

Abstract
Human GSTpi, an important detoxification enzyme, has been shown to modulate the activity of JNKs by inhibiting apoptosis and by causing cell proliferation and tumor growth. In this work, we describe a detailed analysis of the interaction in vitro between GSTpi and JNK isoforms (both in their inactive and active, phosphorylated forms). The ability of active JNK1 or JNK2 to phosphorylate their substrate, ATF2, is inhibited by two naturally occurring GSTpi haplotypes (Ile105/Ala114, WT or haplotype A, and Val105/Val114, haplotype C). Haplotype C of GSTpi is a more potent inhibitor of JNK activity than haplotype A, yielding 75–80% and 25–45% inhibition, respectively. We show that GSTpi is not a substrate of JNK, as was earlier suggested by others. Through binding studies, we demonstrate that the interaction between GSTpi and phosphorylated, active JNKs is isoform specific, with JNK1 being the preferred isoform. In contrast, GSTpi does not interact with unphosphorylated, inactive JNKs unless a JNK substrate, ATF2, is present. We also demonstrate, for the first time, a direct interaction: between GSTpi and ATF2. GSTpi binds with similar affinity to active JNK + ATF2 and to ATF2 alone. Direct binding experiments between ATF2 and GSTpi, either alone or in the presence of glutathione analogs or phosphorylated ATF2, indicate that the xenobiotic portion of the GSTpi active site and the JNK binding domain of ATF2 are involved in this interaction. Competition between GSTpi and active JNK for the substrate ATF2 may be responsible for the inhibition of JNK catalysis by GSTpi.
Keywords: MAP kinases, GSTpi, ATF2, JNK, protein-protein interactions, signaling cascades
Introduction
The cytosolic glutathione S-transferases (GSTs) protect cells against various xenobiotic, electrophilic compounds through conjugation with glutathione, rendering these toxins less harmful to the cells, and considerably more readily excreted.1 Mammalian GSTpi is the most ubiquitous and highly expressed GST and is especially abundant in the lung, placenta, and the esophagus.2,3 Interestingly, GSTpi is not found in the liver, which points to the possibility that GSTpi may have other functions besides detoxification.4 It is also the predominant GST in a wide range of malignant tumors; elevated levels of GSTpi have been found in colon, bladder, stomach, skin, breast, and lung cancers.3 GSTpi is able to catalyze the conjugation of various anticancer drugs with glutathione, which impairs their efficacy and enhances their excretion. Therefore, increased levels of GSTpi in tumors have been linked to both cancer drug resistance and malignancy.4,5
Like all mammalian GSTs, the active site of GSTpi consists of two components: a mostly hydrophilic G-site (glutathione-binding site) and a mostly hydrophobic H-site (xenobiotic-binding site).6 GSTpi exists both as a monomer of 23.5 kDa (at low protein concentrations) and as a dimer (at higher protein concentrations); each monomer contains one active site.6–9
Human GSTpi is polymorphic, with differences in residues 105 and 114. Four haplotypes involving variation between these two residues have been determined and characterized, as follows: GSTpi haplotype A (WT), with Ile105 and Ala114; haplotype B, with Val-105 and Ala-114; haplotype C, with Val-105 and Val-114; and haplotype D, with Ile-105 and Val-114.1,3,10,11 The presence of valine at position 105, which is part of the H-site, was shown to disrupt the water hydrogen-bonding network, thereby allowing GSTpi to accommodate less bulky substrates.12 In addition, the presence of valine at position 105 yields lower enzyme activity toward normal GSTpi substrates, while the presence of valine at position 114 does not affect the standard GSTpi activity; the significance of V114 has not yet been pinpointed. Overall, haplotype C has been linked to higher risk of many cancers, such as those of the testis and bladder.3
GSTpi has also been shown to play an important role in modulating activities of other enzymes through protein-protein interactions. One such example is the heterodimer formation between GSTpi and 1-Cys-peroxiredoxin (1-Cys-Prx). GSTpi was shown to form a complex with the inactive form of 1-Cys-Prx (both in vivo and in vitro), which accounts for reactivation of 1-Cys-Prx.4,13
Mitogen-activated protein kinases (MAPKs) are eukaryotic enzymes at the core of the MAPK cascade. They transduce signals sent by cell surface receptors to important intracellular targets (both cytosolic and nuclear), controlling cell proliferation, differentiation, inflammation, and apoptosis.14,15 In mammals, at least three types of MAPKs are known: extracellular signal-regulating kinases (ERKs), p38 kinases, and c-Jun N-terminal kinases (JNKs).16 MAPKs are found downstream of MAP2K (MKKs) and MAP3K (MEKKs, ASK1, MLK), where MAP3Ks activate MAP2Ks, which in turn activate the MAPKs.17–19
JNKs are one class of MAPK that are activated by UV or osmotic shock, as well as by cytokines and growth factors.20,21 Three JNK genes exist: JNK1, JNK2, and JNK3. Both JNK1 and JNK2 are expressed ubiquitously, while JNK3 is located in the brain, testis, and heart tissue.20 Differential splicing of the JNK genes gives rise to 10 isoforms. These isoforms vary in the length of their N- and C-termini. There are four isoforms for JNK1, four for JNK2, and two for JNK3.22,23 There is 85% sequence identity between these JNK enzymes (JNK1 vs. JNK2 vs. JNK3) and more than 90% sequence identity between JNK isoforms (for example, JNK1α1 vs. JNK1α2).19 Just like GSTs, JNKs share a series of substrates, with varying kinetic properties and binding affinities.15,24
JNKs are activated by MKK4 and MKK7 (MAP2Ks), through dual phosphorylation of threonine and tyrosine in the TPY motif found in the activation loop.17,25,26 Once phosphorylated, JNKs in turn phosphorylate their cytosolic substrates (such as Bcl-2), or enter the nucleus, where they activate c-Jun, ATF2, Elk1, c-Myc, and p53 transcription factors.19,20,27–30 Two of JNK substrates, ATF2 and c-Jun, are part of the AP-1 (Activator Proteins 1) family of transcription factors.29 Once phosphorylated by JNK, they form homo and heterodimers and bind to TPA Response Elements (TRE) or to c-AMP Response Elements (CRE) in various gene promoters, triggering gene transcription and protein synthesis.31
GSTpi has been reported to be an inhibitor of JNK activity in vivo.32 It was noted that in nonstressed cells, the activity of JNK toward c-Jun was basal. Upon UV irradiation and H2O2 treatment, an increase in JNK activation and subsequent c-Jun phosphorylation were seen. These studies led to the purification and identification of GSTpi as the inhibitor of JNK activity. GSTpi was shown to inhibit JNK in a dose-dependent manner with up to 80% inhibition of kinase activity toward c-Jun.32 Increased activity of JNK and consequent phosphorylation of c-Jun was also noted in mice that were lacking GSTpi.33 Evidence was presented showing that the extended C-terminus of JNK is important for GST binding, indicating that only the longer isoforms of JNK can interact with GSTpi.34 It was also demonstrated that haplotype C (V105/V114 haplotype of GSTpi) was the only GSTpi haplotype that was able to inhibit JNK apoptotic activity in vivo.1
To date, it is unclear if the interaction between GSTpi and JNK is isoform specific, or if both JNK1 and JNK2 can interact with GSTpi. Some studies have utilized active forms of JNKs while others used inactive, unphosphorylated JNKs. Therefore, it is still unknown if GSTpi has a preference for active, phosphorylated JNK when compared with inactive, unphosphorylated JNK. Moreover, the majority of the previous work has been qualitative in nature since most studies were performed in vivo. Since the in vivo studies unavoidably involve complex systems, we considered that it was important to establish directly the feasibility of such interactions by in vitro studies, and this approach allowed the examination of the characteristics of the interactions in more detail.This study describes an in vitro investigation of the interaction between human GSTpi and two long isoforms of JNK (JNK1α2 and JNK2α2) in both their active and inactive forms. Here, we tested two GSTpi haplotypes, A and C, for their ability to inhibit JNK activity toward ATF2. We then evaluated GSTpi binding to JNKs in the absence and the presence of a JNK substrate, ATF2. These detailed studies revealed the requirements for modulation of JNK activity by GSTpi.
Results
Haplotype C GSTpi is a better JNK inhibitor than haplotype A GSTpi
Preformed complexes of active JNK1/ATF2 or active JNK2/ATF2 were incubated with either Haplotype A or Haplotype C GSTpi for 30 min at 25 °C, and the ability of JNK to phosphorylate its substrate, ATF2, was measured.‡ As shown in Figure 1, Haplotype C inhibits ATF2 phosphorylation by both active JNK1 [Fig. 1(A)] and JNK2 [Fig. 1(B)] by 75–80%, as judged by Western blotting analysis using antibodies to ATF2 doubly phosphorylated at Thr-69 and Thr-71. The amount of inhibition produced by Haplotype A is significantly less for both active JNK1 (25%) and active JNK2 (45%).
Figure 1.
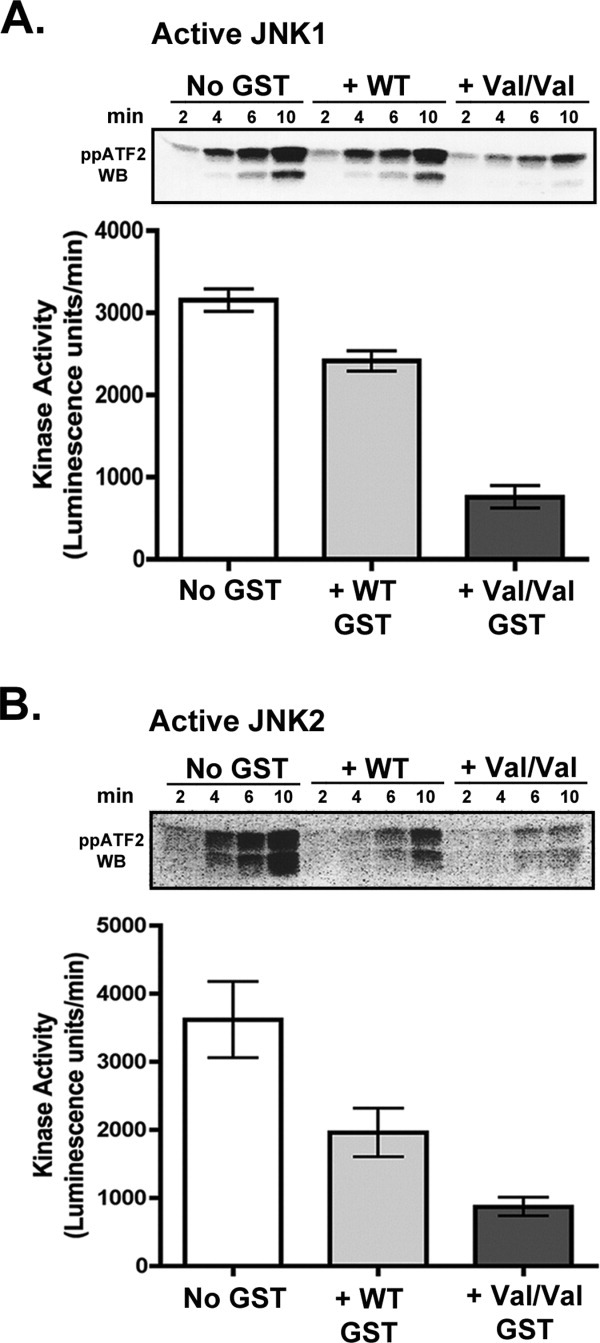
Inhibition of JNK activity by Haplotype A and C GSTpi. Preformed active JNK/ATF2 complexes (in 1:1 molar ratio) were incubated either alone or with 10 μM WT (Haplotype A) or V105/V114 GSTpi (Haplotype C) for 30 min at 25°C. ATP and MgCl2 were added to initiate the JNK catalytic reaction, and the amount of phosphorylation of ATF2 by JNK was monitored as a function of time by Western blot analysis using antibodies against ATF2 phosphorylated at both Thr-69 and Thr-71 (See footnote on page 3). Representative Western blots are shown. Western blot data was analyzed by densitometry in ImageJ to calculate kinase activity in each reaction (in Luminescence units/min). The rates shown in the bar graphs were an average of at least two independent inhibition assays. A, Inhibition of active JNK1. B, Inhibition of active JNK2.
GSTpi is not phosphorylated by JNK
GSTpi purified from Kato III cancer cells was shown to be phosphorylated in the C-terminal region on Ser-196, while GSTpi from normal fibroblasts was not phosphorylated. Since the C-terminus of GSTpi was determined to be the site of JNK binding, JNK was postulated to be responsible for this post-translational modification of GSTpi.35 Therefore, we tested directly whether the inhibition by GSTpi of JNK activity toward ATF2 is due to GSTpi acting as an alternate substrate of JNK. To do this, we monitored the ability of active JNK2 to catalyze the transfer of the γ32-phosphate from [γ32P]ATP to GSTpi. Active JNK2 was incubated with either the WT or the V105/V114 haplotype of GSTpi in the presence of [γ32P]ATP and MgCl2 at room temperature for 2 h. Incorporation of phosphate into MBP-ATF2 from [γ32P]ATP catalyzed by JNK2 was used as a positive control since ATF2 is a known JNK substrate. As shown in Figure 2(A), no ATP incorporation into either of the GSTs was observed, while a strong signal was seen for MBP-ATF2. A Coomassie Blue stained SDS-PAGE gel is also shown to demonstrate the protein amounts used in this experiment.
Figure 2.
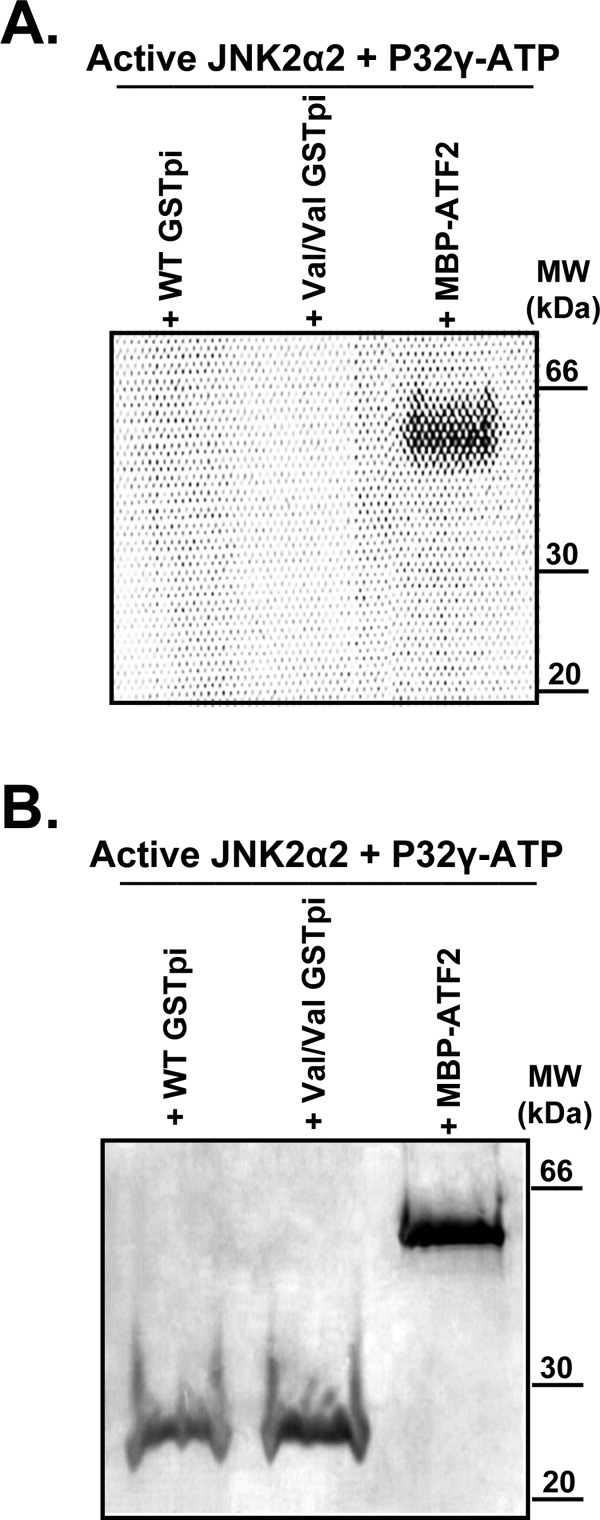
Does JNK catalyze the phosphorylation of GSTpi? To test if active JNK2 was able to phosphorylate GSTpi, incorporation of 32P from [γ32P]ATP into GSTpi was monitored. MBP-ATF2 was used as a positive control since it is known to be phosphorylated by JNK. A, An image generated by the PhosphorImager from a P32 phosphor storage screen. B, Coomassie Blue stained SDS-PAGE gel that was utilized to generate the image in A.
GSTpi binding to inactive JNKs was not observed in the absence of ATF2
GSTpi binding experiments were carried out to determine the conditions needed to detect any GSTpi bound to JNK and eluting together with JNK from the Ni-NTA resin. Since haplotype C GSTpi is the more effective inhibitor of JNK (Fig. 1), this type of GSTpi was used for most of the binding experiments. Initially, we conducted experiments with both active and inactive JNKs bound to the Ni-NTA resin with or without MBP-tagged ATF2. Small amounts of bound GSTpi were detected only in the samples containing both JNK and MBP-ATF2, but none was seen in the samples containing only JNK (data not shown).
We utilized untagged ATF2 to test the effect of ATF2 on GSTpi binding to JNK. When untagged ATF2 was used, readily detectable amounts of GSTpi were bound to the Ni-NTA resin samples containing bound His-JNK1 or His-JNK2; in contrast, very low or undetectable amounts of GSTpi were noted in samples that contained only His-JNKs but no ATF2. Figure 3(A) shows a representative example of such a binding experiment, where haplotype C GSTpi (at 7 μM initial concentration) bound only to Ni-NTA resin samples containing inactive His-JNK (JNK1 or JNK2) plus ATF2. A sample of a known concentration (0.05 μM) of V105/V114 GSTpi was used as a standard. The Western blotting data was analyzed by densitometry; the value for the GSTpi standard was used together with the known volume of the eluate to estimate the amount of GSTpi in various eluates (in nmol). Figure 3(B) compares the amounts of GSTpi bound to either inactive His-JNK1 (with or without ATF2) or inactive His-JNK2 (with or without ATF2) obtained from the Western blot in Figure 3(A). GSTpi alone did not have any significant binding affinity to Ni-NTA resin, and only a basal amount of GSTpi binding was observed without any other proteins present [Fig. 3(B)]. These results clearly demonstrate that, while no interaction takes place between inactive JNKs and GSTpi, the presence of ATF2 results in increased GSTpi binding to these inactive JNKs. Inactive JNK1 is the preferred isoform for interaction with GSTpi when ATF2 is present (0.09 nmol of eluted GSTpi).
Figure 3.
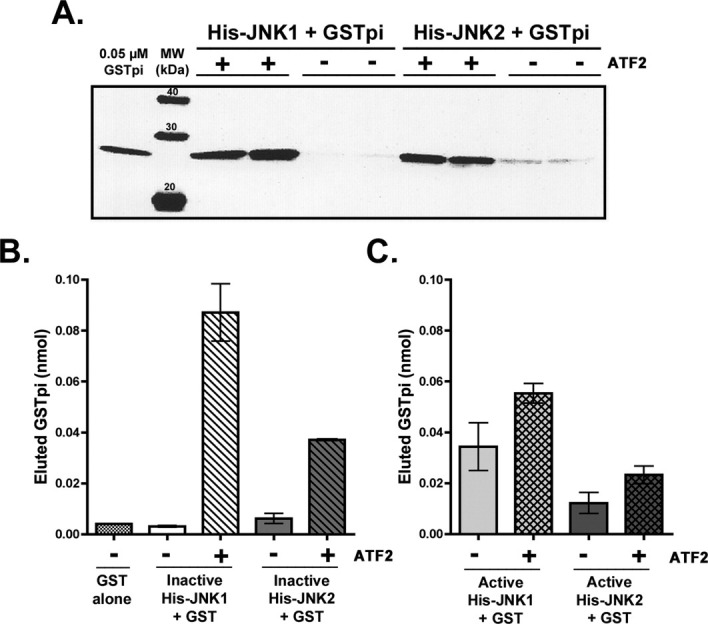
Effect of ATF2 on GSTpi interaction with inactive and active JNKs. Binding experiments were conducted in duplicate, in which either 3.5 μM inactive His-JNK1 or inactive His-JNK2 with or without 3.5 μM untagged ATF2 was used. The binding of V105/V114 GSTpi (7 μM initial) was monitored in the imidazole elutions (as described in the Experimental Procedures). A, Inactive JNKs ± ATF2. Western blotting analysis using antibodies against human GSTpi of the imidazole elutions of the Ni-NTA resin. B, Bar graph of the densitometry results from A, as well as the data for the negative control: GSTpi bound to the Ni-NTA resin lacking any JNK or ATF2. The densitometry value from the GSTpi standard in the Western blot in A (at 0.05 μM), was used (along with volume of the elution) to calculate the amount of GSTpi in the imidazole eluate (in nmol). The total elution volume for samples containing His-JNK1 was 1 mL, while the total elution volume of samples containing His-JNK2 was 0.5 mL. C, Active JNKs ± ATF2. Bar graph of the densitometry analysis of GSTpi binding assays to determine the ability of V105/V114 GSTpi to interact with active JNK1 or active JNK2 in the absence or presence of ATF2. The results are an average of at least two independent binding experiments and were obtained similarly to the results of panel B.
Some GSTpi binding to active phosphorylated JNKs was observed
If active, phosphorylated JNKs [Fig. 3(C)] were used without ATF2, GSTpi amounts in the imidazole elutions were ∼0.03 nmol (JNK1) and ∼0.01 nmol (JNK2). These values are significantly higher than those observed for inactive JNKs [Fig. 3(B)] in the absence of ATF2. Only a small increase in GSTpi bound was observed when ATF2 was included with the active JNKs [Fig. 3(C): to 0.05–0.06 nmol for active JNK1 and 0.02 nmol for active JNK2. Overall, the highest amount of GSTpi was bound in samples containing both inactive JNK1 and ATF2 (0.07–0.1 nmol). The second highest GSTpi amount bound was found when inactive JNK2 was used together with ATF2 (0.03–0.04 nmol). A smaller [Fig. 3(C)], but a significant amount of bound GSTpi was found in the samples containing active JNKs (both without and with ATF2).
GSTpi binds similarly to active JNK/ATF2 complexes and inactive JNK/ATF2 complexes
To determine whether a particular JNK was the preferred binding partner for GSTpi, we measured the GSTpi concentration dependence of GSTpi binding to ATF2/JNK complexes. Using either JNK1α2 or JNK2α2, in both their unphosphorylated, inactive and phosphorylated, active forms, the JNK, and ATF2 concentrations were kept constant (3.5 μM initial), while the initial concentration of the V105/V114 GSTpi was varied from 0.5–10 μM. Figure 4(A) shows a silver stained SDS-PAGE gel of the imidazole elutions corresponding to samples containing varying GSTpi concentrations. All three proteins (active His-JNK1a2, GSTpi, and ATF2, albeit weakly) are visible by the silver stain, with constant amounts of the kinase. GSTpi found in the elutions reached a constant amount at initial concentrations greater than 4.2 μM. Since some silver stained bands were faint, we also employed Western blotting analysis [Fig. 4(B)] using GSTpi antibodies to detect GSTpi across the entire range of concentrations. A standard of known haplotype C GSTpi concentration (0.05 μM) was also loaded on all GSTpi western blots [Fig. 4(B)]. The samples displaying a constant eluted [GSTpi] (above 4.2 μM initial concentration) were only slightly more intense than the GSTpi standard. This is in agreement with the eluted [GSTpi] measured with the binding experiments shown in Figure 3(A). The GSTpi standard allowed for averaging of the data from two independent sets of binding experiments.
Figure 4.
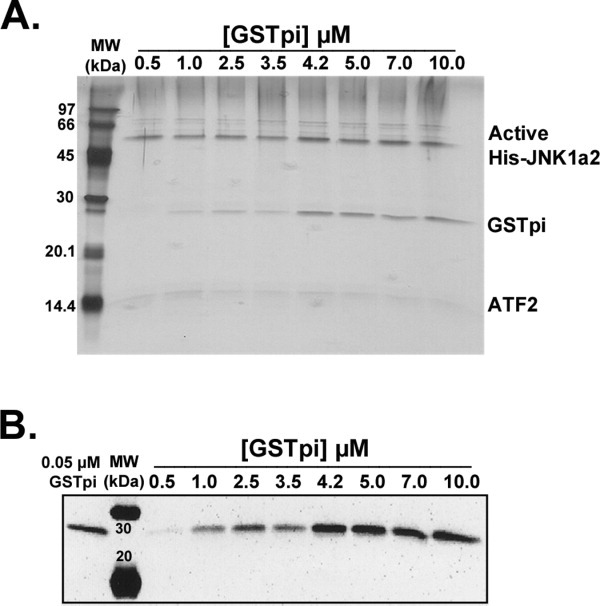
GSTpi binding to active JNK1/ATF2 complexes. Binding experiments in which 3.5 μM ATF2 and 3.5 μM active JNK1 were added to the Ni-NTA resin, and the V105/V114 GSTpi was added in increasing concentrations (from 0.5 to 10 μM). A, Silver staining of the SDS-PAGE gel of the eluted samples of all initial GSTpi concentrations tested. B, Western blot analysis using antibodies against GSTpi of the eluted samples described in panel A.
Such GSTpi binding curves were generated for all four different types of JNK: active JNK1, active JNK2, inactive JNK1, and inactive JNK2 over the same range of initial GST concentration. The maximum luminescence intensity values, which are proportional to the amount of GSTpi bound (and eluted), were lower for the active phosphorylated JNKs. These values are summarized in Table I.
Table I.
Summary of GSTpi Binding Results
| Components (GSTpi with:) | Maximum luminescence intensity (luminescence units)a |
|---|---|
| Inactive JNK1 + ATF2 | 40,100 ± 3800 |
| Inactive JNK2 + ATF2 | 20,600 ± 2600 |
| Active JNK1 + ATF2 | 27,600 ± 1700 |
| Active JNK2 + ATF2 | 9,200 ± 900 |
| ATF2 alone | 20,700 ± 2600 |
Maximum luminescence intensity values were normalized using the standard [GSTpi] shown in Figures 3(A), 4(B), and 6(B). The normalized maximum luminescence intensity is proportional to the amount of bound and eluted GSTpi.
GSTpi interacts with ATF2 in the absence of inactive JNK
We were surprised to see that the amounts of GSTpi bound were relatively high in samples containing both ATF2 and inactive, unphosphorylated JNKs. In addition, when experiments were conducted in which the ATF2 concentration was varied, while the [inactive JNK] was held constant, we noticed that high amounts of ATF2 bound to the Ni without corresponding to the amounts of bound inactive His-JNK (data not shown). Therefore, we carried out a control experiment to ascertain whether ATF2 had affinity for the Ni-NTA resin in the absence of His-JNK and if there was any interaction between GSTpi and ATF2. ATF2 does, in fact, bind to the Ni-NTA resin and is eluted by imidazole. When ATF2 (at 3.5 μM) was bound to the Ni-NTA resin and GSTpi (7 μM) was later added, following the same binding protocol used previously, a relatively high amount of eluted GSTpi was seen in the imidazole elutions (∼0.04 nmol) (Fig. 5). This amount of bound and eluted GSTpi was within standard error of the amount of GSTpi found in samples that had both ATF2 and inactive JNK2 and was only 2-fold lower than the samples that had both ATF2 and inactive JNK1 [Fig. 5(B)]. These results suggest that GSTpi can interact directly with ATF2.
Figure 5.
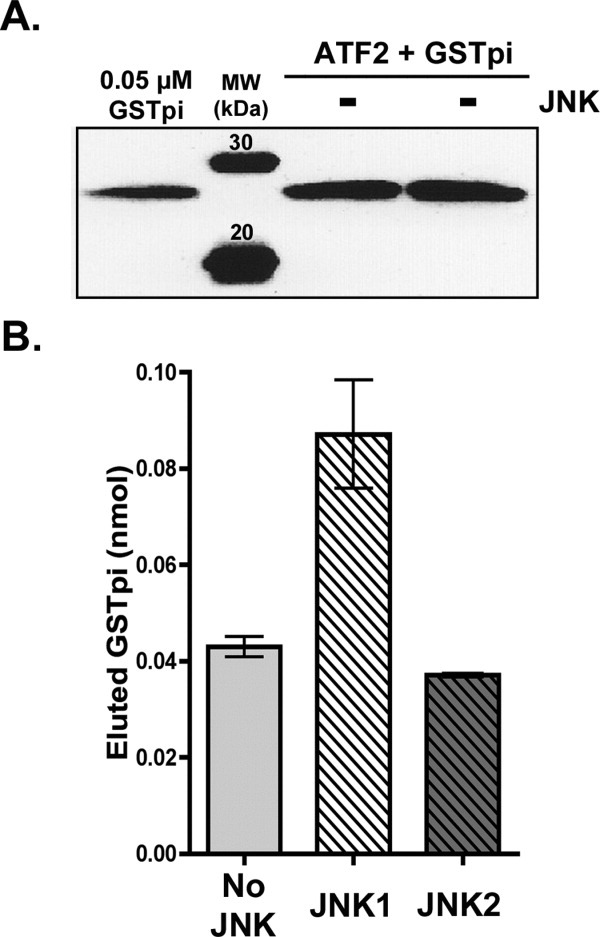
GSTpi interaction with ATF2 in the absence of JNKs. Resin equilibrated with 3.5 μM ATF2 was mixed with initial 7 μM V105/V114 GSTpi, as was described in the Experimental Procedures. A, The amount of bound GSTpi in the eluted fractions was monitored by Western blotting analysis using antibodies against GSTpi. B, Plots of the densitometry analysis from panel A (no JNK) as well as the results from binding experiments when inactive JNKs were present [from Fig. 3(B)]. The total nmol of eluted GSTpi were calculated as was described for Figure 3.
To study this interaction further, we measured the binding of GSTpi as the initial concentration of ATF2 was held constant and the initial concentration of GSTpi was varied from 0.5–10 μM. The silver stained gel in Figure 6(A), shows a clearly visible ATF2 band in all the eluted samples, indicating that ATF2 has a very strong affinity for the Ni-NTA resin, even though it does not contain a His-tag. In both the silver stained gel and the GSTpi Western blot (Fig. 6), a constant amount of eluted GSTpi was observed above an initial 4.2 μM concentration of GSTpi. The maximum luminescence intensity value is similar to the values obtained when inactive JNK2 was included (Table I), suggesting that the inactive JNK2 had no direct interaction with either GSTpi or ATF2. In addition, if GSTpi binding experiments were conducted with samples containing increasing amounts of ATF2 (0.5-3.5 μM initial), but with a constant concentration of active JNK1 (2.0 μM initial) and GSTpi (3.5 μM initial), an 8-fold increase of GSTpi binding was observed over the range of ATF2 concentrations. In contrast, if inactive JNK1 was included (3.5 μM initial), only a 1.2-fold increase in GSTpi binding was seen (data not shown). These results indicate that the presence of inactive JNK1 does not have any effect on the interaction between GSTpi and ATF2, while active JNK1 may be competing with GSTpi for the JNK-binding domain (JBD) of ATF2.
Figure 6.
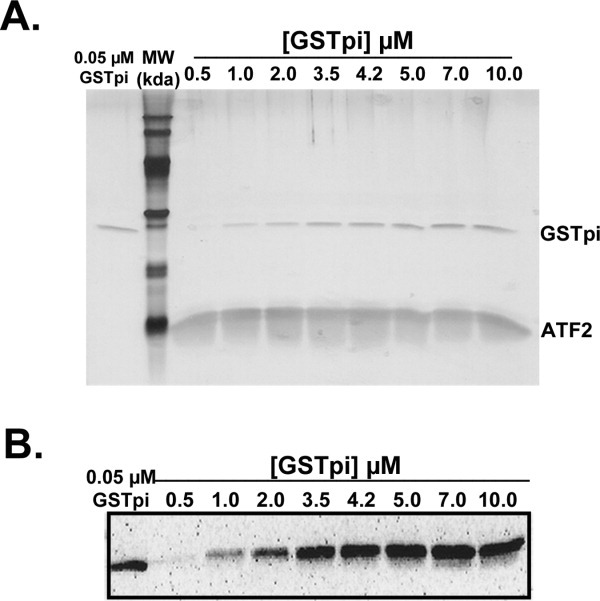
GSTpi binding to ATF2 with determination of maximum luminescence intensity values. In binding experiments, 3.5 μM ATF2 was added to the Ni-NTA resin, and the V105/V114 GSTpi was added to the resin in increasing initial concentrations (from 0.5 to 10 μM). A. Silver staining of the SDS-PAGE gel of the eluted samples of all initial GSTpi concentrations tested. B. Western blot analysis using antibodies against GSTpi for the eluted samples described in panel A.
Thr-69 and Thr-71 of ATF2 are not involved in GSTpi binding
To test whether the phosphorylation sites (T69 and T71) of ATF2 were involved in GST binding, thus leading to decreased ability of JNK to phosphorylate ATF2, we compared GSTpi binding with unphosphorylated ATF2 and with ATF2, which was fully phosphorylated at these residues. Complete ATF2 phosphorylation was confirmed by Western blotting analysis using antibodies against ATF2 phosphorylated at both T69 and T71 (data not shown). Both phosphorylated and unphosphorylated ATF2 bound well to the Ni-NTA resin, as can be seen from the silver stain of the imidazole elutions. Differences between phosphorylated ATF2 samples and the unphosphorylated ones are evident from the fact that the phosphorylated ATF2 runs slower on the SDS-PAGE gel than the unphosphorylated ATF2 [Fig. 7(A)]. The amount of GSTpi bound to unphosphorylated or to phosphorylated ATF2, were similar, as judged by both silver staining and Western blotting, using antibodies to GSTpi (Fig. 7). The ability of GSTpi to interact equally with unphosphorylated and phosphorylated ATF2 indicates that GSTpi does not inhibit JNK activity by blocking the phosphorylation sites of ATF2 (T69 and T71).
Figure 7.
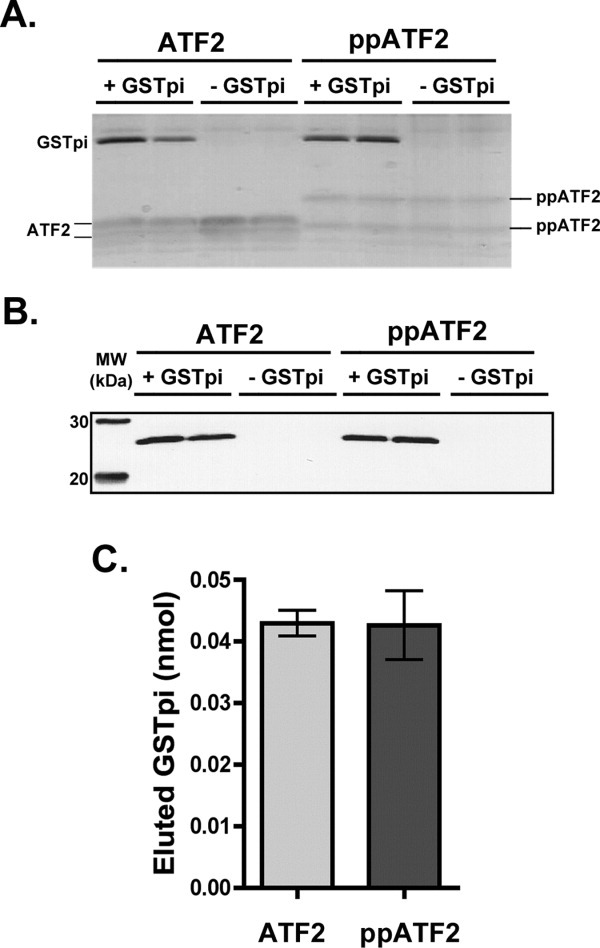
Interaction of GSTpi with unphosphorylated and phosphorylated ATF2. Binding experiments between 3.5 μM unphosphorylated or phosphorylated ATF2 (in duplicates) were carried out as was described for previous experiments. A, Silver stained SDS-PAGE gel of the eluted samples from the binding experiment. B, Western blot using GSTpi antibodies of the gel shown in panel A. C, Bar graph of the densitometric analysis of the GSTpi band from panel B to estimate the total nmol of eluted GSTpi.
Effect of S-methyl and S-hexylglutathione on the interaction between GSTpi and ATF2
To determine if the active site of GSTpi is involved in the interaction with ATF2, we tested the effect of glutathione analogs (such as S-methyl or S-hexylglutathione) on the ability of GSTpi to bind to ATF2. These compounds are known to function as competitive inhibitors of GSTpi.36 Similar amounts of ATF2 are able to bind to the Ni-NTA resin in the presence of S-methylglutathione, while smaller amounts of ATF2 (about half) bound to the resin in the presence of S-hexylglutathione [Fig. 8(A), silver stain]. GSTpi Western blot analysis indicated that S-methylglutathione decreased GSTpi and ATF2 interaction by about 50% [Fig. 8(B)]. No bound and eluted GSTpi was observed in the samples eluted from the Ni-NTA resin, which contained S-hexylglutathione, indicating that the GSTpi and ATF2 interaction was fully blocked [Fig. 8(B)]. These results indicate that the xenobiotic substrate-binding site of GSTpi is the primary site of interaction with ATF2, while the involvement of the glutathione site is only indirect.
Figure 8.
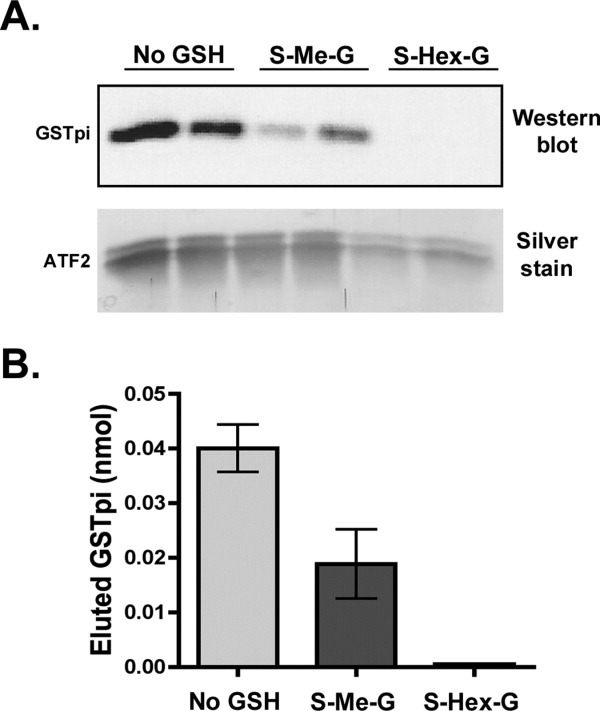
Effect of S-methyl and S-hexylglutathione on the interaction between GSTpi and ATF2. Binding experiments between GSTpi and ATF2 were carried out either in the regular buffer or in the buffer containing 2.5 mM S-methylglutathione (S-Me-G) or 2.5 mM S-hexylglutathione (S-Hex-G) (as described in the Experimental Procedures). A, Western blot analysis using antibodies against GSTpi of the imidazole eluates. Silver staining was also performed on the gel to demonstrate the corresponding amounts of ATF2 that were found in the imidazole elutions. B, Plot of the densitometry analysis of the Western blot in panel A to show the total eluted GSTpi amounts (nmol).
Similar amounts of the V105/V114 and WT GSTpi bind to ATF2
Since the V105/V114 haplotype of GSTpi exhibited higher inhibition of JNK activity when compared with the WT GSTpi (Fig. 1), binding to ATF2 of the WT and the V105/V114 haplotype GSTpi was compared. In the first experiment, saturating concentration (7 μM) of GSTpi was used. As can be seen from the silver stained gel in Figure 9(A), approximately equal amounts of the V105/V114 and the WT GSTpi bound to ATF2. In the second experiment, only 2.5 μM GSTpi was used and yet again, equal amounts of the V105/V114 and the WT GSTpi bound to ATF2 [Fig. 9(B)], indicating that the V105/V114 and the WT GSTpi have similar affinity for ATF2.
Figure 9.
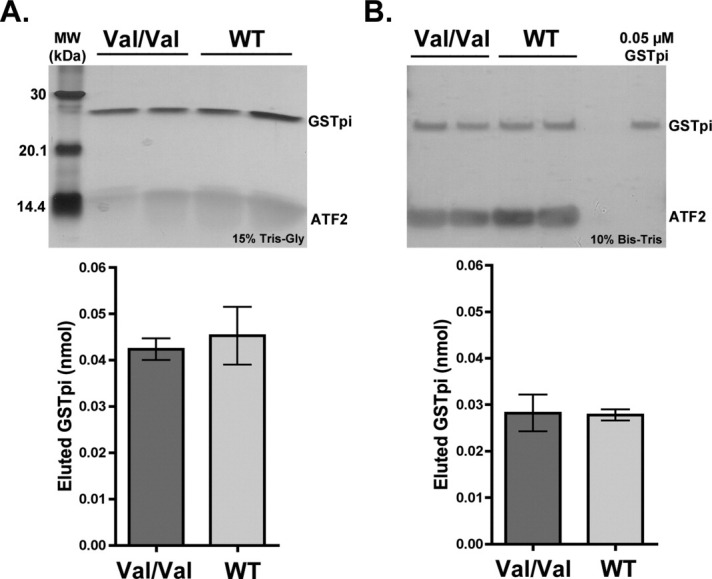
Comparison of the V105/V114 versus WT GSTpi binding to ATF2. Binding experiments between ATF2 (3.5 μM) and two haplotypes of GSTpi (WT or Haplotype A and V105/V114 or haplotype C) were carried out at two different initial GSTpi concentrations (7 and 2.5 μM). Silver stains of the imidazole elutions, as well as the corresponding plots of the densitometry analysis of GSTpi bands are shown (in nmol of eluted GSTpi). A, 7 μM GSTpi. B, 2.5 μM GSTpi.
Discussion
GSTpi, an enzyme characterized to play a key role in phase II detoxification, has recently been in the spotlight as an important partner involved in protein-protein interactions and modulation of activities of other enzymes.4,5,31–34 Although the interaction between GSTpi and c-Jun N-terminal kinases has been described by others using a variety of approaches, many aspects of this interaction still remained unknown. For example, it was unclear if a GSTpi-JNK interaction occurs with phosphorylated or unphosphorylated JNKs, or if one family member binds more strongly than the other (JNK1 vs. JNK2). It was unknown if the addition of a JNK substrate had a large effect on interaction between JNK and GSTpi. Even though the effects of the two most common GSTpi polymorphisms (haplotype A and C) on apoptosis and cellular proliferation have been evaluated,1 the level of inhibition of JNK activity by these haplotypes had not been assessed or compared quantitatively. The work described here addresses some of the unexplored aspects listed above. In addition, our studies of GST interaction with the inactive JNKs led us to discover an entirely new interaction: between GSTpi and ATF2.
When it was first discovered that GSTpi was an inhibitor of JNK, it was shown that GSTpi inhibited up to 80% of JNK activity toward its substrate c-Jun.32 We have now compared the ability of the two haplotypes of GSTpi to elicit inhibition of two different active isoforms of JNK: JNK1α2 and JNK2α2. Our results demonstrate that haplotype C is a much better inhibitor of both active JNK1 and active JNK2 when compared with the haplotype A (Fig. 1). Up to 80% inhibition of ATF2 phosphorylation by either of the two JNKs was seen when the haplotype C GSTpi was utilized. However, the level of inhibition by haplotype A was decreased, with about 25–30% inhibition of active JNK1 and about 45% inhibition of active JNK2. Our results, using ATF2 as a substrate, are consistent with the previously published work that used c-jun.32 To date, the inhibitory effect of the two GSTpi haplotypes on the activity of two different JNK family members (JNK1 and JNK2) has never been quantitatively compared. Interestingly, since haplotype C of GSTpi is the more prevalent GSTpi in some solid tumors, being a better inhibitor of JNK fits well with the greater antiapoptotic effect that was previously demonstrated for this haplotype.1
The mechanism of inhibition by GSTpi of JNK-catalyzed ATF2 phosphorylation is another issue that must be addressed. Others have suggested that GSTpi could potentially serve as a substrate of JNK in vivo. This hypothesis was based on the finding that GSTpi from Kato III gastric cancer cells was phosphorylated on Ser-196, while GSTpi from normal fibroblasts was not post-translationally modified. Intriguingly, Ser-196 is followed by a proline residue and Ser-Pro motifs are the preferred sites of phosphorylation by MAPKs.35 In addition, the C-terminal portion of GSTpi (residues 194-201) was found to be critical for binding to JNK.32,37 To test whether GSTpi serves as a putative substrate of JNK, we monitored incorporation of the γ32-phosphate of ATP into GSTpi by phosphorylated JNK2. Our results definitively demonstrated that GSTpi does not serve as a JNK substrate in vitro (Fig. 2). Therefore, inhibition of JNK activity by GSTpi likely takes place through a different mechanism.
JNK1 and JNK2, in both their active and inactive forms, were tested for their ability to interact with GSTpi to address the requirements for this interaction. The V105/V114 GSTpi haplotype was used in all subsequent experiments since it produced higher JNK inhibition than did the WT GSTpi. We found that GSTpi alone is unable to interact with inactive, unphosphorylated JNKs; significant interaction between GSTpi and JNK requires the JNK substrate, ATF2 [Fig. 3(A–B)]. When ATF2 was included, samples with inactive JNK1 had the highest amounts of bound GSTpi; inactive JNK2 in the presence of ATF2, bound about half as much GSTpi [Fig. 3(B)]. Thus, the isoform of JNK is a determinant of its interaction with GSTpi. More GSTpi binds in the absence of protein substrate in the case of the active JNKs (when compared with the inactive JNKs), and addition of ATF2 leads to only a modest increase in the amounts of bound GSTpi [Fig. 3(C)]. Overall, JNK1 (active and inactive) is the preferred isoform for the interaction with GSTpi.
Measurement of the amount of GSTpi bound allows a semiquantitative comparison between the various JNKs in the presence of ATF2. The amount of maximum GSTpi bound differed significantly (Table I). More GSTpi was bound to inactive JNKs with ATF2 when compared with active JNKs with ATF2. This result, along with the observation that GSTpi was able to interact with inactive JNKs only in the presence of ATF2 led us to suspect that there may be a direct interaction between GSTpi and ATF2.
To test for this possibility, binding experiments were conducted, in which ATF2 was bound to the Ni-NTA resin and its interaction with GSTpi was monitored (Fig. 5). Indeed, ATF2 alone (without any His-JNK) does interact with the V105/V114 double valine GSTpi, and the amount of bound GSTpi in the imidazole eluate is equivalent to that observed when ATF2 was used with inactive JNK2 (Fig. 5). Examination of the GSTpi concentration dependence and maximum luminescence intensity values confirmed that there is a direct interaction between ATF2 and the V105/V114 haplotype of GSTpi (Fig. 6). The value obtained for the maximum luminescence intensity is the same as the value obtained for the inactive JNK2 + ATF2 (Table I). Thus, for inactive JNK2, the entire interaction with GSTpi appears to be attributable to ATF2.
Regardless of the type of kinase used in the binding studies, the low μM range of the GSTpi concentrations used is similar to the Km values that have been determined for JNK substrates. The Km value for ATP has been reported as 3.6-4.3 μM, while the Km for ATF2 was found to be 0.24-0.8 μM.15,22 The concentration range of GSTpi used here (0.5–10 μM) is also similar to the Ki values (1.1 ± 0.3 μM) that were obtained with a small peptide inhibitor of JNK, I-JIP (Inhibitor of JNK based on JIP1).15 Physiologically, JIP-1 (JNK-Interacting Protein 1) acts as a scaffolding protein that brings MEKKs, MKKs, and JNKs together for fast and efficient JNK phosphorylation;15,38 amino acids 153–163 of JIP were found to act as a competitive inhibitor of JNK with respect to ATF2 and c-Jun. The observation that GST affinities for ATF2, or for active JNKs with ATF2, are in the same range as the Km values for JNK substrates and the Ki values for JNK inhibitor, indicates that GSTpi likely inhibits JNK activity through a transient, rather than a prolonged tight binding interaction. This hypothesis is in agreement with the findings that prolonged JNK activation leads to apoptosis, while a more transient JNK activation leads to cell proliferation and differentiation.39,40 Therefore, it is possible that GSTpi could play a key role in maintaining JNK activity low, without inhibiting JNK completely. Such a role would shift the signaling balance toward cell proliferation and inhibit apoptosis, a phenomenon often present in malignant drug resistant tumors. In addition, the preference of GSTpi for JNK1 over JNK2 is in agreement with the reports that JNK1 is responsible for triggering apoptosis, while JNK2 plays a larger role in cell proliferation.41
We must point out that the amounts of GSTpi found in the imidazole eluates were low (0.01–0.1 nmol or 0.01–0.1 μM), when compared with the high GSTpi concentrations (0.5 mL of 0.5–10 μM or 0.25–5 nmol) that were initially added in each binding experiment. However, the amounts of JNK or ATF2 recovered were also estimated to be relatively low when compared with the amounts added at the start of each binding experiment (0.5 mL of 3.5 μM or 1.8 nmol total). The concentration of active JNK in the silver stained gel in Figure 4 was estimated to be 0.12 μM (a 2-fold difference with respect to the concentration of GSTpi). The concentration of ATF2 in the silver stained gel in Figure 6 was estimated to be 0.6 μM (a 6-fold difference with respect to GSTpi). We consider that the low amounts of all 3 enzymes recovered are attributable to the inefficiency of these batch Ni-binding experiments. Nevertheless, these low concentrations of JNK, GSTpi, and ATF2 are likely representative of physiological concentrations. For example, the intracellular concentration of a JNK-related MAPK, ERK2, was estimated to be 100–500 nM.19 Most importantly, all of the experiments described here were conducted in the same way and were reproducible; therefore, we consider that the results described here are qualitatively comparable, and that the differences we report are valid. In addition, because the conditions used for these experiments do not strictly allow equilibrium between the two component proteins, we are not reporting actual Kd values for these binding experiments.
Since both His-JNK and ATF2 are likely present in excess due to their affinity for the Ni-NTA resin, whether free or bound to GSTpi, it was not possible to estimate (from the concentrations of the recovered proteins) the actual stoichiometry of the proteins within the complex. Moreover, we attempted to determine whether the enzymatic activity of GSTpi was affected in any of the collected elutions. However, due to low concentrations of GSTpi, measuring its enzymatic activity was problematic and provided inconclusive results (data not shown).
To characterize the interaction between GSTpi and ATF2 further, we tested whether the haplotype C of GSTpi was able to interact with the phosphorylated form of ATF2 as well as with the unphosphorylated ATF2. Our results demonstrate that the phosphorylation state of ATF2 did not have any effect on the interaction between GSTpi and ATF2. We therefore conclude that GSTpi likely binds to ATF2 somewhere other than the phosphorylation sites (Thr-69 and Thr-71). Since GSTpi inhibits JNK's overall ability to phosphorylate ATF2, the inhibition could be due to binding of GSTpi in or near the JBD of ATF2 (residues 46-56),42 thereby blocking the interaction between ATF2 and JNK. Similarly, the competition between active JNKs and GSTpi for the binding to ATF2 may explain why the amount of GSTpi bound is lower when active JNKs are used instead of inactive JNKs. This hypothesis is supported by the fact that the presence of inactive JNK2 does not have any effect on the interaction between GSTpi and ATF2, while the presence of active JNKs has a significant effect.
We also sought to evaluate the site on GSTpi involved in binding ATF2. The active site of GSTpi consists of two regions: the glutathione binding region (G-Site) and the xenobiotic binding region (H-site).12 To probe for the glutathione active site, we used S-methylglutathione, with its small alkyl group. To probe for both sites simultaneously, we utilized S-hexylglutathione because the S-hexyl portion extends into the xenobiotic substrate site. While S-methylglutathione only partially decreases the interaction between ATF2 and GSTpi, S-hexylglutathione fully disrupts the interaction (Fig. 8). These results indicate that when the H-site of GSTpi is occupied by S-hexylglutathione, ATF2 binding to GSTpi is fully blocked. In contrast, the involvement of the GSH site is only partial or indirect, since the presence of S-methylglutathione only decreased rather than prevented the interaction between GSTpi and ATF2. Others have demonstrated that treatment of cells with GSTpi inhibitors (such as GSH-conjugate analogs) disrupted the interaction of GSTpi and JNK and led to an increase in apoptosis.43 Our results are in good agreement with these findings.
It might be expected that the Val-105/Val-114 GSTpi (haplotype C) would be able to interact with ATF2 much better than the WT GSTpi (Haplotype A) since it was a better inhibitor of JNK than the WT GSTpi. We were surprised to see that at two GSTpi concentrations tested (a saturating concentration of 7 μM and at the much lower concentration of 2.5 uM), equivalent amounts of haplotypes A and C of GSTpi were able to bind to ATF2 (Fig. 9). The results of binding experiments in the presence of S-methyl and S-hexylglutathione indicate that ATF2 binds in the region of the xenobiotic substrate site of GSTpi. It is possible that residue 105 does not play a significant role in the binding of ATF2, and therefore, the Val-105/Val-114 GSTpi binds ATF2 as well as the WT (as reflected from the results in Fig. 9). Residue 105 is part of the xenobiotic binding site of GSTpi, and the presence of valine at this position allows GSTpi to accommodate smaller, less bulky substrates.12 On the other hand, the residue 114 may confer larger inhibition of JNK binding to ATF2 through a more enhanced allosteric effect than residue 105.
In summary, we showed that the haplotype C of GSTpi is a better inhibitor than haplotype A of both active, phosphorylated JNK1 and JNK2. Our studies of the interaction between GSTpi and active phosphorylated JNKs reveal the formation of a complex. The extent of this interaction is isoform-specific, with JNK1 binding more GSTpi than JNK2. In the presence of ATF2, the interaction between JNK and GSTpi is enhanced and there is a significant increase in GSTpi binding. In contrast, complex formation between inactive JNKs and GSTpi occurs only if the JNK substrate, ATF2 is present. A more detailed investigation revealed that an interaction exists between GSTpi and ATF2, or between inactive JNK1, GSTpi, and ATF2 and not between GSTpi and inactive JNK2. In characterizing the GSTpi and ATF2 interaction further, we determined that the phosphorylation site of ATF2 is not involved in GSTpi binding and the xenobiotic site of GSTpi is the site of ATF2 binding. This study is the first to demonstrate and to characterize this novel interaction between ATF2 and GSTpi.
All experiments described here were done in vitro, and it is important to understand the physiological significance of these results. GSTpi is a transient inhibitor of JNK activity, likely shifting the balance away from JNK apoptotic signaling toward cell proliferation and differentiation. It may, therefore, be advantageous to design inhibitors that would prevent the interaction between GSTpi and JNK, as well as between GSTpi and ATF2. Inhibitors of these interactions may allow cancerous cells to shift away from the proliferation mode and be able to undergo apoptosis more easily, yielding better prognoses for cancer patients.
Materials and Methods
Production of human, recombinant GSTpi
A pUC120-WT-GSTpi plasmid (a gift from Dr. William Fahl, University of Wisconsin) was first transformed into DH5α E. coli cells (Invitrogen) to produce high concentrations of plasmid DNA. I105V and A114V mutations were constructed, one at a time, using QuikChange Mutagenesis kit (Stratagene), with the following forward and reverse primers (listed in the 5'-3' direction). Mutated codons are underlined.I105V: Forward: GACCTCCGCTGCAAATACGTCTCCCTCATCTACACCAAC;Reverse: GTTGGTGTAGATGAGGGAGACGTATTTGCAGCGGAGGTC; A114V:Forward: TCATCTACACCAACTATGAGGTGGGCAAGGATGACTATGTGAAG;Reverse: CTTCACATAGTCATCCTTGCCCACCTCATAGTTGGTGTAGATGAG. All samples were sequenced at the University of Delaware DNA Sequencing & Genotyping Center to confirm proper GSTpi sequences.
pUC120-GSTpi plasmid (Ile105/Ala114, Haplotype A and Val105/Val114, Haplotype C) was transformed into JM105 E. coli cells (ATCC), and GSTpi was expressed and purified as follows. Cultures (2 L) were grown at 37 °C to OD600 of 0.5, expression was induced with 1 mM IPTG and cells were grown overnight at 37 °C. The cell culture was centrifuged at 8000 rpm for 20 min at 4 °C and resuspended in 20 ml of 20 mM Tris HCl, pH 7.5, containing 300 mM NaCl, 1 mM EDTA and 1 mM DTT (GST Wash Buffer). Cells were lysed by sonication and sonicates were clarified by centrifugation at 10,000 rpm at 4 °C for 20 min. The purification was a modification of the previously published procedure.44 Supernatants were then applied to a column prepacked with 60 mL S-hexylglutathione-Sepharose resin (Sigma-Aldrich), which was equilibrated in GSTpi Wash Buffer and washed with 1 L of GST Wash Buffer. Enzymes were eluted using GSTpi elution buffer (GSTpi wash buffer containing either 2.5 mM S-hexylglutathione or 25 mM reduced glutathione). Fractions containing GSTpi, as determined by SDS-PAGE analysis, were pooled, concentrated and dialyzed into 20 mM Tris HCl, pH 7.6, containing 150 mM NaCl, 10% glycerol and 1 mM DTT (Kinase Storage Buffer) and stored in 1 ml aliquots at −80°C.
GSTpi enzymatic assays
The enzymatic activity of GSTpi toward 3 mM 1-chloro-2,4-dinitrobenzene (CDNB) and 2.5 mM reduced glutathione (GSH) was measured at 25°C using a Hewlett-Packard 8453 spectrophotometer, according to the method described by Ralat and Colman.6 Formation of the CDNB-GSH conjugate at 340 nm was monitored in 100 mM potassium phosphate buffer, pH 6.5 containing 1 mM EDTA in a total volume of 1 mL. The rate of spontaneous, nonenzymatic formation of the conjugate was subtracted from all measurements.
Determination of protein concentration
The concentrations of active and inactive His-JNKs, GST-ATF2 and untagged ATF2 were determined as was described previously.45 The protein concentrations of human GSTpi was determined by measuring the absorbance at 280 nm and utilizing the predicted extinction coefficient calculated using the Expasy ProtParam Tool Algorithm: (http://expasy.org/tools/protparam.html).
Gel electrophoresis, protein staining and western blotting analysis
Samples were resolved on a Tris-Glycine SDS-PAGE, pH 8.8 (15% or 4-20% acrylamide) or on a Bis-Tris SDS-PAGE, pH 8.7 (10% acrylamide) at room temperature and 140 volts for 90 min. Gels were either stained with Coomassie Blue stain or silver stained using Silver-Snap staining kit (Pierce) according to the manufacturer's protocol.
For Western blotting analysis, samples were resolved by SDS-PAGE at room temperature and transferred to Hybond ECL nitrocellulose membrane (Amersham) using Transfer Buffer (25 mM Tris, 192 mM glycine, pH 8.3 containing 20%(v/v) methanol) in an XCell-II blotting module (Invitrogen) at 60 mA overnight at 4°C. Membranes were then blocked using 5% dry milk (Fisher Sci.) in Tris-buffered saline, pH 7.6, containing 0.1% Tween-20 (TBS-T buffer) for 1 h. Membranes were washed three times for 10 min each in TBS-T buffer. To test for phosphorylation of ATF2, antibodies specific for ATF2 phosphorylated at both Thr-69 and Thr-71 (Sigma-Aldrich) were utilized. Antibodies against ATF2 (Cell Signaling Technology) were used at 1:1000 dilution, by treating the membrane for 1 h at room temperature. Antibodies to human GSTpi (MBL International) were used at a 1:2,000 dilution. The membrane was then washed in TBS-T buffer three additional times and treated with the appropriate anti-IGg secondary antibody (1:2000 dilution) tagged with horseradish peroxidase (Cell Signaling Technology) for 30 min at room temperature. After extensive washing with TBS-T (six times, 10 min each), the membrane was treated for 5 min with ECL Western Blotting substrate (Pierce) and imaged for 5 min using a luminescence filter on an ImmunoChem™ 8800 imager (AlphaInnotech).
Radioisotope incorporation experiments
To determine whether GSTpi serves as a substrate for JNK, active rat JNK2α2 was incubated with either purified WT GSTpi, double valine GSTpi or maltose binding protein (MBP)-tagged ATF2 (Sigma-Aldrich) in the presence of [γ32P] ATP and MgCl2. An aliquot of 25 μL of the radiolabeled ATP (250 μCi) was mixed with 975 μL of sterile water to prepare the radiolabeled ATP stock. Then 10 μL of the hot ATP stock was mixed with 990 μL of 40 μM unlabeled ATP solution (32P-labeled and unlabeled ATP stock). ATP incorporation reactions were prepared with 500 nM active JNK2α2, 5 μM ATP (labeled and unlabeled ATP stock), with either 2 μM GSTpi or 1 μM MBP-ATF2 in 40 mM Tris-chloride buffer, pH 7.5, with 20 mM MgCl2 and 0.1% BSA in a total volume of 500 μL. Reactions were incubated at room temperature for 1 h and aliquots of each sample (100 μL) were mixed with 20 μL of 6× SDS-PAGE sample buffer and boiled for 5 min. Samples were then separated on an SDS-PAGE gel, the gel was stained with Coomassie Blue stain, destained and soaked in a drying buffer (40% methanol, 10% glycerol) for 1 h and air dried overnight. The dried gel was placed in a 32P imaging cassette (Molecular Dynamics) overnight at room temperature. The cassette was placed in the Molecular Dynamics PhosphorImager SI and the location of the 32P-labeled bands was visualized.
Production of active phosphorylated JNKs, untagged unphosphorylated and phosphorylated ATF2
Preparation of phosphorylated, active human and rat JNKs and of untagged ATF2 (residues 19-96) was described previously.45 Fully phosphorylated ATF2 (10 μM) was prepared as described previously,35 with the following modifications. Nonphosphorylated ATF2 (10 μM) was incubated with 300 nM active His-JNK1α2, 2 mM ATP and 10 mM MgCl2 for 1 h at 25 °C. Full ATF2 phosphorylation was confirmed by Western blot analysis with antibodies to dually phosphorylated ATF2.
GSTpi binding experiments on Ni-NTA resin
For each sample, Ni-NTA resin (400 μL, settled) was placed in a 2 ml microcentrifuge tube and equilibrated with seven washes (1.6 ml each) of 20 mM Tris HCl buffer, pH 7.6, containing 150 mM NaCl and 10% glycerol (Complex Formation Buffer). After each wash, the resin was centrifuged at 3,500 rpm for 1 min. An aliquot of 3.5 μM of untagged ATF2 was incubated with or without 3.5 μM JNK (preincubated in Complex Formation Buffer) in a total volume of 500 μL at 25°C for 5 min, and added to the equilibrated resin. Proteins were allowed to bind to Ni-NTA resin for 15 min at room temperature with gentle rocking. Resin samples were then washed seven times (1.6 ml each) in a similar fashion as was done during resin equilibration. Human GSTpi (in 500 μL total volume, at various protein concentrations) was added to each resin sample containing bound ATF2 or His-JNK/ATF2 mixtures, and proteins were allowed to bind for 30 min at room temperature with gentle rocking. Protein/resin mixtures were then centrifuged at 3,500 rpm and unbound GSTpi was collected. Resin samples were then washed nine times with the Complex Formation Buffer (as described above) and complexes were eluted at room temperature for 10 min with gentle rocking using 250 μL of the Complex Formation Buffer containing 500 mM imidazole. For experiments conducted with ATF2 and GSTpi or with ATF2/His-JNK2 and GSTpi, two elutions were collected, while for experiments with ATF2/His-JNK1 and GSTpi, a total of four elutions were collected. Aliquots from each elution (50 μL) were pooled together, and 40 μL of each pool were mixed with 40 μL of 2× SDS-PAGE sample buffer for subsequent SDS-PAGE, silver stain and Western blot analysis. The concentration of GSTpi is proportional to the luminescence intensity in the Western Blots. To calculate the total nmol amount of eluted GSTpi (total elution volume of 0.5 or 1.0 mL), a 40 μL aliquot was used in a SDS-PAGE analysis.
Effect of S-methyl and S-hexylglutathione on complex formation
To determine if the presence of S-methyl or S-hexylglutathione had an effect on complex formation between GSTpi and ATF2, binding experiments were carried out as described in the previous section with the following modifications. After ATF2 (3.5 μM) was bound to the Ni-NTA resin (six samples) and washed four times (with 1.6 ml of Complex Formation Buffer), two samples were designated as negative control (no GSH treatment), two were designated as S-methylglutathione treatment, and two were designated as S-hexylglutathione treatment. Negative control samples were washed three times with complex formation buffer, while S-methylglutathione and S-hexylglutathione samples were washed three times with complex formation buffer containing either 2.5 mM S-methyl or 2.5 mM S-hexylglutathione, respectively. GSTpi solutions (7 μM, prepared in 500 μL volume of the appropriate complex formation buffer – no GSH, S-methyl or S-hexylglutathione) were incubated with the Ni-resin and washed with the appropriate buffer, as described above for the other GSTpi binding experiments. Two elutions with the Complex Formation buffer containing 500 mM imidazole (no GSH) were carried out for each sample. Elutions 1 and 2 for each sample were then pooled and mixed with SDS-PAGE sample buffer as described above for the GSTpi binding experiments,, and analyzed by SDS-PAGE, silver staining, and Western blot analysis.
Preparation of active JNK/ATF2 complexes
An aliquot of 3.5 μM active His-JNK (either JNK1α2 or JNK2α2) was mixed with 3.5 μM untagged ATF2 in the Complex Formation Buffer and incubated at 25 °C for 5 min. Active JNK/ATF2 complexes (500 μL) were bound to 400 μL of settled Ni-NTA resin and washed as described above (for GSTpi binding experiments on Ni-NTA). Complexes were eluted with two elutions of 200 μL Complex Formation Buffer containing 500 mM imidazole, and elutions were pooled together. Active JNK/ATF2 complexes were dialyzed overnight at 4°C against Complex Formation Buffer to remove imidazole.
JNK inhibition studies
Active JNK/ATF2 complexes (120 μL) were incubated with 10 μM WT or double valine GSTpi in a total volume of 150 μL for 30 min at 25 °C. ATP (6 mM) and MgCl2 (10 mM) were added to the inhibition reaction and phosphorylation of ATF2 (See footnote on page 3) by active JNK was allowed to take place for 15 min. Aliquots (30 μL) were withdrawn at various time points and mixed with 30 μL of 2× SDS-PAGE sample buffer to quench the kinase reactions. Samples were resolved on an SDS-PAGE gel and analyzed by Western blot using antibodies specific to doubly phosphorylated ATF2 (See footnote in Results).
Acknowledgments
The authors thank Dr. Thomas Hanson (Dept. of Marine Biosciences, University of Delaware) for insightful discussions. The authors are grateful to Dr. Daniel Simmons (Dept. of Biological Sciences, University of Delaware) for use of the Western blotting imaging equipment.
Glossary
Abbreviations:
- AP1
activator proteins 1
- ATF2
activating transcription factor 2
- CDNB
1-chloro-2,3-dinitrobenzene
- 1-Cys-Prx
1-Cystine Peroxiredoxin
- haplotype A
I105/A114 of GSTpi
- haplotype B
V105/A114 of GSTpi
- haplotype C
V105/V114 of GSTpi
- G-site
glutathione-binding site
- H-site
xenobiotic-binding site
- haplotype D
I105/V114 of GSTpi
- GSH
glutathione
- GST
glutathione S-transferase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- I-JIP
inhibitor of JNK based on JIP
- JIP-1
JNK-interacting protein 1
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- MBP
maltose-binding protein
- Ni-NTA
nickel-nitrilotriacetic acid
- SDS-PAGE
sodium dodecyl sulphate polyacrylamide gel electrophoresis.
Footnotes
The ATF2 used in this study was prepared from commercial ATF2 (residues 19-96) (BPS Bioscience), which is available as an N-terminal fusion with GST. This commercial preparation of GST-tagged ATF2 is expressed and purified as a full-length fusion, as well as a slightly shorter molecular weight fusion truncated at the ATF2 C-terminus. Because it is clearly undesirable to use GST-tagged ATF2 in studies designed to test the influence of GSTpi on JNK function, we removed the GST-tag using PreScission protease, and purified the untagged ATF2 as we have previously described.35 Like the original commercial GST-ATF2 fusion, the untagged ATF2 consists of two molecular weight species (one containing residues 19-96 of ATF2, and the second several amino acids shorter at the C-terminus). However, we have shown that both species were able to become phosphorylated on Thr-69 and Thr-71. Therefore, both ATF2 bands were included in all measurements of JNK catalytic activity.35
References
- 1.Holley SL, Fryer AA, Haycock JW, Grubb SE, Strange RC, Hoban PR. Differential effects of glutathione S-transferase pi (GSTP1) haplotypes on cell proliferation and apoptosis. Carcinogenesis. 2007;28:2268–2273. doi: 10.1093/carcin/bgm135. [DOI] [PubMed] [Google Scholar]
- 2.Henderson C, Wolf CR. Disruption of the glutathione transferase Pi class genes. In: Sies H, Packer L, editors. Hoboken, NJ: Methods in enzymology, Vol.401 Elsevier Academic Press; 2005. pp. 116–137. [DOI] [PubMed] [Google Scholar]
- 3.Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 4.Ralat LA, Manevich Y, Fisher AB, Colman RF. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45:360–372. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 5.Townsend D, Findlay V, Tew K. Hoboken, NJ: Methods in enzymology, Vol.401 Elsevier Academic Press; 2005. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets; pp. 287–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ralat LA, Colman RF. Monobromobimane occupies a distinct xenobiotic substrate site in glutathione S-transferase pi. Protein Sci. 2003;12:2575–2587. doi: 10.1110/ps.03249303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang YC, Misquitta S, Blond SY, Adams E, Colman RF. Catalytically active monomer of glutathione S-transferase pi and key residues involved in the electrostatic interaction between subunits. J Biol Chem. 2008;283:32880–32888. doi: 10.1074/jbc.M805484200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, Olin B, Mannervik B, Jones TA. Structure determination and refinement of human alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 9.Stenberg G, Abdalla AM, Mannervik B. Tyrosine 50 at the subunit interface of dimeric human glutathione transferase P1-1 is a structural key residue for modulating protein stability and catalytic function. Biochem Biophys Res Commun. 2000;271:59–63. doi: 10.1006/bbrc.2000.2579. [DOI] [PubMed] [Google Scholar]
- 10.McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams TA, Mars AE, Buyske SG, Stenroos ES, Wang R, Factura-Santiago MF, Lambert GH, Johnson WG. Risk of autistic disorder in affected offspring of mothers with a glutathione S-transferase P1 haplotype. Arch Pediatr Adolesc Med. 2007;161:356–361. doi: 10.1001/archpedi.161.4.356. [DOI] [PubMed] [Google Scholar]
- 12.Ji X, Tordova M, O'Donnell R, Parsons JF, Hayden JB, Gilliland GL, Zimniak P. Structure and function of the xenobiotic substrate-binding site and location of a potential non-substrate-binding site in a class pi glutathione S-transferase. Biochemistry. 1997;36:9690–9702. doi: 10.1021/bi970805s. [DOI] [PubMed] [Google Scholar]
- 13.Ralat LA, Misquitta SA, Manevich Y, Fisher AB, Colman RF. Characterization of the complex of glutathione S-transferase pi and 1-cysteine peroxiredoxin. Arch Biochem Biophys. 2008;474:109–118. doi: 10.1016/j.abb.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 15.Niu L, Chang KC, Wilson S, Tran P, Zuo F, Swinney DC. Kinetic characterization of human JNK2alpha2 reaction mechanism using substrate competitive inhibitors. Biochemistry. 2007;46:4775–4784. doi: 10.1021/bi602423e. [DOI] [PubMed] [Google Scholar]
- 16.Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 17.Heise CJ, Cobb MH. Expression and characterization of MAP kinases in bacteria. Methods. 2006;40:209–212. doi: 10.1016/j.ymeth.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, Cobb MH. Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J Biol Chem. 1997;272:11057–11062. doi: 10.1074/jbc.272.17.11057. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 20.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 21.Shaw D, Wang SM, Villasenor AG, Tsing S, Walter D, Browner MF, Barnett J, Kuglstatter A. The crystal structure of JNK2 reveals conformational flexibility in the MAP kinase insert and indicates its involvement in the regulation of catalytic activity. J Mol Biol. 2008;383:885–893. doi: 10.1016/j.jmb.2008.08.086. [DOI] [PubMed] [Google Scholar]
- 22.Ember B, LoGrasso P. Mechanistic characterization for c-jun-N-terminal kinase 1alpha1. Arch Biochem Biophys. 2008;477:324–329. doi: 10.1016/j.abb.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Tsuiki H, Tnani M, Okamoto I, Kenyon LC, Emlet DR, Holgado-Madruga M, Lanham IS, Joynes CJ, Vo KT, Wong AJ. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63:250–255. [PubMed] [Google Scholar]
- 24.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 25.Cui J, Holgado-Madruga M, Su W, Tsuiki H, Wedegaertner P, Wong AJ. Identification of a specific domain responsible for JNK2alpha2 autophosphorylation. J Biol Chem. 2005;280:9913–9920. doi: 10.1074/jbc.M412165200. [DOI] [PubMed] [Google Scholar]
- 26.Cuenda A. Mitogen-activated protein kinase kinase 4 (MKK4) Int J Biochem Cell Biol. 2000;32:581–587. doi: 10.1016/s1357-2725(00)00003-0. [DOI] [PubMed] [Google Scholar]
- 27.Nishina H, Wada T, Katada T. Physiological roles of SAPK/JNK signaling pathway. J Biochem. 2004;136:123–126. doi: 10.1093/jb/mvh117. [DOI] [PubMed] [Google Scholar]
- 28.Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, Caron PR, Wilson KP, Su MS. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure. 1998;6:983–991. doi: 10.1016/s0969-2126(98)00100-2. [DOI] [PubMed] [Google Scholar]
- 29.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 30.Cui J, Han SY, Wang C, Su W, Harshyne L, Holgado-Madruga M, Wong AJ. c-Jun NH(2)-terminal kinase 2alpha2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 2006;66:10024–10031. doi: 10.1158/0008-5472.CAN-06-0136. [DOI] [PubMed] [Google Scholar]
- 31.Marks F, Klingmuller U, Muller-Decker K. Cellular signal processing: an introduction to the molecular mechanisms of signal transduction. New York, NY, and Abington, OX14 4RN, UK: Garland Science, Taylor and Francis Group LLC; 2009. [Google Scholar]
- 32.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J Biol Chem. 2003;278:22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 35.Ranganathan PN, Whalen R, Boyer TD. Characterization of the molecular forms of glutathione S-transferase P1 in human gastric cancer cells (Kato III) and in normal human erythrocytes. Biochem J. 2005;386:525–533. doi: 10.1042/BJ20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley AJ, Lo Bello M, Battistoni A, Ricci G, Rossjohn J, Villar HO, Parker MW. The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J Mol Biol. 1997;274:84–100. doi: 10.1006/jmbi.1997.1364. [DOI] [PubMed] [Google Scholar]
- 37.Monaco R, Friedman FK, Hyde MJ, Chen JM, Manolatus S, Adler V, Ronai Z, Koslosky W, Pincus MR. Identification of a glutathione-S-transferase effector domain for inhibition of jun kinase, by molecular dynamics. J Protein Chem. 1999;18:859–866. doi: 10.1023/a:1020679229110. [DOI] [PubMed] [Google Scholar]
- 38.Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 40.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 41.Chen N, She QB, Bode AM, Dong Z. Differential gene expression profiles of Jnk1- and Jnk2-deficient murine fibroblast cells. Cancer Res. 2002;62:1300–1304. [PubMed] [Google Scholar]
- 42.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 43.Turella P, Cerella C, Filomeni G, Bullo A, De Maria F, Ghibelli L, Ciriolo MR, Cianfriglia M, Mattei M, Federici G, Ricci G, Caccuri AM. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res. 2005;65:3751–3761. doi: 10.1158/0008-5472.CAN-04-3903. [DOI] [PubMed] [Google Scholar]
- 44.Ralat LA, Colman RF. Glutathione S-transferase Pi has at least three distinguishable xenobiotic substrate sites close to its glutathione-binding site. J Biol Chem. 2004;279:50204–50213. doi: 10.1074/jbc.M407445200. [DOI] [PubMed] [Google Scholar]
- 45.Thévenin AF, Zony CL, Bahnson BJ, Colman RF. Activation by phosphorylation and purification of human c-Jun N-terminal kinase (JNK) isoforms in milligram amounts. Protein Expr Purif. 2010;75(2011):138–146. doi: 10.1016/j.pep.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]