

Abstract
Location of functional binding pockets of bioactive ligands on protein molecules is essential in structural genomics and drug design projects. If the experimental determination of ligand-protein complex structures is complicated, blind docking (BD) and pocket search (PS) calculations can help in the prediction of atomic resolution binding mode and the location of the pocket of a ligand on the entire protein surface. Whereas the number of successful predictions by these methods is increasing even for the complicated cases of exosites or allosteric binding sites, their reliability has not been fully established. For a critical assessment of reliability, we use a set of ligand-protein complexes, which were found to be problematic in previous studies. The robustness of BD and PS methods is addressed in terms of success of the selection of truly functional pockets from among the many putative ones identified on the surfaces of ligand-bound and ligand-free (holo and apo) protein forms. Issues related to BD such as effect of hydration, existence of multiple pockets, and competition of subsidiary ligands are considered. Practical cases of PS are discussed, categorized and strategies are recommended for handling the different situations. PS can be used in conjunction with BD, as we find that a consensus approach combining the techniques improves predictive power.
Keywords: peptide, binding site, drug, complex, solvent, co-factor, free energy, scoring, equilibrium
Introduction
Location of functional binding pockets on protein molecules is a cornerstone of structural genomics1–4 and targeted drug design projects. The advancement of experimental techniques, such as high throughput crystallography5,6 allows the atomic level determination of ligand structures bound to their protein pockets at an increasing rate. However, there are still many cases where the determination of the structure of the complex of a protein with its known ligand fails (1) or even the ligand is unknown (2), and still the knowledge of location of the functional pocket(s) is necessary.
The blind docking (BD) method has been introduced7,8 as an extension of the use of the very powerful docking engine AutoDock9,10 for the above-mentioned case 1 where the chemical identity of the ligand is known. During BD, the entire surface of the protein target is scanned for putative binding pockets of the ligand, and an atomic resolution complex structure is resulted. It was shown7,8 that in many cases, the primary, functional binding pocket of the ligand can be selected from among the identified pockets according to the binding free energy (ΔG) values corresponding to the interactions of the ligand with the different pockets. Notably, ΔG is produced on-the-fly by a scoring function during the docking procedure. Besides the location of the pocket of primary ligands, numerous studies11–15 have shown that the BD approach is useful in the solution of delicate problems such as the detection of subsidiary binding pockets containing e.g. exosites or allosteric binding sites.
In case 2, where the ligand is not known, only the protein sequence and/or structure can be used as input information. There are various site detection16 and pocket search17 (PS) methods available to accomplish this task. In Table I, a short summary is given on some PS methods used in this study. These methods are citation-classics (Q-SiteFinder24 and Pass26), and a novel, promising program Sitehound23 is also included. The PS algorithms use either geometrical or simplified, chemical grid-based search routines, and represent the putative binding pockets as a cluster of probe spheres. Since a PS does not use ligand information, it cannot provide the atomic resolution ligand-protein complex and the corresponding ΔG. Instead of ΔG, PS methods calculate other type of scores for ranking and selection of the most probable pockets. Such scores are based on the depth of the pocket or a sum of interaction energy values of the clustered probes with the protein.
Table I.
Overview of Blind Docking (BD) and Pocket Search (PS) Methods involved in this Study
| Name | Class | Search method | Scoring | References |
|---|---|---|---|---|
| AutoDock4 | BD without PS | Genetic algorithm | ΔG = EvdW+EH-bond+Eelec+ΔGsolv-TΔStors | 18,19 |
| EADockSF | BD with PS | LIGSITE-based PS and a subsequent local search using an evolutionary algorithm | ΔGSF=Eintra,L+Eintra,P+EvdW+Eelec | 20–22 |
| EADockFF | ΔGFF=ΔGSF+ΔGsolv | |||
| SITEHOUNDX | PS (chemical) | Chemical probes are placed on evenly spaced grid points, their IEs are calculated and binding pockets are defined as clustered grid points with highest TIE. | IEX=EvdW+Eelec TIEX=ΣEX,cluster (X=C or OP) | 23 |
| Q-SiteFinder | PS (chemical) | IEC= EvdW+EH-bond+EelecTIEC=ΣIEC | 24,25 | |
| Pocket-Finder | PS (geometrical) | Probe spheres are placed on evenly spaced grid points, and clustered into putative pockets. | The count of grid points which are well-buried in the protein (exceeding a pre-defined threshold). | 25 |
| PASS | PS (geometrical) | Protein surface is covered with layers of probe spheres. Pockets are predicted as active site points (ASP) having the largest weight among all probe spheres. | The weight of an ASP is proportional to the count of probe spheres in the vicinity and the extent to which they are buried. | 26 |
ΔG: free energy of binding. E: interaction energy between all ligand (L) and protein (P) atoms except cases where “intra” refers to intra-molecular interactions inside L or P. vdW: van der Waals-interactions. H-bond: hydrogen bonding interactions. Elec: electrostatic interactions. ΔGsolv: change of solvational free energy during ligand binding. T: thermodynamic temperature. ΔStors: change of entropy of internal rotations during ligand binding. SF: SimpleFitness scoring. FF: FullFitness scoring. IEX: Interaction Energy of a probe X with the protein target. TIE: Total Interaction Energy for probes in a cluster. C: probe mimicking a methyl group. OP: probe mimicking a phosphate group.
In BD calculations based on AutoDock, the docking of the ligand structure can be performed in parallel in, for example, 100 trials starting from 100 different random positions around the entire protein surface and this global search results in 100 putative binding modes (pockets) and the corresponding ΔG values. Thus, preliminary PS is not necessary in principle, as the numerous global search trials scan the entire protein surface at atomic resolution. However, in other docking packages such as EADock20,21 or GOLD, the PS is a necessary prerequisite of BD as the atomic level docking calculations are focused only on the pockets previously identified by PS. A recent study27 also suggests that a preliminary PS can improve BD by AutoDock, as well.
Despite the above-mentioned increasing knowledge on the application of BD, PS, and their combinations, detection of functional pockets and atomic level binding modes is still challenging for the following reasons. Generally, BD and PS methods identify many putative binding modes and pockets including the real one(s), but the scoring schemes cannot select the real, functional pockets in all cases. Ideally, the aim of BD and PS is the location of the primary pocket. However, in reality, there are subsidiary ligands (co-factors, solvent additives, ions, etc.) available for the same protein target. Together with the hydrating water molecules, the primary and subsidiary ligands compete with each other for the available pockets and can interfere with the equilibrium binding process of each other. Similarly, one ligand can bind to subsidiary, e.g. allosteric pockets besides its primary pocket on the protein.
To address the above problems and formulate some rules on the applicability of the BD and PS methodology, a comparative analysis was conducted using different search engines and scoring schemes (Table I) as follows.
The entire surface of the ligand-bound (holo) and primary-ligand-free (where available, apo) conformations of protein targets (Table II) were subjected to all BD and PS methods studied, and the results with the closest hits are summarized in the Supporting Information. From among the closest hits, Figures 1 and 2 list the top five rank numbers where the root mean squared deviation (RMSD) or the distance measured from the crystallographic ligand is smaller than 5 Å.
Only amino acid residues of the proteins were involved as target structures, that is, waters, ions, and all ligands were removed from the target during BD and PS. Importantly, even the modifications of native amino acids were removed in all cases to mimic the situation when a protein is built using only sequence data by means of structural genomics (homology modeling).
The most important part of our test set was composed of 10 protein targets which had been found problematic in previous studies7,8 using BD driven by AutoDock3.9 In these cases, BD had not been able to reproduce the correct crystallographic pose of the primary ligand (Ligand 1 or L1), and/or the scoring scheme had not been able to distinguish the accurately reproduced pose of L1 from the other, nonrelevant poses. In the latter cases, the correct pose had been incorrectly sorted into a higher (>1), energetically less favorable rank and, therefore, the identification of the appropriate pose as a 1st rank had failed. In this study, the above problematic complexes were considered as negative test cases and marked as “BD-failed” in Table II. There were also other two complexes for which BD had been successful (BD-passed) previously, and four proteins with drug ligands included as untested cases.
The methods were tested not only for finding the 17 primary ligand pockets (L1) but also for the rather difficult detection of 23 pockets of subsidiary ligands (Lj≥2), which turned out to be a real challenge for the methods. Thus, altogether 40 different structural complexes were considered. In cases where two or more binding pockets were available for the same ligand in the Protein Databank (PDB) complex, the binding poses in the different pockets were distinguished by small letters (Table II) after the numeric code of the ligand and used as separate references in comparisons.
Interference of ligands and hydrating water molecules on BD search was also explored to analyze the results of failed predictions.
Table II.
Protein-Ligand Complexes used for Evaluation
| Protein Codeb | Name | AA Countc | Water Countd | Apo structuree | RMS (Å)f | Ligand Code (j) | Nameg | Volume (Å3) | Categorya |
|---|---|---|---|---|---|---|---|---|---|
| PDB codes | |||||||||
| 1b70 | phenylalanyl tRNA synthetase | 1039 | 134 | 1pys | 0.42 | 1 | phenylalanine | 203 | BD-passed |
| 1cea | recombinant kringle 1 domain of human plasminogen | 79 | 148 | 1pkr | 0.50 | 1 | aminocaproic acid | 181 | Drug complex |
| 1dy4 | cellobiohydrolase I | 434 | 342 | 1cel | 0.30 | 1 | s-Propranolol | 338 | BD-failed |
| 2a | NAG 435 | 240 | |||||||
| 2b | NAG 436 | 240 | |||||||
| 1e7a | human serum albumin | 577 | 120 | 1ao6 | 0.89 | 1a | propofol 4001 | 255 | BD-failed |
| 1b | propofol 4002 | 255 | |||||||
| 1eqg | prostaglandin h2 synthase-1 | 550 | 251 | 1prh | 0.38 | 1 | Ibuprofen | 294 | BD-failed |
| 2a | NAG 661 | 240 | |||||||
| 2b | NAG 681 | 240 | |||||||
| 2c | NAG 662 | 240 | |||||||
| 2d | NAG 671 | 240 | |||||||
| 2e | NAG 1672 | 240 | |||||||
| 3a | BOG 802 | 383 | |||||||
| 3b | BOG 801 | 383 | |||||||
| 4 | HEME | 732 | |||||||
| 1h61 | pentaerythritol tetranitrate reductase | 364 | 545 | 1h50 | 0.21 | 1 | prednisone | 427 | Drug complex |
| 2 | FMN | 456 | |||||||
| 1hvy | human thymidylate synthase | 287 | 596 | 1hw3 | 0.77 | 1 | Raltitrexed (Tomudex) | 521 | Drug complex |
| 2 | UMP (covalently bound) | 293 | |||||||
| 1hz4 | transcription factor malt domain iii | 366 | 408 | 1 | benzoic acid | 147 | BD-failed | ||
| 2 | GOL | 114 | |||||||
| 1ivb | influenza virus b/lee/40 neuraminidase | 390 | 0 | 1 | 4-(acetylamino)- 3-hydroxy- 5-nitrobenzoic acid | 245 | BD-failed | ||
| 2 | NAG | 240 | |||||||
| 1ju4 | cocaine esterase | 569 | 436 | 3i2j | 0.22 | 1 | benzoic acid | 147 | BD-failed |
| 1lna | thermolysin | 316 | 158 | 1l3f | 0.62 | 1 | Val-Lys | 322 | BD-failed |
| 1m2z | human glucocorticoid receptor ligand-binding domain | 254 | 205 | 1 | Dexamethasone | 459 | Drug complex | ||
| 2a | BOG 501 | 383 | |||||||
| 2b | BOG 778 | 383 | |||||||
| 2c | BOG 779 | 383 | |||||||
| 1ngp | n1g9 (igg1-lambda) fab fragment | 431 | 131 | 1ngq | 0.29 | 1 | 2-(4-hydroxy-3- nitrophenyl) acetic acid | 204 | BD-failed |
| 1pth | prostaglandin h2 synthase-1 | 550 | 1 | 1prh | 0.36 | 1 | salicylic acid | 158 | BD-failed |
| 2a | NAG 661 | 240 | |||||||
| 2b | NAG 671 | 240 | |||||||
| 2c | NAG 672 | 240 | |||||||
| 2d | NAG 681 | 240 | |||||||
| 3 | BOG | 383 | |||||||
| 4 | HEME | 732 | |||||||
| 3pcn | protocatechuate 3,4-dioxygenase | 436 | 1374 | 2pcd | 0.39 | 1 | 2-(3,4-dihydroxy phenyl) acetic acid | 194 | BD-failed |
| 3tpi | trypsinogen-bpti complex | 287 | 152 | 1 | Ile-Val | 314 | BD-passed | ||
Categories of complexes according to previous investigations. BD-passed/BD-failed: BD of the ligand to the protein was successful/failed with AutoDock3 in the previous studies7,8. Drug complex: complexes with drug as ligand molecule bound to protein.
Protein code used as a reference in this study for both the holo and apo target forms (identical to the PDB ID of the holo conformation of the protein).
Number of amino acid residues in the protein target.
Number of crystallographic water molecules found in the holo PDB file and used for evaluation.
PDB ID of the primary-ligand-free (apo) conformation of the protein target (not used as a reference code in the text).
Root Mean Square Deviation between the Cα atoms of the holo and apo conformations of the target protein.
Abbreviated names of ligand molecules. NAG: N-acetyl-d-glucosamine. BOG: β-octylglucoside. FMN: flavin mononucleotide. GOL: glycerol. UMP: 2′-deoxyuridine 5′-monophosphate.
Figure 1.
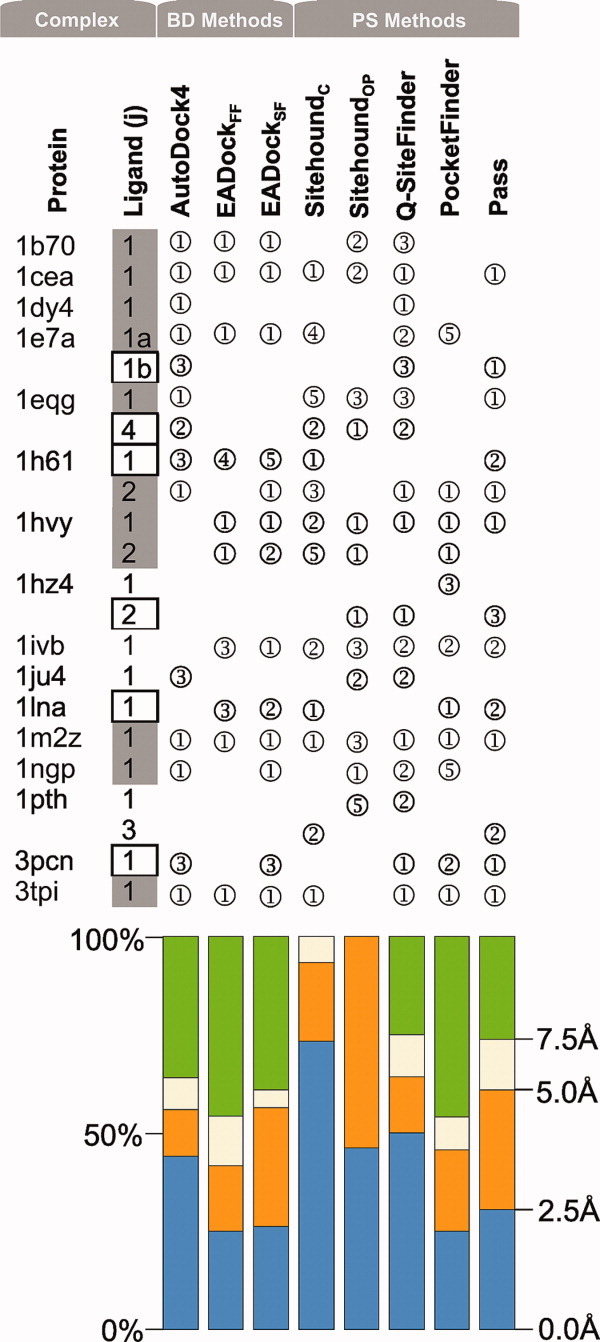
Successful predictions using the ligand-bound conformation of proteins as targets. Rank serial numbers of the top five Ranks with an RMSD/distance <5Å (compared with the crystallographic ligand pose) are listed in circles. Grey-filled boxes mark ligands with Category 1 predictions. Empty boxes denote Category 2 predictions (see Section Discussion for categories.) Color bars represent the precision of the methods in terms of distribution of the above RMSD/distance for complexes where the closest solution was found in the top five Ranks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
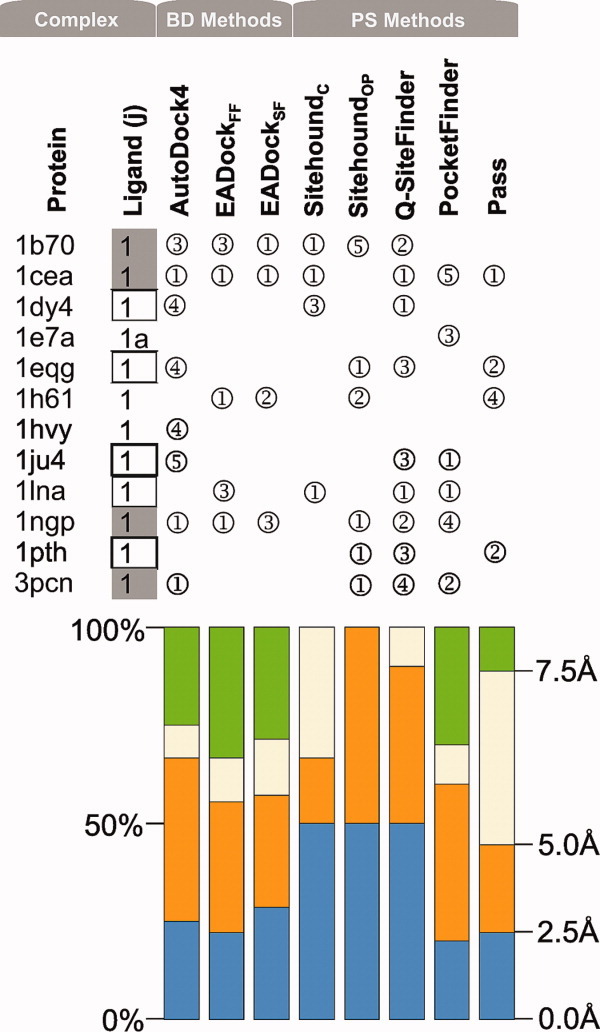
Successful predictions using the primary-ligand-free conformation of proteins as targets. Rank serial numbers of the top five Ranks with an RMSD/distance <5Å (compared with the crystallographic ligand pose) are listed in circles. Grey-filled boxes mark ligands with Category 1 predictions. Empty boxes denote Category 2 predictions (see Section Discussion for categories.) Color bars represent the precision of the methods in terms of distribution of the above RMSD/distance for complexes where the closest solution was found in the top five Ranks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The aim of this study is to give an estimate on the reliability of the methods in the case of problematic complexes. We are particularly interested if the PS methods of different background can help in the verification of binding pockets found by BD. We discuss whether a consensus (BD+PS) approach may show the way ahead toward functional protein pockets.
Results
Easy cases with primary ligands
Among the positive (BD-passed) test cases, the 3tpi protein is relatively small (Table II), its ligand is a dipeptide and, therefore, the holo protein form of this complex is an easy job for BD or PS. The RMSD of Rank 1 (Supporting Information) was an excellent 0.9 Å with AutoDock418,19 and 2.6 Å using EADock. The PS methods also found the pocket centrums as Rank 1 (Fig. 1) with good accuracy except SitehoundOP (Sitehound with phosphate probe), and the best algorithm in this case was Q-SiteFinder with a 0.8 Å distance. Notably, SitehoundOP has been recommended23 for phosphate-containing compounds originally. BD with both AutoDock4 and EADock successfully passed the other positive test case of holo form of 1b70, a very large target and also of two small drug-binding proteins 1cea and 1m2z. However, PS methods except SitehoundOP and Q-SiteFinder failed for 1b70 and Pocket-Finder failed in the case of 1cea. BD and PS were also successful in finding the L1 pocket on the apo form of 1cea and partly of 1b70 (Fig. 2, notably the PDB ID of the holo form of the protein is used as a code also for the apo form in this study). It was somewhat unexpected, that the two BD methods had only partial success in the cases of the holo target forms of two additional drug complexes 1h61-L1 and 1hvy-L1 in both ranking and precision. The only top 1 BD result was found with EADock, the 1hvy-L1, but the corresponding precision was still moderate with a 3.5 Å RMSD. For the apo forms, the success was also limited. The PS methods provided good hints for the holo target forms: SitehoundC, identified the L1 pocket as 1st rank for target 1h61, as well as SitehoundOP did for 1hvy. The other PS methods (Q-SiteFinder, Pocket-Finder and Pass) identified the L1 pockets correctly as a 1st rank at 1hvy and less correctly for 1h61 and for the apo target forms where the distance between the pocket center identified by PS and that of the real pocket was either above 5 Å or it was ranked too high (>5).
Problematic cases with primary ligands
Figure 1 shows that two BD-failed groups can be distinguished according to the performance of AutoDock4 on L1 complexes on the holo target form. In the first group, there are L1-complexes with 1dy4, 1e7a, 1eqg, and 1ngp. In these cases, the present AutoDock4 ranking of the correct pose improved to the 1st rank compared with our previous studies7,8 with AutoDock3, where they had not been found in the best rank. For example, the primary L1a pocket of propofol on 1e7a had been identified7 as Rank 2, whereas now it is located in Rank 1 [Fig. 2(a)] with a nice structural match. AutoDock4 produced a correct RMSD for all four cases. EADock reproduced the crystallographic L1 structures at targets 1e7a and 1ngp (the other two complexes were mis-ranked). In general, the PS methods were not successful in these four cases as they provided only some isolated good hits for the 1st rank at 1dy4 (Q-SiteFinder), 1eqg (Pass), and 1ngp (SitehoundOP). There were also some cases with the correct pose located in the 2nd and 3rd ranks by Q-SiteFinder and once by Pocket-Finder. For the apo target forms, BD generated a top 1 rank only for 1ngp, whereas for 1eqg and 1dy4 only Rank 4 was produced. The above-mentioned second group includes six targets (1hz4, 1ivb, 1ju4, 1lna, 1pth, 3pcn), where AutoDock4 could not improve the ranking/RMSD precision for L1 on the holo target forms compared with previous studies7,8 and failed. EADockSF found the correct pose in only one of six cases (1ivb) as Rank 1. At 1lna, EADockSF found the crystallographic ligand structure as Rank 2, which is remarkable as the Co2+-ion important in ligand binding was not used in this study due to our strict criteria of BD (Introduction). Unexpectedly, the PS methods performed fairly well for two-third (1ivb, 1ju4, 1lna, 3pcn) of this challenging group placing the real pocket into the first three ranks (Fig. 1). In the case of the apo target forms, BD failed to predict the pocket for this group with an exception of 3pcn. The PS methods provided good hints for the apo forms too (Fig. 2).
Subsidiary ligands and pockets
In the case of 1e7a, the same primary ligand (L1, Propofol) binds at two different pockets in the crystallographic structure [Fig. 3(a)]. The second binding pocket (L1b) of Propofol was identified as a 3rd rank by AutoDock4, and two PS methods (Q-SiteFinder and Pass) also placed it in the top three ranks using the holo target form (Fig. 1). No method found L1b on the apo protein structure. Besides the primary ligands, for eight (1dy4, 1eqg, 1h61, 1hvy, 1hz4, 1ivb, 1m2z, 1pth) of the sixteen protein targets of this study, there are also subsidiary ligands (Lj≥2) some of them having multiple binding pockets. The subsidiary ligands (Table II) can be divided into two groups: functional or structural partners (1) and small molecules or solvent additives (2).
Figure 3.
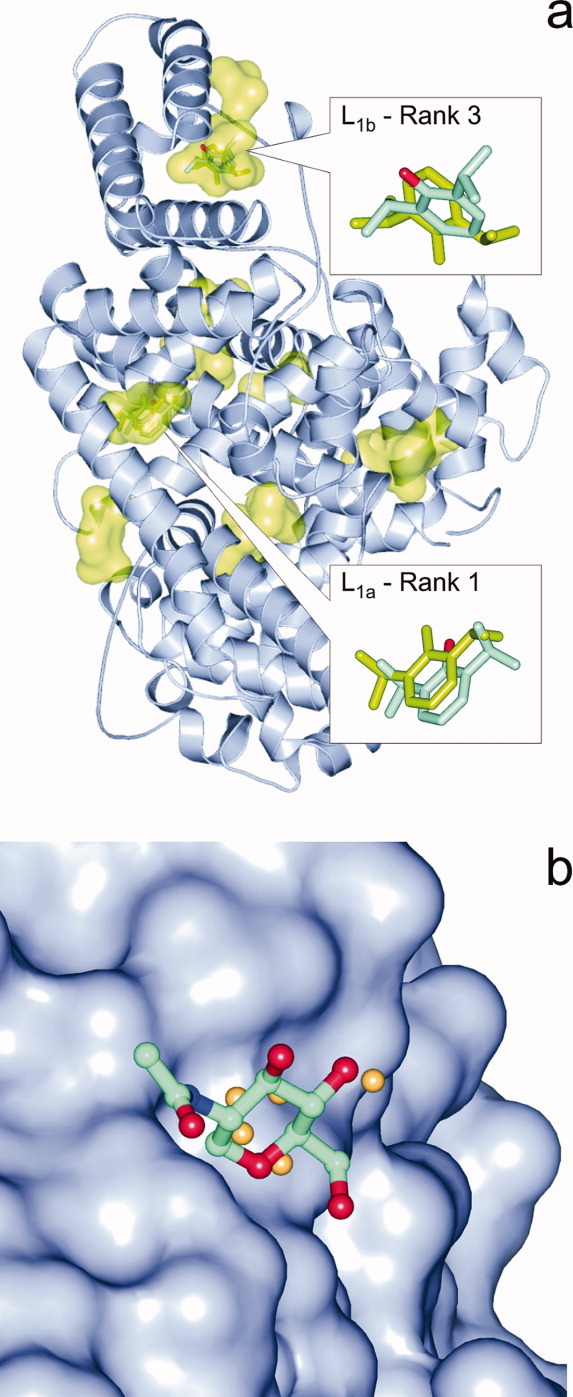
(a) In PDB structure 1e7a two binding pockets of the primary ligand (L1) propofol had been detected by crystallography. Whereas the L1a pocket and the binding mode was identified precisely by BD (shown in inset as green sticks) as Rank 1 and Q-SiteFinder, the L1b pocket was located by PS methods and BD found it as Rank 3 (green sticks) with a rather high deviation from the crystallographic position (sticks colored by atom type). Other pockets found by BD are also shown as green surfaces. (b) Sitehound identified the shallow pocket (protein shown as surface) of NAG in the complex 1eqg-L2d in a 2.9 Å distance from the crystallographic ligand position (balls and sticks colored by atom type) by placing a few Carbon probes (beige spheres) into the proposed pocket. The small number of probes resulted in a low TIE value and a mis-ranking of this real binding pocket into the 88th of 112 ranks. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The first group contains strong binders such as the nucleotides (FMN, UMP) and the HEME. In the complexes of FMN and UMP (1h61-L2 and 1hvy-L2), the co-factors are located close to L1 discussed above. Thus, in both complexes, L1 and L2 interact with each other and their binding pockets are not separated influencing the docking results (see also next section). In the case of FMN as L2, the BD methods performed better for the L2 than for the L1 at target 1h61 (only holo protein structures were used as targets for subsidiary ligands). However, the only acceptable solution was produced for 1h61-L2 by AutoDock4. SitehoundOP identified the 1st rank for the phosphate containing L2 (UMP) of 1hvy similar to its L1 earlier and SitehoundC ranked the L2-s into worse ranks than the L1-s (Fig. 1). The other PS methods (Q-SiteFinder, Pocket-Finder and Pass) at most identified the L2 pockets for both complexes correctly. HEME is the largest ligand investigated with a 732 Å3 molecular volume (Table II). It is part of two target-ligand complexes, the 1eqg-L6 and the 1pth-L7. Although these complexes were not re-produced perfectly by BD, a 2nd rank at 1.4 Å RMSD (1eqg) and a 1st rank with a 7 Å RMSD (1pth) were obtained with AutoDock4. PS methods Sitehound and Q-SiteFinder worked well in the case of 1eqg but none of them were really successful for 1pth.
Molecules of the second group are loose binders (BOG, GOL, NAG), and/or they sit in a shallow surface pocket [Fig. 3(b)] of the protein in question. Ligands of this group can be found in various complexes (Table II). Neither BD nor PS methods were successful in correct identification of their binding poses or pockets. There were few isolated cases where the methods produced good hints, such as Pass (1m2z-L2c, 1pth-L3, 1hz4-L2), SitehoundC (1pth-L3), SitehoundOP, and Q-SiteFinder (1hz4-L2).
The influence of subsidiary ligands
Subsidiary ligands of a target protein are not just test cases of BD and PS (previous section), but they also introduce difficulties due to their inherent competition for the pockets in real life (Introduction). Thus, the scoring function of BD has to be selective enough for a ligand j (Lj) to distinguish its primary crystallographic pocket not only from its subsidiary pockets but also from pockets occupied by other, competing ligands n (Ln≠j) on the same target protein. To gain information on this kind of selectivity of BD, it is useful to check whether the crystallographic binding poses of Lns on the same target indeed interfere with the (mis)docked Lj poses. Ideally, if the BD method is precise and selective enough, then no interference should be measured between the docked Lj pose in the 1st rank and the crystallographic poses of other Lns. In other words, Lj should occupy its crystallographic conformation with the lowest ΔG (Rank #1) and bind to crystallographic pockets of other Lns (or waters, see next section) at Ranks>1 of higher ΔG values. Such interferences were checked at both BD methods and ranks on all holo target forms, by the measurement of the distances between the docked Lj and the crystallographic Ln poses (see Methods for details). The Lj ranks with significant Ln interferences (<5 Å distance) found are listed in the Supporting Information.
As it was expected, a comparison with the results (Fig. 1, Supporting Information) shows no interference at Rank 1 in the cases where the BD method identified the crystallographic pocket of Lj correctly. Some interferences can still be found for these positive cases but only at Ranks >1. To illustrate one of the examples mentioned, Figure 4(a) depicts prostaglandin h2 synthase-1 (1eqg) with 10 binding poses of L1 (Ibuprofen) representing the 10 ranks found by AutoDock4 (Supporting Information). There is only one point of interference between the 10 rank-representatives of L1 and the other ligands (Ln): L4 (HEME) sits at the same place as Rank 8 of L1 [Fig. 4(a)]. Naturally, as this interference occurs at Rank 8, it corresponds to a higher ΔG of L1, and therefore, it can be concluded that BD could discriminate between the binding of L1 to its real site and to the HEME-site by assigning a lower ΔG for the real one.
Figure 4.
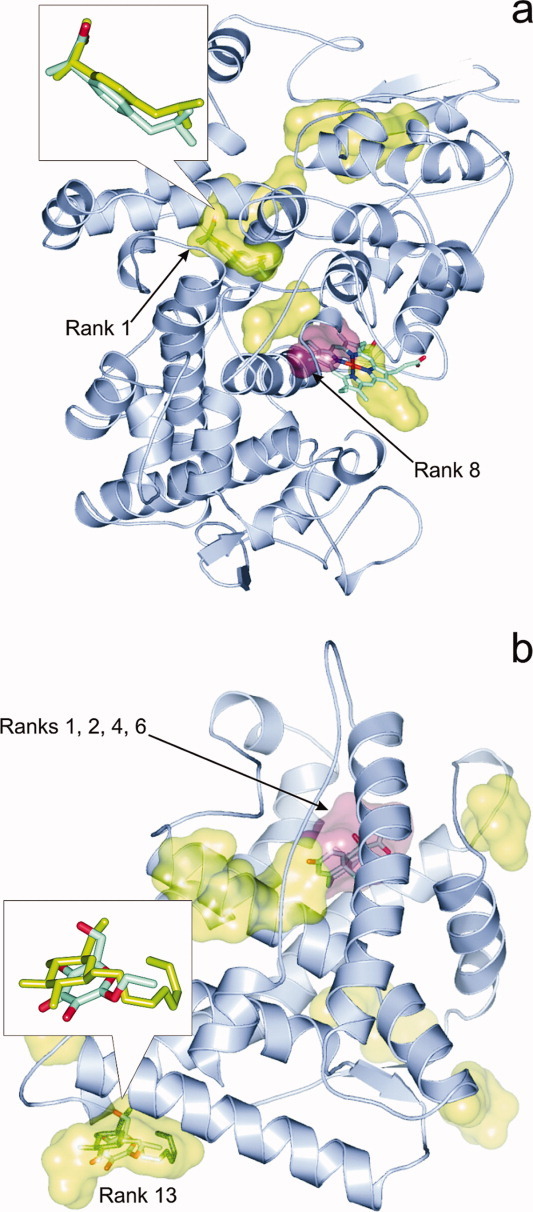
(a) Prostaglandin H2-synthase-1 (1eqg, cartoon) and the binding pockets (green surface) of the primary ligand Ibuprofen corresponding to the 10 ranks found by AutoDock4. In the inset, the overlap between crystallographic (sticks colored by atom type) and docked (sticks in green) Rank 1 conformations of Ibuprofen is featured. Interference of Rank 8 pocket (pink surface) with HEME (sticks colored by atom type) did not affect the results of BD. (b) The binding pockets of BOG (green) identified by AutoDock4 on the surface of human glucocorticoid receptor (1m2z, cartoon). In the inset, the overlap between the crystallographic (sticks colored by atom type) and docked (sticks in green) Rank 13 conformations of BOG is shown. Notably, the octyl group of BOG was not assigned in the crystal structure and the B-factors of the assigned atoms are relatively high (76-90). Predicted pockets of BOG corresponding to Ranks 1, 2, 4, and 6 (pink surface) interfere with the pocket of the primary ligand dexamethasone (sticks colored by atom type), which is partly responsible for the mis-ranking of the correct pocket only as Rank 13. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Besides analyses of the above-mentioned positive examples, checking ligand-interferences may provide even more important information in the negative cases where the crystallographic pocket of Lj was not identified correctly as Rank 1. For example, at complexes 1dy4-L2, 1eqg-L3a(n = 4), 1h61-L1, 1m2z-L2c (AutoDock4), and at 1h61-L1,2, 1m2z-L2c, 1pth-L3 (EADock), there are interferences (Supporting Information) with Lns at ranks with serial numbers equal to/smaller than the best ranks listed in Figure 1. The 1m2z-L2 complex is such an example [Fig. 4(b)], where the binding site of BOG was identified on the surface of human glucocorticoid receptor (1m2z) at the L2c pocket with a reasonably good fit to the crystallographic conformation at 2.2 Å RMSD. However, predicted pockets of BOG corresponding to Ranks 1, 2, 4, and 6 overlap with the pocket of the competing primary ligand dexamethasone (L1), and this interference is partly responsible for the mis-ranking of the correct pocket only as Rank 13 of 14 (Supporting Information).
Another example is 1h61-L1, where the solution for prednisone (L1) with the best RMSD (1.0 Å) was placed to the 3rd rank of the total of 10 ranks found by AutoDock4 as a consequence of L1 interference with FMN [L2, Fig. 5(a)] in the cases of Ranks 1 and 2. As the mis-docked L1 poses at Ranks 1 and 2 adopted a lower ΔG at the L2 binding site (not shown in the figure), the crystallographic pocket was identified only as Rank 3 suggesting that BD was not energetically selective enough to favor the real pocket of L1 over the actual pockets of another ligand (L2).
Figure 5.
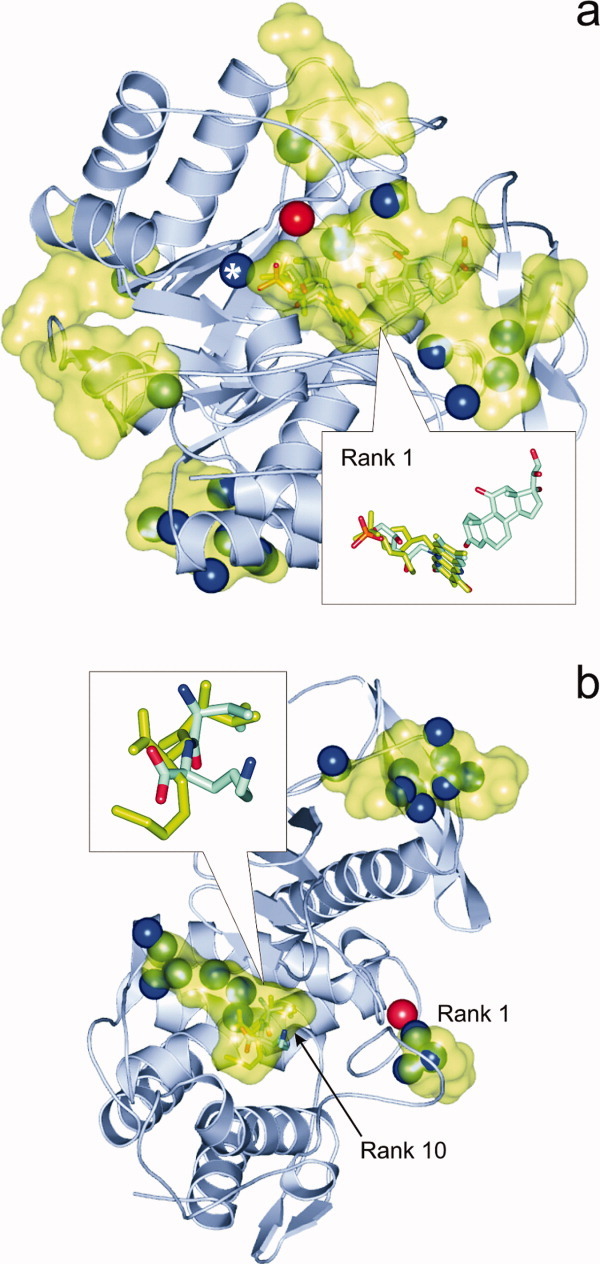
(a) Pentaerythritol tetranitrate reductase (1h61, cartoon) and the binding pockets (green surface) of subsidiary ligand FMN corresponding to the 10 ranks found by AutoDock4. In the inset, the overlap between crystallographic (sticks colored by atom type) and docked (green sticks) Rank 1 conformations of FMN is featured. Notably, the primary ligand prednisone (sticks colored by atom type, in the right corner of inset) is located in an adjacent pocket very close to FMN resulting in ligand-interference and mis-ranking during the docking of prednisone. Blue and red spheres depict the positions of crystallographic water oxygen atoms inside (Type 1) and the bottom (Type 2) of the binding pockets, respectively. The oxygen atom marked with an asterisk represents the only type 1 water interfering with Rank 1 (see main text for details). (b) In the case of docking of ValLys to thermolysin (1lna, cartoon), the binding pocket of the ligand was found as a 10th rank, whereas, for example, in the 1st rank identified by docking two crystallographic water molecules (blue spheres) are sitting in reality. In pockets of Ranks 2…9 (green surfaces), there are 18 water molecules (blue spheres) occupying the binding positions instead of the ligand. In the inset, the overlap between crystallographic (sticks colored by atom type) and docked (green sticks) Rank 10 conformations of ValLys is featured showing a large deviation in the position of the charged side-chain. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The influence of hydration
Like the subsidiary ligands discussed above, solvent molecules can also compete with the binding of a ligand in question. Due to their different locations related to the (docked) ligand and the target protein, there are two types of water molecules distinguished in the present investigation. As BD is usually performed for “dry” protein target, it is possible that predicted ligand binding sites in fact are occupied by solvent molecules. These water molecules sit inside the pocket and are classified as Type 1 water in this study (see also Methods). There are also other water molecules located at the interface between the (docked) ligand and the protein target, at the bottom of the pocket (Type 2) not occupying the docked ligand position. Whereas the Type 1 waters surely compete with the ligand for the pocket, and in the real situation hinder its binding, Type 2 water molecules are not obviously expected to block ligand binding to the actual pocket as they can also assist the ligand-protein interaction via bridging or as spacers. Water-interferences with the representative docked ligand structures at all ranks for both BD methods are tabulated in the Supporting Information.
Similar to the previous section in most of the positive cases of Figure 1, there are no Type 1 water molecules interfering with the docked Lj positions at Rank 1 except three cases (1h61-L2, 1ngp-L1 at AutoDock4 and 3tpi-L1 at EADock) each of them with only one water molecule inside the pocket. At the same time, in Ranks=2, there are several waters located at the same place as the docked ligand. This finding shows that the scoring function was able to distinguish between the real (Rank 1) pocket of an Lj ligand and the others filled with Type 1 water molecules assigning higher ΔG a for the latter pockets. For example, in the case of 1h61-L2 169 Type 1 water molecules can be found (Supporting Information) at different Ranks=2 of higher ΔG values calculated by AutoDock4 and only one Type 1 water molecule [Fig. 5(a)] is located in the middle of the pocket corresponding to Rank 1. The excellent 1.5 Å RMSD between the Rank 1 and the crystallographic L2 conformation also shows that interference of only one Type 1 water molecule may not destroy the correct ranking of the pocket. In these cases, the absence of one Type 2 water molecule also did not influence the correct ranking order during docking (robustness).
Further comparison of the docking results and the table on water interferences (Supporting Information) shows that there are several negative cases, where a ligand was mis-docked into pockets corresponding to lower ranks filled with Type 1 water, in reality. Such examples are 1h61-L1, 1hvy-L1,2, 1hz4-L2, 1ju4-L1, 1lna-L1, 1m2z-2c, 3pcn-L1 (AutoDock4) and 1h61-L2, 1hvy-L1, 1lna-L1, 3pcn-L1 (EADock). For example, at 1lna-L1 [Fig. 5(b)] Rank 10 holds the closest docked ligand with an RMSD of 2.8 Å. Interferences of the docked L1 poses of all higher Ranks (1…9) with Type 1 waters are shown among the AutoDock4 results. Remarkably, in Rank 4, there are four water molecules sitting in the place of L1. In the case of, for example, 3pcn-L1 (AutoDock4) the lack of three Type 2 water molecules (Rank 3) also contribute to the mis-docking of the corresponding pose.
Discussion
The results of this study are categorized according to the success of BD and PS methods in the following sections.
Category 1: at least two different BD or at least one BD and one PS method provides a successful, consensus prediction in Rank 1
Figure 1 shows that in 11 of all 40 complexes (9 of 17 L1-complexes) a valid Category 1 prediction could be produced in case of the holo target forms. Remarking that our test set contained mainly problematic complexes, it can be concluded that in the case of primary ligands, for half of the complexes a good consensus prediction can be achieved. This ratio is much less (4 of 17) for apo targets (Fig. 2). In four BD-failed cases (1dy4, 1e7a, 1eqg, 1ngp), the crystallographic position of L1 was identified correctly using the holo targets as Rank 1 in this study, whereas in the earlier papers7,8 using AutoDock3 these complexes were listed in higher ranks. This improvement may be due to the modified solvation term19 of the scoring function (Table I) of AutoDock4. However, in case of the apo targets BD was successful at 1ngp only (Fig. 2).
In general, the ratio of the top five ranks at <2.5 Å with AutoDock4 dropped to almost the half at the apo forms (Fig. 2) compared with the holo targets (Fig. 1). At the same time, the ratio of the top five ranks with lower precision (2.5…5 Å) increased for the apo targets with AutoDock4 resulting that >50% of the top five ranks is below a 5 Å precision limit. The precision of EADock is low for both target forms. Notably, for subsidiary ligands only one consensus prediction was achieved 1h61-L2 (Fig. 1). The reason of the low success rate at these ligands can be attributed to their disturbing interference with the primary ligand and their higher (less specific) binding energy.
In practice, consensus pockets of Category 1 are the most reliable BD predictions as they are supported by a different BD and/or PS methods.
Category 2: only PS methods provide successful predictions in Rank 1, and BD methods fail
The precision of PS methods is fairly independent on the target form. The ratio of the best (<2.5 Å) solutions dropped with about 20% (Fig. 2) at the apo forms in the case of SitehoundC, but the precision of the other four PS methods remained practically the same if compared with the holo forms (Fig. 1). Considering that PS methods are generally based on a simplified search and scoring scheme (Table I) it may be somewhat surprising that they outperform the atomic-level BD calculations producing Rank 1 hits for an additional 6/40 (holo form) and 5/17 (apo form) complexes of this category (Figs. 1 and 2).
We have previously demonstrated7,8 that AutoDock reproduces many protein-ligand complexes faithfully using holo forms of the proteins. Here we find that, despite the above-mentioned (Section Category 1) improvements in the energy function of AutoDock version 4, some targets remain difficult especially their apo forms. Since the BD protocol tries to dock the entire ligand, a somewhat smaller/closed cavity in an apo structure may preclude insertion of the ligand. In some cases, the decline of ranking precision of BD methods on the apo forms (Fig. 2 vs. Fig. 1) can be attributed to the large change in overall protein conformation as indicated by the RMS Cα distances (1e7a, 1hvy) or by local conformational change of the binding pocket residues (1cel, 1h61). PS algorithms might still detect the cavity however.
Similar to the docking methods, the scoring schemes of the PS methods are also generally cumulative (Table I), that is, the total interaction energy (TIE) score is the sum of individual interaction energy values (Sitehound, Q-SiteFinder) of probes or weighted count of probes (Pocket-Finder, Pass). Therefore, it can be expected, that large pockets corresponding to large ligands (with many interactions) will be found easily by PS at it was the case for 1eqg-L4, with the largest ligand HEME (732 Å3, Table I) or the still considerable 1hvy-L1 (raltitrexed, 521 Å3). However, in the other (1pth) complex of HEME its pocket was not found, and small pockets of, for example, ligands 3pcn-L1 (194 Å3), 1hz4-L2 (114 Å3), and 1ju4-L1 (147 Å3) were identified correctly as Rank 1 or 2 showing that pocket size is not the only factor which contributes to the good performance of PS in this category.
A possible reason of the success of PS methods may arise from the nature of the grid of the probes used for the above cumulative scoring. Whereas in BD only the few atoms of a ligand molecule are used for calculation of interaction energies, in PS the count of probes/grid points of the identified pocket can exceed the number of atoms of the actual ligand [Fig. 6(a)]. In contrast with the connected atoms in the molecules, the location of grid points is determined by their even spacing and all of them are retained within a cluster without any concern on their possible connections. Thus, if the clustering algorithm of a PS works accurately, a functional pocket with large number of probe interactions/grid points will be ranked with a large energy difference [Fig. 6(b)] into the first rank rather than into the lower ranks by PS. In the case of PS, the TIE scores the general functionality of the pocket and not only its suitability/availability for the possible binding conformations of a ligand as is done by ΔG in the case of BD. A recent study suggests28 that these kinds of robust scores and approaches representing multiple binding modes at a pocket may reflect the inherently dynamic nature of ligand binding. In this sense, the TIE may be considered as a score of pocket functionality in certain situations [Fig. 6(a,b)], where the small ΔG differences lead to mis-ranking of pockets in BD even if its ΔG scoring is more sophisticated (multiple atom types, 3D geometry, connectivity, etc. considered) than the PS scoring like TIE, which is generally based on a single grid/probe type. Notably, this benefit is highly dependent on the robustness of the clustering scheme (the selection procedure of relevant probes/grid points for the proposed pocket) of PS and has mostly geometrical and no physical background. Obviously, the success of the PS scoring is not guaranteed. For the tricky situation of 1e7a-L1 where the same ligand occupies two different pockets (a and b), two PS methods (Q-SiteFinder and Pass) with different background (Table I) provided a good hint (Fig. 1) for the second pocket L1b [Fig. 3(a)] using the holo target, and at the same time L1a was not identified by Pass (only by Q-SiteFinder). On the apo target, no PS methods ranked pocket L1b in the top five. This example of complex 1e7a-L1 shows the limitations of PS methods and the necessity of consensus pocket identification by at least two PS methods for a reasonable prediction.
Figure 6.
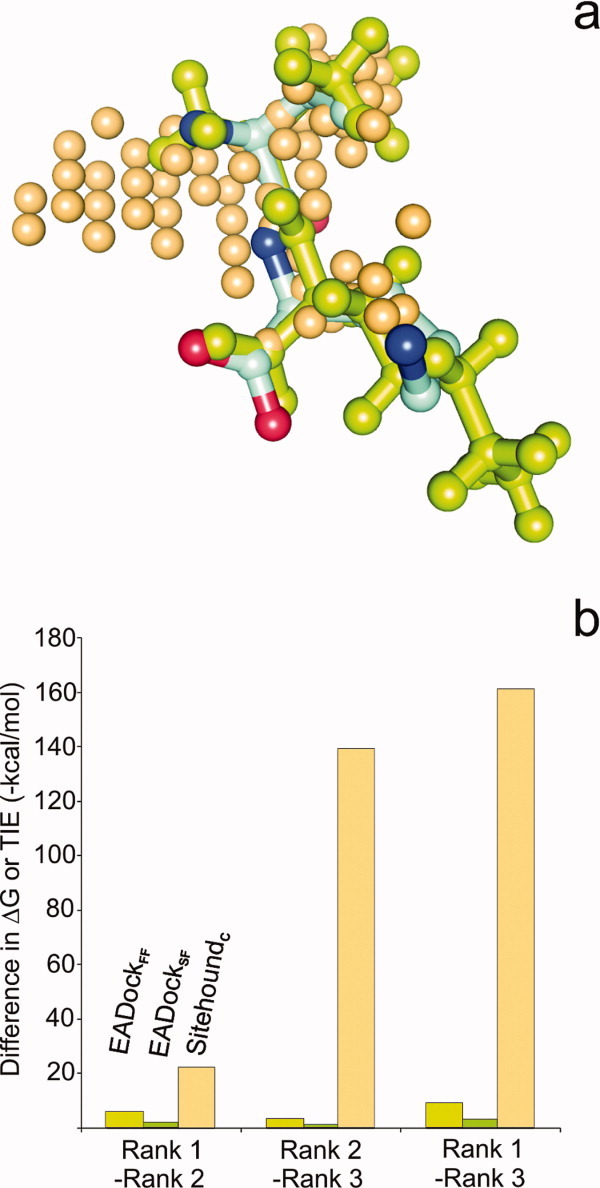
(a) The number of evenly and tightly spaced probes (beige spheres) representing the pocket according to SitehoundC is significantly larger than the number of atoms of the ValLys primary (L1) ligand molecule docked by EADock (green balls and sticks). Protein thermolysin (1lna) is not shown and the crystallographic ligand conformation is represented by balls and sticks colored by atom type. Although the docked and crystallographic ligand conformations match with each other, this correct solution was placed to only Rank 3 according to EADockFF scoring. (b) Pairwise differences of the first three ranks in terms of binding free energy ΔG values calculated by EADock scoring schemes and the TIE values obtained by SitehoundC for the 1lna-L1 complex. According to summation of interaction energy values corresponding to the relatively large number of probes shown in part (a) the differences in TIE values is larger than the differences between the ΔG values obtained for the few atoms of the docked ligand. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In summary, Figures 1 and 2 suggest that independent top (1…3) rankings of the same pocket by 2-3 PS methods with different scoring and search schemes serve as an indication of a functional pocket even if BD fails to identify the pocket in question.
Category 3: all BD and PS methods fail to identify the pocket in Rank 1
Whereas pockets of the main ligands (L1) were identified at least by one BD and/or PS method (Fig. 1), this was not the case for the co-factors and weak binders, and therefore, most of the Lj≥2 ligands fall into Category 3. A possible reason of the low performance of the BD methods in this category could be that these ligands are mostly weak binders and, therefore, the afore-mentioned energy difference between the Ranks is even smaller than it was in the cases of Category 2. Furthermore, as these ligands bind generally small and/or shallow pockets on the protein surface the above advantage of the cumulative grid scoring of the PS methods also cannot prevail due to the small number of selected (clustered) grid points/probes [Fig. 3(b)], defining the pocket, if any. Interferences with other ligands and hydrating water molecules detailed in section Results provide additional explanation for the failure of BD. The results on the influence of subsidiary ligands show that mis-docked Lj≥2 ligands may find the well-defined binding pocket of, for example, the main ligand L1 [Fig. 4(b)] or of a large ligand. BD methods similarly failed, in the cases where the ligand was mis-docked in the pocket of two or more hydrating water molecules. Both types of interferences are results of the inadequate scoring function, which cannot distinguish energetically between the fine differences occurring at binding to different pockets. In the particular case of interferences of hydrating water molecules, the inappropriate solvation term (Table I), whereas for ligand interferences the whole scoring scheme is responsible for the lack of energy differences between the Ranks.
Category 4: only BD methods provide successful predictions in Rank 1 and all PS methods fail
This is a pseudo-category as the above situation did not occur in any of the 40 complexes. Once a BD method produced a successful (Rank 1) prediction, there was always at least one PS method providing the same pocket in Rank 1 and in one case in Rank 2 (Figs. 1 and 2). This finding is very important for the verification of Rank 1 hits in future BD studies showing that a true Rank 1 prediction by BD should be accompanied with at least one positive Rank 1…3 PS prediction for the same pocket. If it is not the case then it is suspicious that indeed the pocket was mis-found by BD and we face a Category 3 situation.
Conclusions
Recommendations
To estimate the reliability of a future search for the main (lowest energy) functional binding pocket on the entire protein surface, some rules can be concluded. (1) A Category 1 consensus pocket can be considered as a reliable prediction in most of the cases as it is based on positive Rank 1 results obtained by at least two different methods (sufficient and satisfactory conditions). (2) It is a suspicious situation if the pocket found by BD is not verified by at least one of the PS methods (pseudo-Category 4). This case may easily be indeed a Category 3, where the binding pocket is mis-found by inappropriate modeling or interferences of other binding partners. (3) If at least 2-3 different PS methods result in the same pocket which does not match the BD prediction (Category 2), then further investigations are necessary. For example, a local re-docking may be necessary including, for example, molecular dynamics calculations with explicit water surrounding to obtain an improved complex structure. As PS methods are very fast (some seconds), their above use for verification of BD results or prediction will not slow down the work.
Methodological aspects
The main problem with the BD and PS methods is that they produce a large number (=10) of possible ranks and corresponding pockets (Supporting Information) with very small differences (BD) in their scores in many cases. In this study, it was investigated whether among the many ranks, the consensus top ranks from BD and PS contain the real, functional pocket represented in the crystallographic structure or not. For this, a test set containing many problematic and/or weakly bound complex structures was used. It was also shown how other factors such as the interference of subsidiary ligands and/or a group of two or more Type 1 hydrating water molecules can negatively influence the BD results. The comparison of the quality of the methods for a non-BD (focused docking) problem was not the aim of the study, as there are numerous thorough analyses available, for focused (restricted) docking search. Similar to other studies,7,28 the problem of protein flexibility was addressed in this study by the involvement of apo structures with ligand-free pocket conformations.
Multiple functionality
Beyond the crystallographic pocket (ideally corresponding to Rank 1, the global energy minimum), there may be others with equal or even more important function (e.g., allosteric binding sites) located in BD Ranks>1. Moreover, it can happen that the same ligand has more than one experimentally determined binding pockets [e.g., propofol on 1e7a, Fig. 3(a)]. The above recommendations were not meant for these pockets. In the case if the detection of these pockets is necessary, then (consensus) BD and PS Ranks>1 should be also considered and the corresponding sites checked by, for example, experiments.
Methods
Preparation of protein and ligand molecules
All protein-ligand complexes including the holo protein form and primary ligand–free (apo) protein structures (where available) were obtained from the PDB. All apo structures were superimposed on the holo structures and the respective RMSD measured between the Cα atoms of the holo and the superimposed apo protein structures are listed in Table II. This superimposition step allowed a comparison of the results on the apo structures with the holo-bound crystallographic ligand position. A list of the PDB codes is provided in Table II and used for identification of the protein in this study. All chains available in the PDB file were processed except the following cases where only the first copy of identical chains was used (chain identifiers listed in brackets): 1cea, 1e7a, 1eqg, 1hvy, 1m2z, 1pth (chain A), 1ngp (chains L and H), 3pcn (chains A and M). All nonamino acid (AA) residues and ligands were removed from the target proteins. AA side-chains containing post-translational modifications and non-AA (HETATM) groups were changed to contain only AA parts by deletion of the HETATM parts. That is, the first residue of 1dy4 was deleted, the Cme43 residue of 1hvy was mutated to Cys, and the Oah530 residue of 1pth was mutated to Ser. The use of only AA-containing targets allowed the study of real situations where no information on post-translational modifications is available. For BD with AutoDock4 and EADock the protein molecules were equipped with H-atoms using AutoDock Tools.18 The ligand molecules (Table II) including co-factors and solvent additives were equipped with H-atoms and energy-minimized using Mopac 629 with a PM3 Hamiltonian and eigenvector following routine for energy minimization (except of HEME for where the crystal structure was used). In all cases, the force constant matrices were positive definite. For comparison, ligand volumes (Table II) were calculated by an analytical algorithm.30
AutoDock 4 calculations
BD jobs including 100 runs each were set up as described previously.7 Briefly, the target and ligand molecules were equipped with Gasteiger charges using AutoDock Tools. Grid maps were calculated at 0.55 Å spacing and covered the entire surface of the target proteins. Docking runs were started with a random ligand position and orientation. The Lamarckian genetic algorithm and the pseudo-Solis and Wets local search with a maximum number of 20 million energy evaluations, 250 population size, 2 Å translation and 50–50° rotation and quaternion steps were applied. All sigma bonds of the ligand except rings and amide bonds were released during the flexible docking. Protein target was kept rigid, that is, protein flexibility was not considered during the calculation.
EADock calculations
The EADock calculations were performed using the SwissDock server (http://swissdock.vital-it.ch/). The ligand molecules were converted to Sybyl mol2 format using UCSF Chimera software as required by the server. The target molecules were provided as PDB files. Docking type was set to “accurate” in BD mode. The DOCK4-type outputs of the server containing 250 docked ligand conformations in each were used for subsequent ranking evaluations.
Ranking of BD results
A uniform procedure7 was applied to rank the 100 and 250 docked ligand structures of each complex produced by AutoDock 4 and EADock jobs, respectively. Briefly, in consecutive cycles, the structure of lowest ΔG (AutoDock 4) or “FullFitness” (EADock) was selected and the neighboring docked ligand structures within 5 Å RMSD were collected in the rank, then a new rank was opened with the lowest energy of the remaining docked structures, etc. The ranking was continued until all 100 (AutoDock 4) or 250 (EADock) docked ligand structures were used up in a rank. RMSDs from the crystallographic ligand structures were calculated for the lowest energy (representative) members of each rank. The ranks of the lowest RMSD values are listed in the tables of the Supporting Information. A full list including all ranks is also provided in the Supporting Information. For comparison, the ranking was performed with the “SimpleFitness” scoring of EADock, as well.
Pocket search
The heavy atoms of the protein structures were used as inputs in all cases. The off-line version of Sitehound was applied. In Sitehound and Q-SiteFinder, grid maps are calculated for the probes covering the entire proteins with 1 and 0.9 Å spacing, respectively. Sitehound was tested with both carbon and phosphate probes, whereas Q-SiteFinder applies a methyl probe. In case of Q-SiteFinder and Pocket-Finder the server produced the top 10 binding sites. Sitehound and Q-SiteFinder ranks the results according to the TIE, which is the sum of nonbonded interaction energy of all probe points with the protein atoms in the detected binding site. Pocket-Finder and Pass use probe spheres, measuring how much the spheres are geometrically buried in protein pockets and ranks the pockets according to the number of well-buried probe spheres. For all methods, the default parameters were used. For the present evaluations, distances between the centers of predicted pockets and the crystallographic ligand structures were measured for all ranks and methods and the smallest distances and the corresponding ranks are listed in the tables of the Supporting Information. In cases of Q-SiteFinder and Pocket-Finder precision is calculated as the percentage of the probes of a site that are within 1.6 Å of an atom of a particular ligand, that is, it is a measurement of how well the predicted site maps onto the ligand coordinates (ideally at least 25%).
Water and ligand interferences
The interferences of docked ligands with hydrating water molecules were investigated as follows. All hydrating crystallographic water molecules (Table II) were classified as sitting inside (1), at the bottom of (2) or outside the pocket corresponding to a BD rank. For this, the distances between the crystallographic water oxygen atom and all heavy atoms of the representative ligand structure of the BD rank were measured and the shortest distance was selected. If the shortest distance was smaller than 2.5 Å, that is, the oxygen atom practically overlapped the ligand structure then this water molecule was considered to sit middle-inside the pocket (Type 1). Similarly, the shortest distance of the crystallographic water oxygen atom was measured to the protein and if the distance was smaller than 3.5 Å and the distance from the representative ligand was larger than/equal to 2.5 Å, then the water molecule was considered to sit at the bottom of the pocket, that is, on the protein surface, below the ligand (Type 2). The thresholds 2.5 and 3.5 Å were selected as typical covalent and H-bond lengths between heavy atoms, respectively, with some tolerance. This selection procedure was repeated for all crystallographic water molecules and BD ranks at each complex of Table II. The number of water molecules were counted by rank and summarized by type and complex (Supporting Information). The usefulness of the distinction between the two types of in-pocket water molecules is discussed in the main text. All coordinates of Type 1 and 2 waters are provided as Supporting Information. The interferences of docked ligands (Lj) with other known interfering ligand molecules (Ln≠j) were also studied. For this the distance between the centrum of the representative Lj structure of the BD rank and the centrum of a Ln crystallographic ligand structure was measured. If the distance was smaller than 5 Å, then the rank number and distance of interference was tabulated for both BD methods (Supporting Information).
Supplementary material
References
- 1.van Voorhis WC, Hol WGJ, Myler PJ, Stewart LJ. The role of medical structural genomics in discovering new drugs for infectious diseases. PLoS Comput Biol. 2009;5:e1000530. doi: 10.1371/journal.pcbi.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weigelt J, McBroom-Cerajewski LDB, Matthieu Schapira M, Zhao Y, Arrowmsmith CH. Structural genomics and drug discovery: all in the family. Curr Opin Chem Biol. 2008;12:32–39. doi: 10.1016/j.cbpa.2008.01.045. [DOI] [PubMed] [Google Scholar]
- 3.Mirkovic N, Li Z, Parnassa A, Murray D. Strategies for high-throughput comparative modeling: applications to leverage analysis in structural genomics and protein family organization. Proteins. 2007;66:766–777. doi: 10.1002/prot.21191. [DOI] [PubMed] [Google Scholar]
- 4.Goldsmith-Fischman S, Honig B. Structural genomics: computational methods for structure analysis. Protein Sci. 2003;12:1813–1821. doi: 10.1110/ps.0242903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joachimiak A. High-throughput crystallography for structural genomics. Curr Opin Struct Biol. 2009;19:573–584. doi: 10.1016/j.sbi.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nat Rev Drug Discov. 2002;1:45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 7.Hetényi C, van der Spoel D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006;580:1447–1450. doi: 10.1016/j.febslet.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 8.Hetényi C, van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 10.Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 11.Gutierrez LJ, Enriz RD, Baldoni HA. Structural and thermodynamic characteristics of the exosite binding pocket on the human bace1: a molecular modeling approach. J Phys Chem A. 2010;114:10261–10269. doi: 10.1021/jp104983a. [DOI] [PubMed] [Google Scholar]
- 12.Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci USA. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Othman R, Kiat TS, Khalid N, Yusof R, Newhouse EI, Newhouse JS, Alam M, Rahman NA. Docking of noncompetitive inhibitors into dengue virus type 2 protease: understanding the interactions with allosteric binding sites. J Chem Inf Model. 2008;48:1582–1591. doi: 10.1021/ci700388k. [DOI] [PubMed] [Google Scholar]
- 14.Iorga B, Herlem D, Barré E, Guillou C. Acetylcholine nicotinic receptors: finding the putative binding site of allosteric modulators using the “blind docking” approach. J Mol Model. 2006;12:366–372. doi: 10.1007/s00894-005-0057-z. [DOI] [PubMed] [Google Scholar]
- 15.Espinoza-Fonseca LM, Trujillo-Ferrara JG. The existence of a second allosteric site on the M1 muscarinic acetylcholine receptor and its implications for drug design. Bioorg Med Chem Lett. 2006;16:1217–1220. doi: 10.1016/j.bmcl.2005.11.097. [DOI] [PubMed] [Google Scholar]
- 16.Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand binding site prediction and functional annotation. Proc Natl Acad Sci USA. 2008;105:129–134. doi: 10.1073/pnas.0707684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laurie ATR, Jackson RM. Methods for the prediction of protein-ligand binding sites for structure-based drug design and virtual ligand screening. Curr Prot Pept Sci. 2006;7:395–406. doi: 10.2174/138920306778559386. [DOI] [PubMed] [Google Scholar]
- 18.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 20.Grosdidier A, Zoete V, Michielin O. Blind docking of 260 protein–ligand complexes with EADock 2.0. J Comput Chem. 2009;30:2021–2030. doi: 10.1002/jcc.21202. [DOI] [PubMed] [Google Scholar]
- 21.Grosdidier A, Zoete V, Michielin O. EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins. 2007;67:1010–1025. doi: 10.1002/prot.21367. [DOI] [PubMed] [Google Scholar]
- 22.Hendlich M, Rippmann F, Barnickel G. Ligsite: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–363. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 23.Hernandez M, Ghersi D, Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009;37:W413–W416. doi: 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laurie ATR, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 25.Jackson RM. Q-fit: a probabilistic method for docking molecular fragments by sampling low energy conformational space. J Comput Aided Mol Des. 2002;16:43–57. doi: 10.1023/a:1016307520660. [DOI] [PubMed] [Google Scholar]
- 26.Brady GP, Stouten PFW. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des. 2000;14:383–401. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- 27.Ghersi D, Sanchez R. Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins. 2009;74:417–424. doi: 10.1002/prot.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skolnick J, Brylinski M. FINDSITE: a combined evolution/structure-based approach to protein function prediction. Brief Bioinform. 2009;10:378–391. doi: 10.1093/bib/bbp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart JJP. Mopac: a general molecular orbital package. Quant Chem Prog Exch. 1990;10:86. [Google Scholar]
- 30.Connolly ML. Computation of molecular volume. J Am Chem Soc. 1985;107:1118–1124. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.