

Abstract
There are many questions in brain science, which are extremely interesting but very difficult to answer. For example, how do education and other experiences during human development influence the ability and personality of the adult? The molecular mechanisms underlying such phenomena are still totally unclear. However, technological and instrumental advancements of electron microscopy have facilitated comprehension of the structures of biological components, cells, and organelles. Electron crystallography is especially good for studying the structure and function of membrane proteins, which are key molecules of signal transduction in neural and other cells. Electron crystallography is now an established technique to analyze the structures of membrane proteins in lipid bilayers, which are close to their natural biological environment. By utilizing cryo-electron microscopes with helium cooled specimen stages, which were developed through a personal motivation to understand functions of neural systems from a structural point of view, structures of membrane proteins were analyzed at a resolution higher than 3 Å. This review has four objectives. First, it is intended to introduce the new research field of structural physiology. Second, it introduces some of the personal struggles, which were involved in developing the cryo-electron microscope. Third, it discusses some of the technology for the structural analysis of membrane proteins based on cryo-electron microscopy. Finally, it reviews structural and functional analyses of membrane proteins.
Keywords: structural physiology, electron crystallography, high resolution electron microscopy, cryo-electron microscope, water channel, ion channel, gap junction channel, membrane proteins
Introduction
For understanding brain functions and also other biological functions, ion channels are crucially important and Roderick MacKinnon's group published many important papers based on X-ray crystallography. These studies made the subject of “structural physiology” meaningful through their structural and functional studies of K+ channels.1–5 We are struggling to study structure and function of bacterial and mammalian Na+ channels, which are similarly important voltage-gated cation channels. Low resolution structure analysis by the single particle method using cryo-electron microscopy (cryo-EM) revealed the complex structure of the voltage-sensitive Na+ channel purified from the electric organ of electric eel.6 Although no three-dimensional (3D-) structure has been determined yet for bacterial Na+ channels, mobility of voltage-sensor domains has been observed mainly by electrophysiology.7,8 Simultaneously, we are studying water channels, which should be important for brain functions, because water content in brain is, remarkably, up to 85% and ion permeation has to be blocked to ensure the ion channel function, even when water molecules rapidly pass through the channels.
A predominant water channel in brain is aquaporin-4 (AQP4). However, its physiological function is not clear. Structural and functional analyses of AQP4 revealed that this water channel has a weak cell adhesive property.9 We therefore named such channels as belonging to the “adhennel” family (obtained by concatenating the words, adhesive and channel). We are very much interested in this type of multifunctional channel because adhennels could be responsible for complex biological functions, such as higher order brain functions and also other biological functions. A typical adhennel could be a gap junction channel.
Electron Crystallography
For structure analyses of membrane proteins such as ion channels and water channels, electron crystallography is a particularly good candidate for the following several reasons.
Structures can be analyzed in membranes, which provide an environment for membrane proteins similar to the native condition. This is shown in the structure analysis of bacteriorhodopsin together with lipid molecules (Fig. 1).10
Structure can be analyzed even using poor crystals, although the resolution is strongly related to the quality of crystals. For example, the structure of proton pump, H+, K+-ATPase was analyzed only from electron micrographs of rather small crystals, which gave no electron diffraction patterns (Fig. 2).11
Both sides of the specimen are kept open, and there can be less influence from artificial crystal packing as shown later for the structure analysis of gap junction channel. Furthermore, this feature enables researchers to challenge the freeze-trapping technique, which was developed and actually used for studying gating mechanism of Nicotinic Acetylcholine receptor (AChR) by N.Unwin.12
Phases for structure analysis are calculated directly from images and give a better map than that of X-ray crystallography when the quality of maps is compared at a same resolution.
Figure 1.
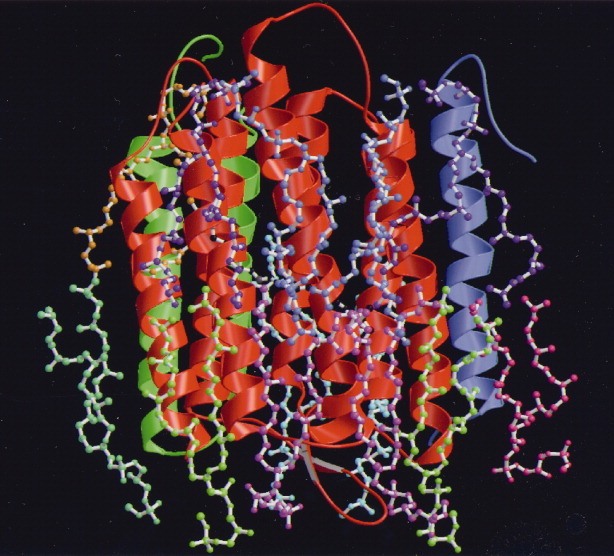
Structure of bacteriorhodopsin together with lipid molecules analyzed by electron crystallography. Figure is represented based on Ref. 10.
Figure 2.
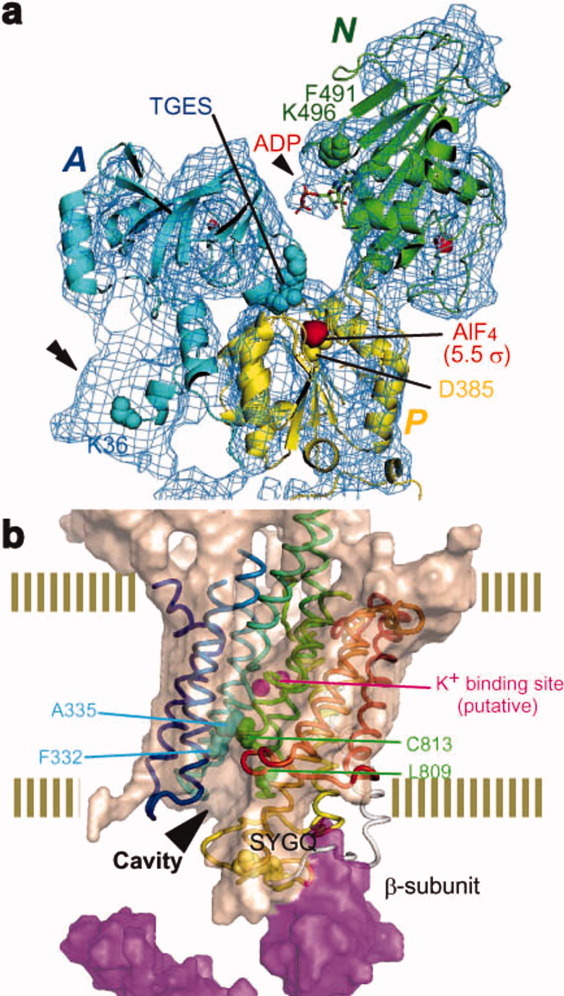
Structure of H+K+-ATPase analyzed only by image data based on electron crystallography. Crytoplasmic structure (a). Trans-membrane structure together with luminal structure (b). Figures are represented based on Ref.11.
It was electron crystallography that was used for the first time to obtain a real image of a membrane protein, bacteriorhodopsin whose structure was analyzed by Henderson and Unwin in 1975.13 On one hand, this method is extremely powerful especially for structural study of membrane proteins and Henderson et al.14 determined for the first time an atomic structure of bacteriorhodopsin based on electron crystallography. On the other hand, this method has been rather slowly spread in structural biology presumably because of technological difficulties as well as the difficulty of crystallizing membrane proteins. I however believe electron crystallography could be the core method for establishing structural physiology because it enables us to analyze the functional structure of membrane proteins in membrane layers at a high resolution at which we can discuss physiological functions based on detailed structures.
High Resolution Electron Microscopy
When I started to learn electron microscopy, researchers in the electron microscopic field argued whether electron microscopy could give an atomic image. We therefore tried to confirm that we could take an atomic image without any ambiguity. For an imaging sample, we used crystals of rather complex organic compounds, although many electron microscopists used inorganic crystals or just dried droplets of heavy atom solutions, presumably because these samples are much easier than organic chemicals to observe in an electron microscope. After many trials, we could observe an image of chlorinated copper phthalocyanine in which the copper atoms, porphyrin rings, and chlorine atoms are clearly discriminated (Fig. 3).15 Although this image proved that electron microscopy could give an atomic image, I took huge numbers of images to get just one best (Scherzer's) focus image, because I had to adjust the focus with a very dark electron beam. The image in Figure 3 is one image taken by chance after many trials.
Figure 3.

High resolution image of chlorinated copper phthalocyanine by which electron microscopy was proved to give an atomic image. The chemical structure is indicated in the image. Figure is related with Ref.15.
To overcome such difficulty, we developed a system for focusing without extra beam on the imaging specimen area and named the system Minimum Dose System (MDS). High resolution images of radiation sensitive organic molecule, such as silver-7, 7′, 8, 8′-tetracyanoquinondimethan complex, Ag-TCNQ, were easily and reproducibly observed by the MDS.16 Unlike organic molecules, biological molecules are extremely radiation sensitive, and the resolution of their images is therefore strongly restricted by radiation damage.
Radiation Damage and Cryo-protection
For high resolution structural study, the radiation and dehydration sensitivities of biological molecules give us a difficult challenge and force us to develop an effective and stable cryo-electron microscope with a helium cooled specimen stage and a cryo-transfer system for ice-embedded samples. The dehydration problem was overcome by the ice-embedding method of the rapid freezing technique.17 For transferring an ice-embedded sample to the cryo-stage of the electron microscope, a cryo-transfer system, which kept the ice-embedded sample at a lower temperature than 140 K (the phase change temperature of amorphous ice) had to be developed. Therefore, we developed a cryo-transfer system for cryo-EM. The remaining most serious problem for high resolution observation of biomolecules is therefore radiation damage. The electron beam inevitably causes breakage of chemical bonds of organic and biological materials. Subsequently, the produced radicals may attack circumference bonds and/or molecular fragments can diffuse away, leading to collapse of the initial structure. Therefore, the radiation damage could effectively be reduced by minimizing movement of the molecular fragments. Minimization of the movement of atoms and fragments may also increase the population of reforming bonds. After many attempts to find a solution to this difficult problem, only cooling of the biological specimen appears to reduce the radiation damage by limiting the diffusion of atoms and molecular fragments.
The irradiation damage to crystals of biological molecules was found to be reduced to about 1/10 and 1/20 of the damage at room temperature when the specimen was cooled below 20 K and 8 K, respectively (Fig. 4). The lattice heat capacity of all materials becomes very small at very low temperature such as liquid helium temperature. The heat conductivity of non-metals is dramatically reduced at such low temperature. Therefore, the temperature of the specimen could rise rapidly in the area being irradiated by electron beam, which tend to change to thermal energy by interacting with specimen. To confirm the actual temperature at the irradiated site, we developed an instrument to introduce gases, such as N2 and Ne. We can then estimate the specimen temperature by recording the electron diffraction patterns of solid N2 and Ne. This indicated that the specimen temperature must have been lower than 20 K and 8 K, respectively, even under the condition used to record the electron diffraction patterns by which we measured radiation damage of crystals of biological molecules. The temperatures indicated in Figure 4 were actually measured by this system.
Figure 4.
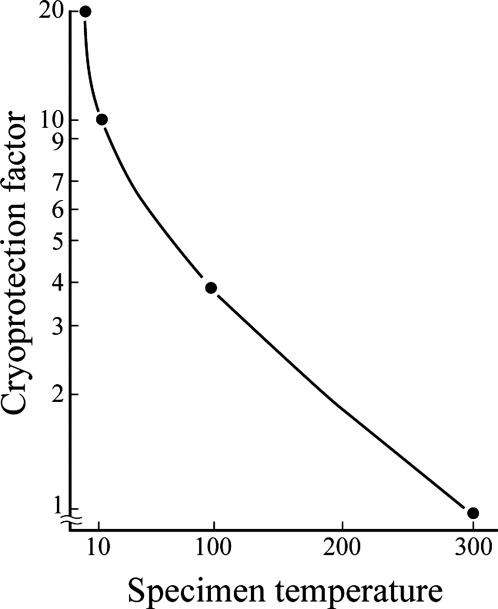
Cryo-protection factors and specimen temperature. Radiation damage of biological specimens is reduced to 1/10 and 1/20 at specimen temperature below 20 K and 8 K, respectively, compared with the damage at room temperature. Figure is represented based on Ref.20.
Based on these observations, we intended to develop an electron cryo-microscope equipped with a specimen stage cooled by liquid helium. Mechanical vibrations; however, could be caused by the boiling of the coolants, liquid N2, and liquid He. Specimen drift induced by temperature changes in the specimen stage also presented difficulty. These problems had to be overcome in the design of a high-resolution cryo-EM.
Development of the Cryo-electron Microscope
For high-resolution data collection, 2D-crystals are embedded in a thin layer of amorphous ice and/or a sugar solution for which trehalose is recommended.18 The specimen is then mounted on the cryo-stage of an electron microscope using a cryo-transfer device. In this case, the thickness of the ice layer has to be optimized for high resolution data collection, because the flatness of 2D-crystals is crucially important especially for data collection with the specimen tilted relative to the electron beam. To make the crystal flat, a thinner ice layer is better, but in contrast, such a thin layer causes dehydration of the sample. The best specimen preparation condition may only occur for a small part of the target and thus many trials are generally required for high-resolution data collection, especially at resolution higher than 3 Å. Without many trials, it is impossible to accurately regulate the best thin layer formation with a less dehydrated condition because sample condition and other conditions, such as density of crystals and temperature as well as humidity, easily change the thickness of water layer on the specimen grid. Specimen preparation for cryo-EM therefore requires us to make many trials to get the very best specimen for high resolution data collection. To try many specimen preparation conditions, cryo-EM with a quick specimen exchange device and a stable specimen stage is inevitable. We then developed a cryo-electron microscope with a stable helium stage as well as an effective cryo-transfer system by which we can exchange and take an image of a new sample at 2 Å resolution just after finishing observation of the previous specimen in shorter than 10 min. Therefore my own record of specimen exchange frequency in a day was 34 times.
The resolution of an image of a biological macromolecule is usually limited to a value much worse than 3 Å due to radiation damage. We can however make an atomic model if we can achieve three-dimensional structure analysis at higher resolution than 3 Å. We have therefore developed a super fluid helium stage that achieved a resolution of 2.6 Å in 1991.19 Thermal shield by liquid Nitrogen and Helium tank is gold plated to minimize radial heat (Upper right in Fig. 5). Small liquid Helium container, which was set at the center of the stage, was connected from the Helium tank by a capillary and named as pot (Upper right in Fig. 5). The pot cooling specimen is cooled down to 1.5 K by superfluid Helium, and we call this as 1.5 K pot. Although our target resolution of electron micrographs is something like 3 Å, an instrument yielding better resolution would be highly beneficial, because biological molecules consist mainly of light atoms that exhibit small atomic scattering factors in a high-resolution range. Higher resolution close to 2.5 Å might be required to identify water molecules. From the second-generation cryo-EM, we made quite a big effort to improve resolution as well as operation of the cryo-EM with a top-entry type helium stage. Then we improved the instrumental resolution to 2.0 Å and overcame the operational difficulties.20 We could develop the third generation cryo-EM, which is a mature instrument for electron crystallography with very stable cryo-stage. We developed an effective cryo-transfer system and modified the system for ice embedded specimens to an automatic one which is user-friendly. After the third generation cryo-EM, the fourth generation cryo-EM was equipped with the automatic cryo-transfer system. The fifth generation cryo-EM was also developed for mainly single particle method. Recently we developed a new cryo-microscope, the seventh generation cryo-EM with an outer-control tilting device for electron tomography and some other analyses (Fig. 5).
Figure 5.

History of development of cryo-electron microscopes with helium stage. From first generation cryo-EM to seventh generation one. Upper right figure shows schematic construction of the helium stage.
Beam Induced Image Shift and Techniques for Specimen Preparation
When we attempted structure analysis of bacteriothodpsin, we encountered a serious difficulty of image shift, which could be induced by specimen charging, especially under tilted specimen conditions. We then took more than 10,000 bad images to get only about 200 good images, which was a terribly tedious task. This was the most serious problem for image data collection from the two-dimensional crystals of biological macromolecules. The image shift extinguishes optical diffraction spots perpendicular to the tilt axis even in a medium or low resolution area. Almost all of the images from a tilted specimen prepared on a one-layer carbon support tend to be deteriorated by the image shift caused by beam-induced charging on specimen. The success ratio for obtaining high resolution images from tilted specimens is therefore only about 2%. To overcome the serious problem, the carbon sandwich preparation method, in which crystals are put between two sheets of carbon film, was investigated. When we used carbon sandwiched specimens, the ratio of images which showed deterioration in diffraction patterns due to the image shift was significantly decreased when compared with specimens supported by a single carbon film. Thus, the carbon sandwich preparation method was confirmed to overcome the most difficult problem and contribute to more prompt structural analysis by electron crystallography.21
Another good side effect of the carbon sandwich technique was observed for AQPO, where the dehydration problem was minimized and the structure analyzed at 1.9 Å.22 At such a high resolution, lipid molecules together with water molecules in the AQP0 crystal were clearly discriminated.
Structure Analyses of Membrane Proteins by Electron Crystallography
Water channel aquaporin-1
Water is the most abundant molecule in any cells on the earth. Ordinary cell membranes therefore require an effective water channel function. In 1992, a 28 kD membrane protein, which was eventually named AQP1, was identified in red blood cells and its function as a water channel was clearly shown in Xenopus oocytes.23 The cell membrane exquisitely regulates entry and exit of ions because ion concentration is strongly related to cell signaling. The water channels, therefore, need to maintain ionic conditions in and out of a cell, even while translocating a large amount of water. The pH regulation in the cell was also well known to be crucially important for cell functions such as signal transduction, proliferation, and survival of cells. An atomic model of AQP1 at 3.8 Å resolution by electron crystallography gave answers to such puzzling questions and revealed the molecular basis of water selectivity.24 This was also the first atomic-resolution structure of a human membrane protein. To accomplish the effective water channel functions, the structure showed a peculiar conformation which we named the AQP fold, as shown in Figure 6(a).
Figure 6.

Ribbon model of AQP1 structure. The side view of the monomer of AQP1 molecule showing the two short helices colored light blue (HB) and dark orange (HE) (a). Structure of AQP1 representing by four different presentation styles. The tetramer structure is shown from the extra cellular side (b). Schematic drawing of water channel AQP1 explaining water selectivity and fast water permeation (c). These figures were prepared based on Ref.24.
The handedness of aquaporin-1 structure was carefully examined by compare with the data of bacteriorhodopsin structure, which we analyzed at atomic resolution10 and shown to be a right-handed helical bundle based on relatively low resolution of 6 Å.25 The handedness was directly confirmed at a higher resolution of 3.8 Å at which an atomic model was constructed as shown in Figure 6(b) in 2000.24
The critical function of AQP1 is high water permeability, about 3 billion water molecules per channel per second. Almost all residues within a central 20 Å zone in the pore are highly hydrophobic, whereas one might have expected that a water channel would be hydrophilic. A narrow part of the pore, of about 3 Å diameter, is located at the middle of the membrane where loops B and E interact with each other especially with proline 77 and 193 (AQP1) of the conserved NPA sequences in all water channels. In addition to the enormous capacity for water conductance, the AQP1 pore also exhibits marked selectivity. Water molecules were observed to be strongly oriented in the channel interior, through alignment of the short helices which form electric dipoles and cause the water molecules to orient with their oxygen atoms facing the NPA side.24 Two major interaction sites for water molecules were identified inside the water channel: the NPA and Ar/R regions. The two highest enthalpic barriers for water molecules are located directly adjacent to the NPA region. This, together with the water orientation that is also centered here, renders the NPA region a major selectivity filter as shown in Figure 6(c). This feature was named the hydrogen bond isolation mechanism.24 Contiguous hydrogen-bonded water chains are known to be efficient proton conductors. Aquaporin must prevent proton conduction along its pore, to maintain the proton gradient across the cell membrane that is essential for energy transduction and signal regulation. The AQP1 structure revealed unexpected features, such as the right-handed helical bundle and the hydrophobic channel which facilitated water transport. These would have been difficult if not impossible to predict without the structure analysis at high resolution.
Structure Determination of AQP4
AQP4 is the predominant water channel in the mammalian brain and is therefore of great interest. We determined the AQP4 structure by electron crystallography of double-layered, two-dimensional (2D-) crystals, which were prepared from AQP4 molecules expressed in Sf9 cells and purified using a detergent, n-octyl-ß-D-glucopyranoside (OG). Analysis of the AQP4 structure was complicated by variations in the double-layered 2D-crystals in lateral alignment and distance between the two layers. Electron diffraction data were not sufficiently sensitive to select one specific crystal type among different crystal variants, despite the high resolution of 3.2 Å. Phase data extracted from images at medium resolution, on the other hand, were sensitive enough to discriminate between crystals with different arrangements of the two layers. The use of a helium-cooled electron microscope20 and the carbon sandwich specimen preparation technique, which significantly increases the yield of good images,21 allowed us to also take an image of each crystal that produced a high-resolution diffraction pattern. Classification based on the image data, which provided phase information to better than 6 Å, identified one predominant crystal type, which accounted for about 70% of the analyzed crystals that yielded high quality electron diffraction pattern. In this crystal type, the two layers have a spacing of 45 Å (molecule center to molecule center). The final 3.2 Å resolution intensity data set was used to determine the AQP4 structure by molecular replacement using the AQP1 structure. The images (recorded after the corresponding electron diffraction patterns and used for classification) were also used to calculate a density map and confirmed the consistency of our structure.
Glial cells contain characteristic orthogonal arrays in the plasma membrane, which are especially prominent in glial end feet surrounding vascular capillaries in the brain. Immunogold labeling experiments showed that these arrays consist of AQP4.26 While AQP4 and AQP1 both function as very fast water-selective pores, AQP4 has distinctive biological characteristics as it forms orthogonal arrays in intact membranes. Furthermore, AQP4 exists in glial cells as a full-length protein starting with Met1 (AQP4M1) and an alternative shorter splicing isoform that starts with Met23 (AQP4M23). Freeze fracture replica labeling technique allows us to discuss how the expression of the long AQP4 splicing variant can inhibit the orthogonal array formation of the short splicing variant.27
AQP4 is also expressed in glial lamellae of the hypothalamus, where it may play a role in osmo-, thermo-, and glucose-sensing. In glial lamellae, the plasma membrane forms large junctions between individual layers, which have been shown to contain AQP4. Interestingly, another water channel, lens-specific AQP0, forms the “thin junctions” between fiber cells. Structure analysis of AQP4 by double layered 2D-crystals revealed that the molecule contains a short 310 helix in an extracellular loop, which mediates weak but specific interactions between AQP4 molecules in adjoining membrane.9 This finding suggests a previously unexpected role for AQP4 in cell adhesion as shown in Figure 7(a). This notion was corroborated by expression of AQP4 in L-cells, which resulted in clustering of the cells. Our AQP4 structure thus enables us to propose models for the size regulation of orthogonal arrays and channel-mediated cell adhesion as observed in glial lamellae of the hypothalamus. AQP4 membrane junctions may reduce the water permeability of glial cell plasma membranes, because the tight tongue-into-groove packing of the two crystalline layers results in a partial blockage of the extracellular pore entrances. While the packing of the AQP4 tetramers in the junctions must create resistance for water flowing across the two membranes, rapid water flow through the channels may also reduce the adhesion between adjoining membranes. This may establish the basis for a role of AQP4 in osmo-sensing. For example, a high AQP4M1/AQP4M23 expression ratio would produce small AQP4 arrays providing weak adhesion between membranes, which could easily be separated and thus react to small water flows resulting from small osmotic differences. A low AQP4M1/AQP4M23 expression ratio, on the other hand, would result in extensive AQP4 arrays providing relatively strong adhesion between membranes that would withstand large water flows associated with large osmotic differences. Although further experiments are needed to elucidate the interplay of the two functions in aquaporins and potentially other membrane channels with adhesive properties, we propose the name “adhennels” for “adhesive water and ion channels.”9
Figure 7.
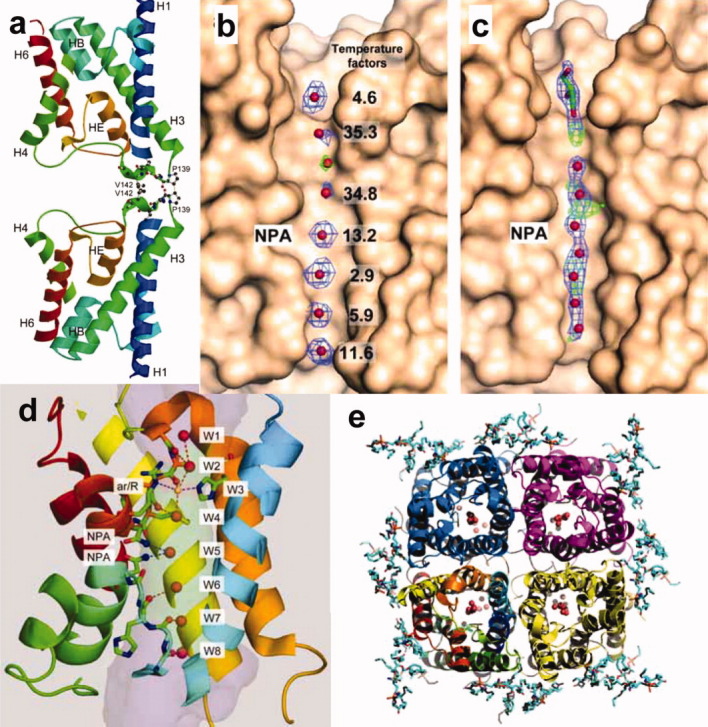
Adhesive structure of AQP4. Two adjoining molecules form an adhesive interaction via 310 helices (a). Figure of (a) is prepared based on Ref. 9. Structural map densities of water molecules in the water channel of AQP4 analyzed by electron crystallography (b) and X-ray crystallography (c). Electron crystallography could give discriminated densities of water molecules in the channel, while X-ray could give only blurred water densities even at higher resolution, presumably because the structure of AQP4 molecules by EM was analyzed in membrane but by X-ray it was done in micelles (b, c). Figures of (b) and (c) were prepared based on Refs. 28 and 29, respectively. Eight water molecules in the AQP4 channel analyzed by electron crystallography at 2.8 Å resolution. The single file arrangement of these water molecules explains the Hydrogen bond isolation mechanism (d). Figure of (d) is represented based on Ref. 28. The end-on view of AQP4 tetramer. Water molecules in the channel and lipid molecules at around the tetramer are observed (e). Figure of (e) is prepared based on Ref. 28.
Higher Resolution Analysis of AQP4
Based on the electron crystallographic structure of AQP1, the hydrogen bond isolation mechanism was proposed to explain how AQPs are impermeable to protons despite their very fast water conduction. The mechanism by which AQPs exclude protons, however, is controversial. Therefore, we present the structure of AQP4 by electron crystallography of double-layered two-dimensional crystals at a resolution of 2.8 Å. The improvement in data quality makes it possible to identify seven individual water molecules in the channel as shown in Figure 7(b).28 In contrast, a map from X-ray crystallography showed only blurred densities of water molecules in the channel even at 1.8 Å resolution [Fig. 7(c)].29 In addition, the Fo-Fc map showed additional spherical density at the ar/R constriction site [the green cage in Fig. 7(b)]. Since the side chains of AQP4 around the ar/R region were represented by clear density and the atoms of the protein molecule had low temperature factors in this region, we assigned an eighth water molecule to the spherical density at the ar/R site [Fig. 7(b) and W3 indicated in yellow in Fig. 7(d)]. The narrow diameter at this constriction would make it an unfavorable position for a water molecule, potentially explaining the weak density at this position. The two neighboring water molecules on either side of the ar/R constriction, which form hydrogen bonds with the unstable water molecule in the constriction, showed higher temperature factors (35 Å2) compared with those of all the other water molecules in the channel (2.9 Å2 to 13.2 Å2) [Fig. 7(b)].
In X-ray crystallography, a resolution of 2.8 Å would be considered too low to see water and lipid molecules, but our density map resolved all water molecules in the channel as well as lipid molecules as shown in Figure 7(e). The B-factors of the water molecules in the pore are lower than 40 Å2 [Fig. 7(b)], significantly lower than those of water molecules seen in membrane protein structures determined by X-ray crystallography. To analyze the 3D structure of a membrane protein by electron crystallography, data have to be collected from hundreds of 2D crystals. This has been considered a disadvantage of electron crystallography, however, our resultant density map clearly, and counter-intuitively, resolved eight spherical densities that could be assigned to water molecules [Fig. 7(b) and Fig. 7(d)]. The perceived weakness of electron crystallography that data are collected from many different crystals may in fact work in its favor. We collected more than a thousand diffraction patterns but selected only the 199 very best patterns,28 so that the final data set includes only information from the very best crystals. Furthermore, while we saw diffraction spots to a resolution of 1.9 Å, we truncated the resolution of our density map to 2.8 Å. Due to these procedures, the final lattice line data were of exceptionally high quality.
Although we tried to prepare the specimens as consistently as possible during the months of data collection, there must have been variations among the specimens. As a consequence, our map represents an average of the specimen under various, slightly different conditions. As we were still able to resolve water molecules in the channel, these are presumably in defined positions. This notion is corroborated by the low temperature factors of the waters, ranging from 2.9 to 35.3 Å2, which are significantly lower than those of the surrounding peptide main chain atoms. The same phenomenon was observed in the 1.9 Å electron crystallographic structure of AQP0.22 The B-factor of the water molecule associated with the amide groups of the NPA motifs is 13.2 Å2, while those of the surrounding main chain atoms are in the range of 30-40 Å2 [Fig. 7(b)]. The large difference in the B factors between the water and protein atoms might be due to the characteristic features of electron crystallography and/or the enhancement of the effect of the helical dipole moments due to the lipid environment. By electron crystallography, lipid molecules were observed as shown in Figure 7(e). In X-ray structures of AQPs in detergent micelles, the water molecules sometimes also displayed lower temperature factors compared with those of the surrounding main chain atoms, but the difference is usually less than 20 Å2. Another possible reason why we could observe water molecules in the AQP4 channel could be attributed to the data collection at liquid helium temperature. We collected all data for this structure analysis at stage temperature of 4.2K and such low temperature could give clearly the lowest energy positions for water molecules in the channel.
The eight water molecules in the AQP4 channel are in a single-file arrangement as indicated by W1 to W8 in Figure 7(d). From the measured distances between successive water molecules in the channel, all water molecules appear to form hydrogen bonds with their neighbors [red dotted lines in Fig. 7(d)], except for the water at the NPA site and the one below it. These two water molecules (W5 and W6) thus seem to be separated from the other water molecules in the channel, lending support to the hydrogen bond isolation mechanism. The inside surface of water channels is largely hydrophobic except for the lining hydrophilic spots formed by the oxygen atoms of the main chain carbonyl groups of Gly209, Ala210, Ser211, His95, Gly94, and Gly93 and the nitrogen atoms of the side chain amide groups of Asn213 and Asn97 of the NPA motifs.28 The line of mechanically stable carbonyl groups provides “a guide rail” of hydrogen bonding partners for the hydrogen atoms of the permeating water molecules as illustrated in Figure 7(d). Each water molecule in the single file can thus form two or three hydrogen bonds. Since water in bulk solution usually forms three or four hydrogen bonds with neighboring water molecules, water molecules entering the channel only have to sacrifice a single hydrogen bond, an energy cost of about 3 kcal. The arrangement of carbonyl and amide groups in the AQP4 channel thus dramatically lowers the energy barrier for water molecules entering the narrow AQP channel and allows for the very fast water permeation through the otherwise hydrophobic channel.
The NPA motifs and the arrangement of the carbonyl groups in the hydrophobic channel together with the arrangement of the two short pore helices HB and HE are crucially important to break the hydrogen bond network, which prevents proton conduction while maintaining fast water permeation. The ar/R constriction site might be important for blocking H3O+ but not for the separation of hydrogen bonds. Our higher-resolution structure of AQP4 supports the hydrogen bond isolation mechanism, which has previously been proposed based on the lower-resolution structure of AQP124 to explain the puzzling mechanism by which water channels can conduct water at very high speed while completely blocking proton permeation.
Gap Junction Channel
We found the cell adhesive function of the water channel AQP4 to be very weak. On the other hand, connexion molecules are known to form rather tight cell adhesive channels. Gap junctions contain intercellular communication channels that allow a wide variety of solutes with different sizes to be transferred between the cytoplasm of adjacent cells. These solutes include ions, metabolites, nucleotides, peptides, and secondary messengers. Gap junction channels have critical roles in many biologically important processes including cardiac development, fertility, the immune system, and electrical signaling in the nervous system. The diversely expressed Connexin26 (Cx26) is the second smallest member of the conserved mammalian gap junction protein family.
We focused on the structure of Cx26 gap junction channels and first used a site specific mutant of human Met34, hCx26M34A, because this mutant expresses in baculovirus infected Sf9 cells at higher quantities than wild type Cx26 infected cells. The hCx26M34A mutant is a single site mutation at the same position as the hCx26M34T mutant, which can cause prelingual non-syndromic hereditary deafness.30 Although the purified hemichannel is hexameric, the 2D arrays obtained by dialysis showed an orthorhombic crystal lattice. A side view of the 3D map at 10 Å resolution reveals that the crystals have a thickness of about 240 Å and contain three lipid bilayers [labeled Mem-1, Mem-2, and Mem-3 in Fig. 8(a)]. Remarkably, the map also shows that the hemichannels re-docked through their extracellular surfaces forming complete gap junction channels. This is consistent with published results proposing extensive hydrophobic surfaces in the gap region. In bilayers Mem-1 and Mem-3, the hemichannels show poorer density than in Mem-2 [Fig. 8(a)], presumably because the large cytoplasmic domains of the connexin subunits are variable and could easily be deformed by their contact with the carbon film to which the crystals are adsorbed in the sample preparation procedure for cryo-electron microscopy. By contrast, the hemichannels in Mem-2 are protected from any forces such as the surface tension upon specimen drying and mechanical interactions with the carbon film. Therefore, the structural features of the hemichannels in Mem-2 should be the most accurate and, in particular, preserve the structure of the flexible cytoplasmic domains of the connexins. Thus, the description of the gap junction structure is based on the hemichannels in Mem-2 unless noted otherwise.
Figure 8.
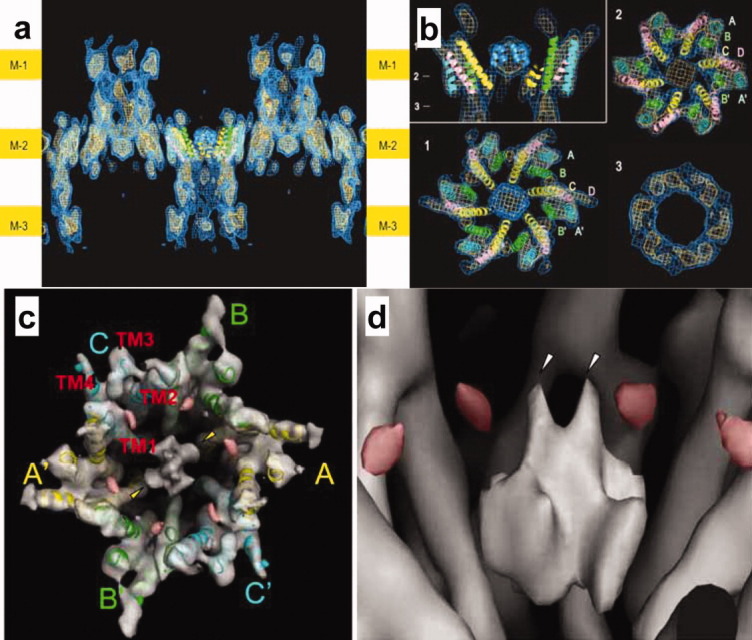
Structure of three membrane layered 2D-crystal of CX26. Three membrane layers are indicated as M-1, M-2, and M3 at the both sides of the figure (a). Plug structure in the gap junction channel. Upper left figure indicates sectional image perpendicular to membrane surfaces and the indicating numbers show the position of each sectional image parallel to the surfaces (b). Figures of (a) and (b) are represented based on Ref. 31. Gap junction structure with two-fold symmetry (c) and two layer arrangement of plug structure in the gap junction channel (d). Red densities could be related with loops between helix 1 to plug helix in each subunit (c, d). Figures of (c) and (d) are represented based on Ref. 33.
The 3D map shows a novel density in the center of the pore [Fig. 8(b)].31 Since this density was dramatically reduced in the structure of N-terminal deletion mutant, it was confirmed that the plug was formed by N-terminal helices of six subunits in the hemichannel. The plug is located inside of the membrane layer and forms contacts with the surrounding channel wall, which at the constricted part of the vestibule is formed by the innermost helices 1. This density strongly suggests that a plug physically blocks the channel within the membrane. Each hemichannel has its own plug, conferring it the ability to gate its pore autonomously. It is possible that the transjunctional voltage sensor and the physical gate reside exclusively within a single hemichannel.
Our Cx26 gap junction crystal structure shows that the channel vestibule is blocked by a physical obstruction, which we call the “plug.” Structure analyzed by X-ray crystallography at higher resolution of the open state32 provide more structural detail and supported the plug gating mechanisms in these widely expressed channels. We recently improved the map of hCx26M34A mutant based on electron crystallography. The analysis revealed the plug forming helices are not arranged with six-fold symmetry at same height in the channel but form a two layer arrangement [Fig. 8(c, d)]. The whole gap junction structure, especially the cytoplasmic parts, has two-fold rather than six-fold symmetry [Fig. 8(c)]. The cytoplasmic loop of the Cx26 molecule interacts with the plug loop and is therefore a key structural determinant for regulation of complex gating mechanism by gap junction channels.33
Conclusions
The ability of electrons to form images, combined with advances in cryo-technology, are enabling us to acquire detailed structural and chemical information about membrane proteins in their physiological lipid and ionic settings as shown in Figure 9. This information complements that obtained by X-ray diffraction of proteins in detergent, where the biological relevance of the structure may be less certain. The development of cryo-electron tomography and real-space averaging methods together with instrumental development such as the seventh generation cryo-EM shown in Figure 5 will extend the possibilities of obtaining high resolution information from increasingly complex protein-lipid arrays, and from membrane specializations in situ.
Figure 9.

Structures of membrane proteins analyzed by utilizing originally developed cryo-EM systems.
Acknowledgments
These studies were performed in nice collaboration with many researchers, whose names were recorded as authors in each referenced paper. The author is grateful to Dr. Brian Matthews for critical reading of the manuscript. Dr. Fujiyoshi is the 2010 recipient of the Protein Society Christian B. Anfinsen Award for significant technical achievements in the field of protein science.
References
- 1.Declan AD, Cabral JHM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 2.Cabral JHM, Zhou Y, MacKinnon R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y, Cabral JHM, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 5.Long SBL, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 6.Sato C, Ueno Y, Asai K, Takahashi K, Sato M, Engel A, Fujiyoshi Y. The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature. 2001;409:1047–1051. doi: 10.1038/35059098. [DOI] [PubMed] [Google Scholar]
- 7.Nagura H, Irie K, Imai T, Shomomura T, Hige T, Fujiyoshi Y. Evidence for lateral mobility of voltage sensors in prokaryotic voltage-gated sodium channels. Biochem Biophys Res Commun. 2010;399:341–346. doi: 10.1016/j.bbrc.2010.07.070. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura T, Irie K, Nagura H, Imai T, Fujiyoshi Y. Arrangement and mobility of the voltage sensor domain in prokaryotic voltage-gated sodium channels. J Biol Chem. 2011;286:7409–7417. doi: 10.1074/jbc.M110.186510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on Array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 10.Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 11.Abe K, Tani K, Nishizawa T, Fujiyoshi Y. Inter-subunit interaction of gastric H+,K+-ATPase prevents reverse reaction of the transport cycle. EMBO J. 2009;28:1637–1643. doi: 10.1038/emboj.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 14.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 15.Uyeda N, Kobayashi T, Ishizuka K, Fujiyoshi Y. High voltage electron microscopy for image discrimination of constituent atoms in crystals and molecules. Chemia Scripta. 1979;14:47–61. [Google Scholar]
- 16.Uyeda N, Kobayashi T, Ishizuka K, Fujiyoshi Y. Crystal structure of Ag-TCNQ. Nature. 1980;285:95–97. [Google Scholar]
- 17.Adrian M, Dubochet J, Lepault J, McDwall AW. Cryo-electron microscopy of vuruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 18.Hirai T, Murata K, Mitsuoka K, Kimura Y, Fujiyoshi Y. Trehalose embedding technique for high-resolution electron crystallography. J Electron Microsc. 1999;48:653–658. doi: 10.1093/oxfordjournals.jmicro.a023731. [DOI] [PubMed] [Google Scholar]
- 19.Fujiyoshi Y, Mizusaki T, Morikawa K, Yamagishi H, Aoki Y, Kihara H, Harada Y. Development of a superfluid helium stage for high-resolution electron microscopy. Ultramicroscopy. 1991;38:241–251. [Google Scholar]
- 20.Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- 21.Gyobu N, Tani K, Hiroaki Y, Kmegawa A, Mitsuoka K, Fujiyoshi Y. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J Struct Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preston GM, Carroll TP, Guggino WB, Agre P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- 24.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 25.Walz T, Hirai T, Murata K, Heymann JB, Mitsuoka K, Fujiyoshi Y, Smith BL, Agre P, Engel A. Three-dimensional structure of aquaporin 1. Nature. 1997;287:624–627. doi: 10.1038/42512. [DOI] [PubMed] [Google Scholar]
- 26.Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci USA. 1998;95:11981–11986. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H, Nishikawa K, Hiroaki Y, Fujiyoshi Y. Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. Biochim Biophys Acta. 2008;1778:1181–1189. doi: 10.1016/j.bbamem.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Tani K, Mitsuma T, Hiroaki Y, Kamegawa A, Nishikawa K, Tanimura Y, Fujiyoshi Y. Mechanism of aquaporin-4's fast and highly selective water conduction and proton exclusion. J Mol Biol. 2009;389:694–706. doi: 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 29.Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WEC, Robbins RA, Miercke LJW, Stroud RM. Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc Natl Acad Sci USA. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 31.Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE. Roles of Met-34, Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci USA. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin-26 gap junction channel at 3.5 Å resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 33.Oshima A, Tani K, Toloue MM, Hiroaki Y, Smock A, Inukai S, Cone A, Nicholson BJ, Sosinsky GE, Fujiyoshi Y. Asymmetric configurations and N-terminal rearrangements in connexin26 gap junction channels. J Mol Biol. 2011;405:724–735. doi: 10.1016/j.jmb.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]