstream of, and including, the first 21 bp of the revR gene, using primers JRP3520 and JRP3521 (see Table S1 in the supplemental material). The BamHI- and Asp718-digested PCR fragment was cloned into the equivalent sites of pJIR2715 (6), upstream of the erythromycin resistance gene erm(Q), to give pJIR3374. A 1,254-bp region downstream of revR was PCR amplified using primers JRP3718 and JRP3719, digested with XhoI and NotI, and cloned downstream of the erm(Q) cassette in pJIR3374 to construct pJIR3429. All recombinant plasmids were confirmed by restriction digestion and sequencing.
A revR deletion mutant was constructed by allelic exchange between pJIR3429 and the C. perfringens JIR325 chromosome. The suicide plasmid pJIR3429 was introduced by electroporation and erythromycin-resistant transformants selected. The resultant revR mutant, JIR12233, was confirmed to be derived from a double-crossover event by PCR analysis and Southern hybridization (data not shown).
To construct the revR complementation plasmid, pJIR3534, the wild-type revR gene, including 500 bp of upstream sequence encompassing the putative revR promoter, was PCR amplified using Pwo polymerase (Roche), purified with a Qiagen PCR purification kit, and cloned into pT7Blue-3 (Novagen). The fragment then was subcloned into the BamHI/Asp718 sites of the E. coli-C. perfringens shuttle vector pJIR750, which confers chloramphenicol resistance (5). The revR mutant was complemented by transformation with pJIR3534, selecting for chloramphenicol resistance.
Microarray analysis of C. perfringens RNA transcripts.
Expression studies were carried out with microarrays that consisted of 500-bp PCR products that were printed in duplicate in different sectors of the array and were designed for optimal binding to all potential coding sequences determined from the C. perfringens strain 13 genome sequence (44). RNA was prepared from 4-h TPYG broth cultures of the wild type and the revR mutant. For each strain, identical microarrays were hybridized separately using cDNA derived from four independent biological replicates, including two dye swaps. The synthesis of cDNA, labeling, and hybridization were carried out using the Gensiphere 3DNA 900MPX kit as performed previously (40), except that the amount of starting RNA was reduced to 4 μg and the microarray slides were prehybridized in a solution containing herring sperm DNA (Promega) instead of salmon sperm DNA. The dried, hybridized, and labeled microarray slides were scanned using a GMS418 array scanner (Affymetrix) and the scans captured using GMS scanner software (V.1.51.0.42) (Affymetrix). Fluorescence intensity was quantified using ImaGene 5.1 (BioDiscovery) software. ImaGene data files were subjected to statistical analysis using the Limma software package for R (59, 60). Spot intensities were normalized first per print tip group and then between arrays using Lowess normalization. Fold ratios were determined by comparison to wild-type spot intensities, and P values were determined using the moderated t test algorithm.
Bioinformatic analysis.
Analysis of the C. perfringens strain 13 genome for putative signal transduction systems was carried out using the Microbial Signal Transduction Database (MIST) (66). Database searches were carried out using the PSI-BLAST algorithm (1). Multiple-sequence alignments were conducted using the ClustalW algorithm (65), and conserved domain searches were conducted using the CD-search algorithm (33, 34). Protein structure models were generated using Swiss-Model (http://swissmodel.expasy.org) with the PhoB (ExPDB template code 1GXQ) and YycF (ExPDB template code 2D1V) structures as templates. Ribbon diagrams were generated using PyMol software verson 0.99 (Delano Scientific).
QRT-PCR analysis of differentially expressed genes.
Quantitative reverse transcriptase PCR (QRT-PCR) was performed to quantify gene-specific mRNA levels from the wild-type, mutant, and complemented derivatives. RNA was isolated from 4-h TPYG broth cultures (three independent biological replicates) of the wild type (JIR325), a revR mutant carrying the shuttle vector pJIR750, and the complemented revR mutant carrying pJIR3534. Reverse transcriptase (RT) reactions were performed using 4 μg of total RNA and Superscript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions, except that 1 μg of random hexamers was used and RT reaction mixtures were incubated at 42°C for 2 h. Reaction mixtures then were heated to 65°C for 15 min to denature the reverse transcriptase. Control samples were prepared by replacing Superscript III reverse transcriptase with nuclease-free water.
For each biological replicate, specific gene expression levels were determined relative to the level of rpoA expression. All QRT-PCRs were performed as described previously (16), with the exception that cDNA preparations were diluted 10-fold. Signal detection was performed on a Mastercycler ep Realplex real-time PCR machine (Eppendorf). Reactions were confirmed as being the result of a single product by disassociation curve analysis.
Quantitative toxin and extracellular enzyme assays.
All assays were carried out in triplicate on three independent biological replicates of C. perfringens cultures grown in either TPYG broth or Todd-Hewitt broth (Oxoid) containing 0.1% (wt/vol) sodium thioglycolate and 0.38% (wt/vol) glucose, added after autoclaving. Culture supernatants or cell lysates were harvested from these cultures at the time of inoculation (T = 0), 4 h postinoculation (logarithmic growth phase, T = 4) and 8 h postinoculation (stationary phase, T = 8). All enzyme assay results are expressed as the initial rate of activity per min per mg total protein. Total protein was determined using a Pierce bicinchoninic acid (BCA) protein assay kit.
Hyaluronidase activity was determined using 4-methylumbelliferyl-N-acetyl-β-d-glucosamide (MUG) as the substrate, as before (14). The cell pellets from 15-ml TPYG broth cultures were washed in sterile DPBS (140 mM NaCl, 2.68 mM KCl, 4.23 mM Na2HPO4, 10 mM KH2PO4, pH 7.5) and resuspended in 1 ml of sterile DPBS. The cell suspension was treated with 100 μl of lysozyme (10 mg/ml) (Amresco) at 37°C overnight and centrifuged at 3500 × g, and the resultant supernatant was diluted 10-fold. Assays were conducted using 25 μl of diluted supernatant and 50 μl of MUG (0.2 mg/ml) in hyaluronidase buffer (100 mM citric acid, 250 mM NaH2PO4, pH 7.5) in black-walled, flat-bottom 96-well microtiter plates (Nunc). The rate of increase in fluorescence intensity obtained after hydrolysis of MUG was followed fluorometrically with excitation and emission wavelengths of 365 nm and 412 nm, respectively. Hyaluronidase assays were conducted at room temperature for 60 min, with data collected every 2 min on a Tecan Infinite 200 plate reader. The amount of 4-methyumbelliferone (4-MU) liberated was determined by comparison to a 4-MU standard curve. Hyaluronidase activity is expressed as pmoles of 4-MU released per min per mg protein.
For sialidase assays, culture supernatants from Todd-Hewitt broth cultures were collected by centrifugation at 5800 × g for 10 min and concentrated 20-fold using Amicon Ultra centrifugal filter devices (Millipore) with a nominal cutoff of 30 kDa. Sialidase activity was determined as described previously (18) except that the absorbance at 620 nm was determined using a Tecan Infinite 200 plate reader. Sialidase activity is expressed as the increase in absorbance at 620 nm per min per mg protein.
For collagenase and protease assays, supernatants were derived from TPYG broth cultures. Collagenase assays were conducted as described previously (4), and results are expressed as the rate of increase in absorbance at 520 nm per min per mg of total protein. Extracellular protease activity was assayed using azocasein as the substrate under reducing conditions (5 mM dithiothreitol [DTT]) in the presence of Ca2+, as previously described (46); these conditions have been shown to activate the major extracellular protease of C. perfringens, α-clostripain (64). Reaction mixtures were incubated at 37°C for 2 h, and the absorbance of the clear supernatant was determined at 450 nm using a Multiskan plate reader (Thermo-Labsystems). Protease activity is expressed as the increase in absorbance at 450 nm per min per mg of total culture supernatant protein. Perfringolysin O assays (2) and phospholipase C assays (58) were carried out as described previously.
Virulence trials with mice.
The virulence of C. perfringens strains was assessed in the mouse myonecrosis model using 6- to 8-week-old female BALB/c mice, as described previously (30). The right hind muscle was injected with 50 μl of washed cells in sterile phosphate-buffered saline (PBS), equal to approximately 109 CFU. Disease progression was then recorded every 30 min by observing limping, swelling of the thigh or footpad, and blackening of the thigh or footpad (30). Disease symptoms were scored on a scale of 0 (no sign of disease), 0.5 (moderate disease), or 1 (severe disease). When a score of 1 was reached for any symptom other than swelling of the thigh, the mice were euthanized humanely for animal ethics reasons. All virulence trials were conducted in accordance with Victorian State Government regulations and were approved by the Monash University SOBS B Animal Ethics Committee. C. perfringens cells were isolated from at least four independent biological replicates, and the virulence for each strain assessed in at least 20 mice. Statistical analysis of Kaplan-Meier survival curves was carried out using a log rank Mantel-Cox test with GraphPad Prism 5 software.
Bacterial strains were recovered from euthanized mice by homogenizing muscle tissues in sterile PBS and allowing large muscle fragments to settle for 20 min. The supernatant was then subcultured onto NA supplemented with either erythromycin, chloramphenicol, or rifampin and nalidixic acid, at concentrations appropriate for C. perfringens strains. Plates were incubated under anaerobic conditions at 37°C. The genotypes of the recovered strains were determined by PCR using primers JRP3954 and JRP3966; under these conditions, a 1.1-kb band is detected in the wild type and a 1.6-kb band in the revR mutant.
Microarray data accession number.
Microarray data were deposited into the Gene Expression Omnibus (GEO) database with the accession number GSE26508.
RESULTS
A revR mutant has an altered cellular morphology.
This study commenced as part of a larger project aimed at determining the functional role of orphan response regulators in C. perfringens. BLAST searches of the C. perfringens strain 13 genome sequence (55) identified a gene, here designated revR, whose putative product (accession number NP_561558, CPE0642) had an N-terminal REC domain (12) and a putative C-terminal winged helix-turn-helix DNA binding domain (22). It had significant similarity to PhoB from Clostridium kluyveri (65% amino acid sequence identity), PhoP from Bacillus subtilis (53% identity), VicR from Streptococcus pneumoniae (52% identity), and YycF (WalR) from B. subtilis (49% identity). Based on the role of VicR in the regulation of several proteins involved in virulence in S. pneumoniae (39, 47, 67) we decided to construct a revR mutant of C. perfringens and examine its role in virulence.
A revR deletion mutant was constructed by homologous recombination and confirmed to be derived from a double-crossover event by PCR analysis and Southern hybridization (data not shown). Gram staining of the revR mutant revealed many long filamentous bacterial cells, unlike the shorter rods observed with the wild-type strain (Fig. 1). No difference in growth rate was observed in TPYG broth, and production of the two major C. perfringens type A toxins, alpha-toxin and perfringolysin O, was unaffected (data not shown). When the revR mutation was complemented by introducing the wild-type revR gene on a multicopy plasmid, no filaments were observed (Fig. 1). This phenotype was consistent with that observed in B. subtilis, where overexpression of the essential yycF gene leads to increased cell division (21).
Fig. 1.

Gram staining of isogenic revR derivatives. Gram-strained smears of the wild-type strain (WT), the revR mutant, and a complemented revR mutant [revR(revR+)] are shown.
Mutation of revR alters the transcription of several genes encoding potential virulence factors.
Microarray analysis was conducted on the wild type and the revR mutant to identify the genes whose expression was regulated by RevR. Initially, the microarray data were filtered to exclude genes that were less than 2-fold up- or downregulated and with a P value of ≤0.05. Using these criteria, over 400 genes were identified as being differentially regulated as a result of the revR mutation. Subsequent tightening of these filtering criteria using a P value of ≤0.001 still revealed over 100 genes that were differentially regulated (see Table S2 in the supplemental material).
Several genes whose products were potentially involved in the early and late stages of sporulation, including genes encoding proteins involved in the formation of the spore septum, cortex, and coat were downregulated in the revR mutant. In addition, differentially regulated genes potentially encoding virulence factors were identified (Table 2). Both the colA gene (encoding collagenase) (4) and the ccp gene (encoding the cysteine protease α-clostripain) (46) appeared to have increased levels of expression in the revR mutant. In contrast, two putative hyaluronidase genes (nagH [14] and nagL [55]) were downregulated. The nanI gene, encoding the major C. perfringens sialidase NanI (18), was overexpressed significantly in the revR mutant, while the gene encoding the minor sialidase NanJ (18) was downregulated significantly in the mutant (Table 2). Finally, expression of the two major virulence factor genes, plc (alpha-toxin) and pfoA (perfringolysin O), was unaltered in the revR mutant, which was consistent with the alpha-toxin and perfringolysin O assay data.
Table 2.
Relevant genes differentially expressed in the revR mutanta
| Category | Gene name | Locus tag | Fold ratiob | P valuec | Function |
|---|---|---|---|---|---|
| Upregulated genes | nanI | CPE0725 | 23.9 | 1.8E−05 | Sialidase (NanI) |
| CPE0185 | 16.9 | 8.5E−06 | Acylneuraminate lyase (sialic acid lyase) | ||
| colA | CPE0173 | 7.02 | 4.1E−04 | Collagenase (kappa-toxin) | |
| ccp | CPE0846 | 6.1 | 5.0E−03 | α-Clostripain | |
| Downregulated genes | nagH | CPE0191 | 0.245 | 0.031 | Hyaluronidase (mu-toxin) (NagH) |
| nanJ | CPE0553 | 0.127 | 3.3E−04 | Sialidase (NanJ) | |
| nagL | CPE1523 | 0.117 | 8.9E−05 | Hyaluronidase (mu-toxin) (NagL) | |
| CPE1423 | 0.253 | 8.8E−04 | Small acid-soluble spore protein C2 | ||
| CPE1753 | 0.081 | 2.39E−05 | Stage IV sporulation protein A | ||
| CPE2218 | 0.062 | 2.4E−06 | Probable spore coat protein | ||
| Other virulence genes | pfoA | CPE0163 | 1.16 | 0.72 | Perfringolysin O (theta-toxin) |
| plc | CPE0036 | 1.1 | 0.733 | Phospholipase C (alpha-toxin) |
RNA was extracted from 4-h exponential-phase cultures. Microarrays were hybridized with cDNA generated from four independent biological replicates with two dye swaps.
Expressed as a fraction of the wild-type expression level.
Determined using a modified t test algorithm.
Complementation restores virulence gene expression to levels similar to those in the wild type.
To validate the microarray data, the revR mutation was complemented in trans with the wild-type revR gene, and the expression levels of several potential virulence genes were determined by QRT-PCR. The expression of the pfoA gene was monitored as a control; the results (Fig. 2 A) showed that pfoA expression was not statistically different in the wild-type, mutant, and complemented strains, which correlated with the microarray results.
Fig. 2.

Expression of selected genes in the isogenic revR strains. RNA was prepared from cells grown in TPYG broth for 4 h, corresponding to exponential growth phase. QRT-PCR analysis was carried out using primer pairs specific for the ccp and pfoA (A) and the colA, nanI, nagH, nagL, and nanJ (B) genes. Expression levels are normalized to the expression of a control rpoA gene. Values are the averages of three independent biological replicates ± standard errors of the means (SEM). Statistically significant differences (P ≤ 0.05 by Student's t test)) are denoted by an asterisk.
Since many potentially RevR-regulated genes were identified (see Table S2 in the supplemental material), confirmatory QRT-PCR experiments were conducted only on genes encoding potential virulence factors, i.e., the colA, nagH, nagL, nanI, nanJ, and ccp genes. In the revR mutant, the expression levels of all of these genes except colA were significantly different from the respective wild-type levels (Fig. 2), with ccp and nanI expression significantly increased in the mutant and nagH, nagL, and nanJ expression decreased. These results were in agreement with the microarray data. Complementation with the wild-type revR gene restored the expression of these genes to levels similar to those in the wild type (Fig. 2). However, for the colA gene, where microarray analysis showed that the gene was upregulated in the mutant (Table 2), QRT-PCR demonstrated that there was no significant difference in the expression of the colA gene in the mutant compared to the wild type (Fig. 2).
Complementation restored extracellular enzyme activity to levels similar to those in the wild type.
To show that these transcriptional changes resulted in changes in extracellular enzyme production, quantitative hyaluronidase (putatively encoded by nagH, nagL, and three other nag genes [55]), collagenase (colA), protease (ccp), and sialidase (nanI and nanJ) assays were carried out on the isogenic wild-type, mutant, and complemented strains at various growth stages.
Hyaluronidase assays showed that there was a significant decrease in activity in cell lysates from the revR mutant compared to those from the wild-type strain at both 4 h (P = 0.03) and 8 h (P = 0.0006) postinoculation (Fig. 3 A), which correlated with both the microarray and QRT-PCR data. The decreased activity could be restored to levels similar to those for the wild type when the revR mutation was complemented with the intact revR gene (Fig. 3A).
Fig. 3.

Extracellular enzyme production in the isogenic revR strains. Quantitative assays were carried out to determine the relative amount of enzyme activity in 4-h cultures grown in TPYG medium or Todd-Hewitt broth (for sialidase assays only). Enzyme activity was measured using cell lysates for hyaluronidase activity (A) and using culture supernatants for protease activity (B), sialidase activity (C), and collagenase activity (D). All results are the averages of three biological replicates ± SEM; a significant difference as determined by Student's t test (P ≤ 0.05) is indicated by an asterisk.
C. perfringens has been shown to produce a cysteine protease, α-clostripain (64), and two sialidases, NanI and NanJ, with NanI being responsible for the majority of the extracellular sialidase activity (18). The revR mutant showed a significant increase in both protease (Fig. 3B) and sialidase (Fig. 3C) activities compared to the wild type at both 4 h and 8 h, in agreement with both the microarray and QRT-PCR data. This increased activity was restored to wild-type levels upon complementation of the revR mutation. Finally, analysis of extracellular collagenase activity confirmed the QRT-PCT data; collagenase production was not affected by the revR mutation (Fig. 3D).
RevR is required for wild-type virulence in C. perfringens.
To investigate the potential role of the RevR regulon in the pathogenesis of C. perfringens infections, the virulence of the isogenic strains was tested in our standard BALB/c mouse myonecrosis model (30). For each strain, 20 mice were injected in the thigh muscle with cells grown from four independent biological replicates. Mice infected with the wild-type strain typically developed observable signs of disease, such as limping, blackening of the thigh, and blackening of the footpad, at approximately 2 to 3 h postinfection (Fig. 4), with disease symptoms severe enough to require euthanasia for animal ethics reasons at approximately 3 to 4 h postinfection (Fig. 5). In contrast, mice injected with the revR mutant took much longer to develop any signs of disease (Fig. 4), and rarely did the disease severity require euthanasia until after 6 h postinfection (Fig. 5). Kaplan-Meier survival curves showed that the survival of mice infected with the revR mutant was significantly attenuated (as determined by a log rank Mantel-Cox test; P < 0.01) compared to that of mice infected with either the wild-type strain or the complemented revR mutant. Complementation restored the disease progression and severity to wild-type levels, with no significant difference in the survival of mice infected with either the wild-type strain or the complemented mutant being detected (Fig. 5). Blackening of the thigh and footpad, which are qualitative measures of ischemia, was not restored to wild-type levels, presumably because the severity of the other virulence criteria required euthanasia for animal ethics reasons before blackening could occur.
Fig. 4.

Progression of disease symptoms in the isogenic revR strains. Development of limping, swelling of the footpad, blackening of the footpad, and blackening of the thigh is shown. Results are the cumulative scores for 20 infected mice.
Fig. 5.
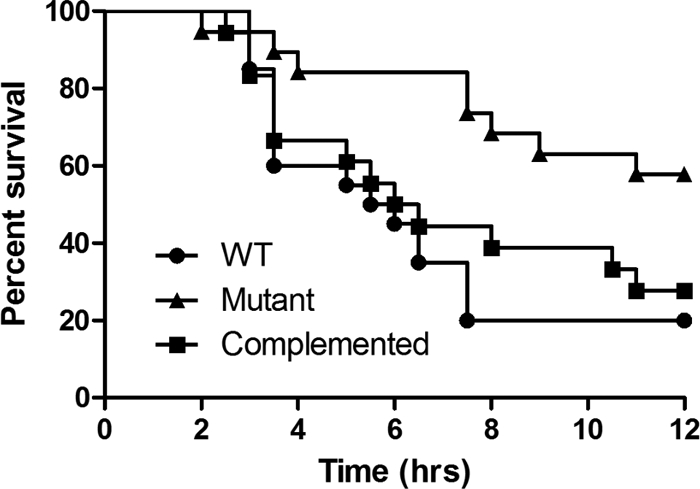
Kaplan-Meier survival curves. Mice (n = 20) were injected with the wild-type, revR, or complemented strain and survival monitored every 30 min for 12 h.
DISCUSSION
In this study we report that a previously unknown response regulator, RevR, regulates extracellular enzyme production and virulence in a manner that is independent of the VirSR two-component signal transduction system, which regulates toxin production either directly via the response regulator VirR (32, 43, 54, 56) or indirectly by regulatory RNA molecules encoded by the vrr, virX, or virT gene (7, 41, 46, 57). Microarray and QRT-PCR experiments confirmed that the expression of genes encoding the putative NagH and NagL hyaluronidases, the NanI and NanJ sialidases, and α-clostripain was altered by the revR mutation, in a manner different from that observed following mutation of virR or vrr (44). The differences in expression of the hyaluronidase, protease, and sialidase structural genes were reflected by differences in the production of these extracellular enzymes. Furthermore, expression of the VirSR-regulated genes plc and pfoA, encoding alpha-toxin and perfringolysin O, respectively, was unaffected by the revR mutation, confirming that gene regulation by RevR is distinct from the VirSR system. The effects on both extracellular enzyme production and virulence were reversed by complementation with the wild-type revR gene, providing evidence that the original phenotypic changes resulted from the revR mutation. RevR represents the first regulatory protein outside the VirSR system to be shown to modulate virulence in C. perfringens. Microarray analysis revealed that the expression of more than 400 genes was altered (P ≤ 0.05) in the revR mutant, making it difficult to determine the gene or genes directly responsible for the change in virulence.
RevR has sequence similarity to PhoB from C. kluyveri (62% amino acid sequence identity) and YycF from B. subtilis (49% identity). These two response regulators generally form part of the classical two-component signal transduction systems PhoBR and YycFG, respectively (31, 68). The predicted RevR sequence contains all the important residues (Asp-11, Glu-12, and Lys-106) required for the formation of an N-terminal phosphoacceptor pocket, with Asp-54 predicted to be the site of phosphorylation. Conserved-domain searches revealed the presence of a potential winged-helix DNA binding domain present within the C-terminal region of RevR, consistent with the DNA binding domain of YycF and PhoB, suggesting that RevR regulates gene expression by binding to a DNA target (45). Modeling of the RevR amino acid sequence on the known structures of the PhoB effector domain from E. coli (11) and on the C-terminal region of YycF from B. subtilis (45) revealed very little difference in the overall structure between the two models (Fig. 6), reflecting the high degree of structural similarity between the two templates (45). The recognition sites of YycF and PhoB also are similar; purified YycF has been shown to bind to known PhoB binding sites (45). Unlike in the phoBR and yycFG systems, no gene encoding a readily identifiable sensor kinase protein could be identified either upstream or downstream of revR. RevR appears to be an orphan response regulator that is phosphorylated by an as-yet-unidentified sensor kinase.
Fig. 6.

Models of the C-terminal domain of RevR. (A and B) The RevR amino acid sequence was modeled on the crystal structure of the PhoB effector domain from E. coli (Protein Data Bank [PDB] template code 1GXQ) (A) or the YycF effector domain from B subtilis (PDB template code 2D1V) (B) using the SWISS-MODEL server (http://swissmodel.expasy.org/). (C) The two hypothetical RevR models are superimposed.
To identify the potential cognate sensor kinase for RevR, BLAST searches using the amino acid sequences of YycG and PhoR, the cognate sensor kinases of YycF and PhoB from B. subtilis, and VicK, the cognate sensor kinase of the related protein VicR from S. pneumoniae, were carried out. These searches identified a putative orphan sensor histidine kinase gene, cpe1757, whose product was previously shown to have 47% identity to PhoR from Clostridium acetobutylicum (20) and which has 25 to 28% amino acid identity to the search proteins YycG, PhoR, and VicK. CPE1757 is predicted to contain one transmembrane domain and HAMP, HisKA, and ATPase domains that are common to all sensor histidine kinases (23). Further experiments involving the purification of both proteins and phosphotransfer studies are required to determine the relationship between RevR and CPE1757, if any.
YycFG (or WalRK) orthologs have been identified in many Gram-positive bacteria, including B. subtilis (19), Enterococcus faecalis (25), Staphylococcus aureus (35), and S. pneumoniae (where they are termed VicKR) (68). Such proteins have been reported to be essential for survival, controlling vital functions such as murein biosynthesis and cell division, as well as virulence factor expression in a number of bacterial species (reviewed in reference 68). Although a yycF or yycG deletion mutant cannot be obtained in B. subtilis (19, 68), when a conditional temperature-sensitive yycF mutant was cultured at a nonpermissive temperature, the cells stopped growing, leading to the accumulation of so-called empty cells (19). In a B. subtilis strain overexpressing yycF, an accumulation of smaller cells was observed (21), which is consistent with the fact that shorter rods also were observed when revR was overexpressed in the revR mutant and suggests that RevR has at least some functional similarity to members of the YycF family. The signal for the YycFG two-component signal transduction pathway of B. subtilis remains to be determined (68).
Genes encoding proteins involved in the formation of the spore coat, septum, and cortex were all downregulated in the revR mutant, in addition to some genes that have been annotated as being associated with the cell wall or cell division. Unfortunately, strain 13 does not sporulate efficiently in culture media, so it was not possible to determine if there was any effect on sporulation. However, phenotypic changes that were related to the cell division process were observed (Fig. 1). A gene encoding a putative rod-shape-determining protein (cpe2143) was downregulated in the revR mutant, and genes encoding a probable cell wall binding protein (cpe0202) and a capsular polysaccharide biosynthesis protein (cpe0250) were both upregulated. In B. subtilis the YycFG system regulates the expression of the ftsAZ operon (21), which controls cell division (70), but the expression of a putative ftsZ homolog in C. perfringens (cpe1765) was unaffected in our experiments. In S. pneumoniae the filamentous phenotype displayed in a vicRK mutant has been attributed to the downregulation of the putative murein hydrolyase encoded by pcsB (38). The function of PcsB in S. pneumoniae remains unknown, although it is known to be essential for correct cellular division (8, 37). BLAST searches of the C. perfringens genome revealed two potential pcsB homologs, cpe1614 and cpe0202, with low-level (22 to 23%) amino acid sequence identity to PcsB (NP_359612.1) from S. pneumoniae. The expression levels of cpe1614 and cpe0202 were not significantly altered in the revR mutant; therefore, the filamentous phenotype seen in this mutant is presumably the result of an as-yet-unidentified mechanism.
RevR also has similarity to PhoB or PhoP proteins, which are involved in phosphate regulation. The Pho regulon has been identified in many bacterial species but has been studied primarily in E. coli and B. subtilis (28, 31). In E. coli it encompasses no fewer than 47 genes that encode proteins involved in phosphate detection, uptake, and metabolism (26, 31). PhoR is a sensor histidine kinase that interacts with the proteins of the Pst system, including PhoU (31). At low phosphate concentrations, PhoR phosphorylates its cognate response regulator PhoB (known as PhoP in B. subtilis), which directly regulates the transcription of the pho regulon (28). In C. perfringens, revR is situated directly downstream of a putative phosphate uptake and metabolism operon, which encompasses genes encoding a potential phosphate transporter system that is composed of the PstABCS proteins, as well as a putative PhoU accessory protein (28, 31). The genetic organization of this gene region, together with the putative structural similarity of RevR to PhoB, suggests that RevR may also function in a phosphate-dependent manner.
In summary, it appears that in C. perfringens the RevR protein, and its putative cognate sensor histidine kinase, may have functional properties that are carried out by both the YycFG (WalRK) and the PhoBR systems in other bacteria. Deletion of the revR gene leads to a cellular morphology that is similar to that of a VicKR mutant of Streptococcus pyogenes (37), and the genetic organization of the revR gene suggests that it is involved in the regulation of phosphate metabolism. Recent studies have shown that the Pho regulon is involved in virulence in Citrobacter rodentium (15). In C. perfringens, RevR regulates virulence in the mouse myonecrosis model by a mechanism that was previously unrecognized in this bacterium. It has been known for some time that the regulation of virulence in C. perfringens was a complex process (42, 46, 50); in this study we have identified yet another layer of hitherto unknown complexity.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by a grant from the Australian National Health and Medical Research Council. T.J.H. was a recipient of an Australian Postgraduate Award.
We thank Paul Harrison and Sally Turner from Monash University for their assistance with the microarray experiments and John Emmins for technical advice and assistance.
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 14 March 2011.
REFERENCES
- 1. Altschul S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Awad M. M., Bryant A. E., Stevens D. L., Rood J. I. 1995. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15:191–202 [DOI] [PubMed] [Google Scholar]
- 3. Awad M. M., Ellemor D. M., Boyd R. L., Emmins J. J., Rood J. I. 2001. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 69:7904–7910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Awad M. M., et al. 2000. Construction and virulence testing of a collagenase mutant of Clostridium perfringens. Microb. Pathog. 28:107–117 [DOI] [PubMed] [Google Scholar]
- 5. Bannam T. L., Rood J. I. 1993. C. perfringens-Escherichia coli shuttle vectors that carry single antibiotic resistance determinants. Plasmid 29:223–235 [DOI] [PubMed] [Google Scholar]
- 6. Bannam T. L., Teng W. L., Bulach D., Lyras D., Rood J. I. 2006. Functional identification of conjugation and replication regions of the tetracycline resistance plasmid pCW3 from Clostridium perfringens. J. Bacteriol. 188:4942–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banu S., et al. 2000. Identification of novel VirR/VirS-regulated genes in Clostridium perfringens. Mol. Microbiol. 35:854–864 [DOI] [PubMed] [Google Scholar]
- 8. Barendt S. M., et al. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J. Bacteriol. 191:3024–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ba-Thein W., et al. 1996. The virR/virS locus regulates the transcription of genes encoding extracellular toxin production in Clostridium perfringens. J. Bacteriol. 178:2514–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beier D., Gross R. 2006. Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 9:143–152 [DOI] [PubMed] [Google Scholar]
- 11. Blanco A. G., Sola M., Gomis-Ruth F. X., Coll M. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701–713 [DOI] [PubMed] [Google Scholar]
- 12. Bourret R. B. 2010. Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13:142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calva E., Oropeza R. 2006. Two-component signal transduction systems, environmental signals, and virulence. Microb. Ecol. 51:166–176 [DOI] [PubMed] [Google Scholar]
- 14. Canard B., Garnier T., Saint-Joanis B., Cole S. T. 1994. Molecular genetic analysis of the nagH gene encoding a hyaluronidase of Clostridium perfringens. Mol. Gen. Genet. 243:215–224 [DOI] [PubMed] [Google Scholar]
- 15. Cheng C., et al. 2009. Contribution of the pst-phoU operon to cell adherence by atypical enteropathogenic Escherichia coli and virulence of Citrobacter rodentium. Infect. Immun. 77:1936–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheung J. K., Awad M. M., McGowan S., Rood J. I. 2009. Functional analysis of the VirSR phosphorelay from Clostridium perfringens. PLoS One 4:e5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheung J. K., Rood J. I. 2000. Glutamate residues in the putative transmembrane region are required for the function of the VirS sensor histidine kinase from Clostridium perfringens. Microbiology 146:517–525 [DOI] [PubMed] [Google Scholar]
- 18. Chiarezza M., et al. 2009. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect. Immun. 77:4421–4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fabret C., Hoch J. A. 1998. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J. Bacteriol. 180:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer R.-J., Bahl H. 2005. Transport of phosphate, p. 287–294 In Durre P. (ed.), Handbook on Clostridia. CRC Press, Boca Raton, FL [Google Scholar]
- 21. Fukuchi K., et al. 2000. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology 146:1573–1583 [DOI] [PubMed] [Google Scholar]
- 22. Galperin M. Y. 2010. Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 13:150–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao R., Stock A. M. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao R., Stock A. M. 2010. Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13:160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hancock L. E., Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J. Bacteriol. 186:7951–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris R. M., Webb D. C., Howitt S. M., Cox G. B. 2001. Characterization of PitA and PitB from Escherichia coli. J. Bacteriol. 183:5008–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoch J. A. 2000. Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol. 3:165–170 [DOI] [PubMed] [Google Scholar]
- 28. Hsieh Y. J., Wanner B. L. 2010. Global regulation by the seven-component Pi signaling system. Curr. Opin. Microbiol. 13:198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoue H., Nojima H., Okayama H. 1990. High-efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28 [DOI] [PubMed] [Google Scholar]
- 30. Kennedy C. L., et al. 2009. Cross-complementation of Clostridium perfringens PLC and Clostridium septicum alpha-toxin mutants reveals PLC is sufficient to mediate gas gangrene. Microbes Infect. 11:413–418 [DOI] [PubMed] [Google Scholar]
- 31. Lamarche M. G., Wanner B. L., Crepin S., Harel J. 2008. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 32:461–473 [DOI] [PubMed] [Google Scholar]
- 32. Lyristis M., et al. 1994. Identification and molecular analysis of a locus that reguates extracellular toxin production in Clostridium perfringens. Mol. Microbiol. 12:761–777 [DOI] [PubMed] [Google Scholar]
- 33. Marchler-Bauer A., et al. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marchler-Bauer A., Bryant S. H. 2004. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32:W327–W331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin P. K., Li T., Sun D., Biek D. P., Schmid M. B. 1999. Role in cell permeability of an essential two-component system in Staphylococcus aureus. J. Bacteriol. 181:3666–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morelle G. 1989. A plasmid extraction procedure on a miniprep scale. Focus 11:7–8 [Google Scholar]
- 37. Ng W.-L., et al. 2003. Constitutive expression of PcsB suppresses the requirement for the essential VicR (YycF) response regulator in Streptococcus pneumoniae R6. Mol. Microbiol. 50:1647–1663 [DOI] [PubMed] [Google Scholar]
- 38. Ng W. L., Kazmierczak K. M., Winkler M. E. 2004. Defective cell wall synthesis in Streptococcus pneumoniae R6 depleted for the essential PcsB putative murein hydrolase or the VicR (YycF) response regulator. Mol. Microbiol. 53:1161–1175 [DOI] [PubMed] [Google Scholar]
- 39. Ng W. L., Tsui H. C., Winkler M. E. 2005. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J. Bacteriol. 187:7444–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. O'Connor J. R., et al. 2006. Construction and analysis of chromosomal Clostridium difficile mutants. Mol. Microbiol. 61:1335–1351 [DOI] [PubMed] [Google Scholar]
- 41. Ohtani K., Bhowmik S. K., Hayashi H., Shimizu T. 2002. Identification of a novel locus that regulates expression of toxin genes in Clostridium perfringens. FEMS Microbiol. Lett. 209:113–118 [DOI] [PubMed] [Google Scholar]
- 42. Ohtani K., et al. 2010. Identification of a two-component VirR/VirS regulon in Clostridium perfringens. Anaerobe 16:258–264 [DOI] [PubMed] [Google Scholar]
- 43. Ohtani K., Kawsar H. I., Okumura K., Hayashi H., Shimizu T. 2003. The VirR/VirS regulatory cascade affects transcription of plasmid-encoded putative virulence genes in Clostridium perfringens strain 13. FEMS Microbiol. Lett. 222:137–141 [DOI] [PubMed] [Google Scholar]
- 44. Ohtani K., et al. 2009. Virulence gene regulation by the agr system in Clostridium perfringens. J. Bacteriol. 191:3919–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okajima T., et al. 2008. Response regulator YycF essential for bacterial growth: X-ray crystal structure of the DNA-binding domain and its PhoB-like DNA recognition motif. FEBS Lett. 582:3434–3438 [DOI] [PubMed] [Google Scholar]
- 46. Okumura K., Ohtani K., Hayashi H., Shimizu T. 2008. Characterization of genes regulated directly by the VirR/VirS system in Clostridium perfringens. J. Bacteriol. 190:7719–7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ren B., Szalai A. J., Hollingshead S. K., Briles D. E. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun. 72:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robinson V. L., Buckler D. R., Stock A. M. 2000. A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 7:626–633 [DOI] [PubMed] [Google Scholar]
- 49. Rood J. I. 1983. Transferable tetracycline resistance in Clostridium perfringens strains of porcine origin. Can. J. Microbiol. 29:1241–1246 [DOI] [PubMed] [Google Scholar]
- 50. Rood J. I. 1998. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52:333–360 [DOI] [PubMed] [Google Scholar]
- 51. Rood J. I., Cole S. T. 1991. Molecular genetics and pathogenesis of Clostridium perfringens. Microbiol. Rev. 55:621–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 53. Scott P. T., Rood J. I. 1989. Electroporation-mediated transformation of lysostaphin-treated Clostridium perfringens. Gene 82:327–333 [DOI] [PubMed] [Google Scholar]
- 54. Shimizu T., Ba-Thein W., Tamaki M., Hayashi H. 1994. The virR gene, a member of a class of two-component response regulators, regulates the production of perfringolysin O, collagenase, and hemagglutinin in Clostridium perfringens. J. Bacteriol. 176:1616–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shimizu T., et al. 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. U. S. A. 99:996–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shimizu T., et al. 2002. Proteome and transcriptome analysis of the virulence genes regulated by the VirR/VirS system in Clostridium perfringens. J. Bacteriol. 184:2587–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shimizu T., Yaguchi H., Ohtani K., Banu S., Hayashi H. 2002. Clostridial VirR/VirS regulon involves a regulatory RNA molecules for expression of toxins. Mol. Microbiol. 43:257–265 [DOI] [PubMed] [Google Scholar]
- 58. Sloan J., et al. 1992. Construction of a sequenced Clostridium perfringens-Escherichia coli shuttle plasmid. Plasmid 27:207–219 [DOI] [PubMed] [Google Scholar]
- 59. Smyth G. K. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:1–25 [DOI] [PubMed] [Google Scholar]
- 60. Smyth G. K., Michoud J., Scott H. S. 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21:2067–2075 [DOI] [PubMed] [Google Scholar]
- 61. Songer J. G. 2010. Clostridia as agents of zoonotic disease. Vet. Microbiol. 140:399–404 [DOI] [PubMed] [Google Scholar]
- 62. Songer J. G. 1997. Clostridial diseases of animals, p. 153–182 In Rood J. I., McClane B. A., Songer J. G., Titball R. W. (ed.), The clostridia: molecular biology and pathogenesis. Academic Press, San Diego, CA [Google Scholar]
- 63. Stock A. M., Robinson V. L., Goudreau P. N. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183–215 [DOI] [PubMed] [Google Scholar]
- 64. Tanaka H., et al. 2008. Construction and characterization of a clostripain-like protease-deficient mutant of Clostridium perfringens as a strain for clostridial gene expression. Appl. Microbiol. Biotechnol. 77:1063–1071 [DOI] [PubMed] [Google Scholar]
- 65. Thompson J. D., Higgins D. G., Gibson T. J. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ulrich L. E., Zhulin I. B. 2007. MiST: a microbial signal transduction database. Nucleic Acids Res. 35:D386–D390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wagner C., et al. 2002. Genetic analysis and functional characterization of the Streptococcus pneumoniae vic operon. Infect. Immun. 70:6121–6128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Winkler M. E., Hoch J. A. 2008. Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in gram-positive bacteria. J. Bacteriol. 190:2645–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wuichet K., Cantwell B. J., Zhulin I. B. 2010. Evolution and phyletic distribution of two-component signal transduction systems. Curr. Opin. Microbiol. 13:219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Young K. D. 2010. Bacterial shape: two-dimensional questions and possibilities. Annu. Rev. Microbiol. 64:223–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.