

Abstract
X-linked neutropenia (XLN, OMIM #300299) is a rare form of severe congenital neutropenia. It was originally described in a three-generation family with 5 affected members and with an L270P mutation in the GTP-ase binding domain (GBD) of the Wiskott-Aldrich-syndrome protein (WASP) (Devriendt, et al 2001).
Here, we report and describe a large three-generation family with XLN, with 10 affected males and 8 female carriers. A c.882T>C WAS gene mutation was identified, resulting in an I294T mutation. The infectious course is variable and mild in view of the deep neutropenia. In addition to the original description, low-normal IgA levels, low to low-normal platelet counts and reduced NK-cell counts also appear as consistent XLN features. However, inverted CD4/CD8 ratios were not found in this family, nor were cases identified with myelodysplastic syndrome or acute myeloid leukaemia. Female carriers exhibited a variable attenuated phenotype. Like L270P WASP, I294T WASP is constitutively active towards actin polymerisation In conclusion, this largest XLN kindred identified to date provides new independent genetic evidence that mutations disrupting the auto-inhibitory GBD of WASP are the cause of XLN. Reduced NK cells, low to low normal platelet counts and low to low-normal IgA levels are also features of XLN.
Keywords: neutropenia, X-linked, Wiskott-Aldrich syndrome protein
Introduction
X-linked neutropenia (XLN, OMIM #300299) due to an L270P mutation in exon 9 of WAS was first described in a Belgian kindred (Devriendt, et al 2001). Since, only two isolated cases with a comparable phenotype have been described, one with an I294T and one with an S272P WAS mutation (Ancliff, et al 2006). We have recently discovered a large Irish kindred with neutropenia and an X-linked pattern of inheritance, in which we demonstrated a WAS I294T mutation in 18 family members. In fact, this family had already been reported in 1988, with severe neutropenia, recurrent bacterial infections, monocytopenia, a maturation delay in the promyelocyte/metamyelocyte stage, a reduced CD4/CD8 ratio in 4/8 patients and low IgA (Cryan, et al 1988). We here report detailed clinical, haematological, immunological and genetic data from this pedigree. In addition, the functional consequences of the I294T WAS mutation were studied.
Material and Methods
Mutational screening
DNA was extracted from peripheral blood using the High Pure PCR Template Preparation kit (Roche Diagnostics GmbH, Basel, Switserland). Exon 9 of WAS was amplified using primers exon9F 5’-tgtctcctcgccttattcctc-3’ and exon9R 5’-ggtgctcccataaggactga-3’ (GeneAmp PCR system 2400, Applied Biosystems, Foster City, California). PCR products were verified with 6% PAGE-gels and sequenced in both directions, using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequencing results were analysed using Sequence Scanner (Applied Biosystems).
Flow cytometry
PB samples were collected in sodium EDTA. Cells were immunostained with flurorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll (PerCP) and allophycocyanin (APC) conjugated monoclonal antibodies: anti-CD3, anti-CD4, anti-CD8, anti-CD19, anti-CD16/56, anti-CD45. Red cells were lysed with FacsLyse (Becton Dickinson Immunocytometry Systems, Erembodegem, Belgium) in a lyse-no wash procedure and analysed on a FACSCalibur flow cytometer (Becton Dickinson Immunocytometry Systems). Analysis of the data was performed with MultiTest (Becton Dickinson Immunocytometry Systems) software. Lymphocyte subsets were identified with the following antibody panels: B-cells (CD3-/CD19+), helper T-cells (CD3+/CD4+), cytotoxic T-cells (CD3+/CD8+), and NK cells (CD3-/CD16+56+).
X-chromosome inactivation study
Leucocytes were isolated from peripheral blood by density centrifugation over Polymorphprep (Axis-Shield, Oslo, Norway). Leucocytes were immunostained with anti-CD3, anti-CD19, anti-CD16 and anti-CD14. T-lymphocytes, B-lymphocytes, polymorphonuclear cells and monocytes respectively were sorted on a FACSvantage (Becton Dickinson Immunocytometry Systems). DNA was extracted from the sorted fractions and from mouth swabs, as previously described (Delforge, et al 1998). Half of the sample was digested overnight in the presence of the methylation specific restriction enzyme HpaII, digesting only unmethylated DNA and leaving the methylated inactive allele untouched. The second half of the sample was incubated without restriction enzymes. PCR was performed as previously described and fragment length was analysed using Genescan 500 ROX Size standard and Genescan computer program (both from Applied Biosystems) (Delforge, et al 1998). For each sample, a corrected ratio (Cr) was calculated. A cell fraction was considered skewed (i.e., asymmetrically inactivated) if the expression of one allele exceeded 75%, corresponding to Cr <0.3 or >3 (Busque, et al 1996).
Biochemistry
To test the activity of I294T WASP in vitro, a truncated WASP construct GBD-VCA (containing WASP residues 230-310-(GGS)2-420-501), GBD-VCA I294T mutant, VCA (WASP residues 420-501), actin, and Arp2/3 complexes were generated.
Circular dichroism spectra (222 nm) were obtained on 10 μM samples in 25 mM phosphate (pH 7), 150 mM NaCl, and 1 mM DTT. Melting temperatures were determined by the first derivative of the curve. Actin polymerisation assays were carried out by measuring the increase in pyrene fluorescence, as previously described. Briefly, pyrene fluorescence at 407 nm (excitation wavelength: 365 nm) was monitored over time. Polymerization assays were performed in KMEI buffer (10 mM imidazole (pH 7), 50 mM KCl, 1 mM MgCl2, 10 mM EGTA) and contained 4 μM actin (6% pyrene), 10 nM bovine Arp2/3 complex, and 500 nM WASP protein (Devriendt, et al 2001).
Results
The clinical, haematological and immunological phenotype of XLN
DNA from the index case (patient II.20 in the pedigree), a young man with severe neutropenia and a family history of neutropenia, was analysed for mutations in exon 9 of the WAS gene. A c.882T>C mutation was identified (according to den Dunnen and Antonarakis 2000), resulting in an I294T substitution in the GBD of WASP. Subsequently, we screened 60 family members (27 males and 33 females) for this mutation and identified 10 affected males (age 7-45y) and 8 female carriers (age 8-61y) (Table I and II, Figure 1). Variable neutropenia was observed in 8 out of 10 affected males, but surprisingly patients II.4 and III.26 had a normal neutrophil count in the absence of G-CSF support (Table I). Supplementary tables 1 and 2 show consecutive and stable values in cases III.10 and III.26. Affected individuals showed considerable variation in infectious history, and the severity of neutropenia does not seem to correlate closely with the susceptibility to infections. Patients I.6, II.14, II.16 and II.20 have been taking G-CSF intermittently because of frequent upper respiratory tract infections (URTI), most frequently bronchitis and otitis, requiring antibiotics several times per year. One patient had had pneumonia and one patient suffered bronchiolitis and chronic ear infections as a child. Patients II.14 and II.16 continue to have recurrent URTI’s as adults (8-10 times a year). Patients II.26 and II.27 are clinically well, without evidence of infections, despite profound neutropenia and in the absence of growth factor support. In addition, two male family members, with a history of recurrent infections, one of whom was neutropenic, died, at age 5 and age 18 years, from meningitis and infectious colitis respectively.
Table I.
Haematological & immunological parameters in affected males
| Case | I.6 | II.4 | II.14 | II.15 | II.16 | II.20 | II.26 | II.27 | III.10b | III.26b | Normal adult range |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (y) | 45 | 32 | 34 | 33 | 32 | 23 | 18 | 16 | 8 | 7 | |
| Leucocytes a | 1.7 | 5.1 | 2.8 | 4 | 2.3 | 1.7 | 2.1 | 3 | 4.6 | 5.2 | 4.0-11 * 109/L |
| Neutrophils | 0.4 | 2.4 | 1.0 | 0.1 | 0.5 | 0.5 | 0.2 | 0.5 | 2.0 | 3.3 | 2.0-7.0 * 109/L |
| Monocytes | 0.4 | 0.4 | 0.1 | 1.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 0.4 | 0.2-1.8 * 109/L |
| Eosinophils | 0.0 | 0.1 | 0.2 | 0.1 | 0.0 | 0.0 | 0.2 | 0.2 | 0.1 | 0.1 | 0.02-0.5 * 109/L |
| Platelets | 133 | 142 | 149 | 137 | 168 | 131 | 133 | 96 | 279 | 184 | 150-400 * 109/L |
| Lymphocytes | 0.7 | 2.2 | 1.4 | 2.6 | 1.5 | 1.1 | 1.6 | 2.2 | 2.0 | 1.4 | 1.0-3.0 * 109/L |
| T-cells (CD3+) | 690 | 1621 | 1153 | 2287 | 1418 | 881 | 1248 | 1861 | 1568 | 957 | 900-1700 * 106/L |
| CD4+ | 196 | 837 | 572 | 1339 | 752 | 517 | 636 | 955 | 946 | 513 | 700-1100 * 106/L |
| CD8+ | 493 | 782 | 646 | 938 | 620 | 337 | 676 | 804 | 692 | 456 | 500-900 * 106/L |
| CD4/CD8 ratio | 0.4 | 1.07 | 0.89 | 1.43 | 1.21 | 1.53 | 0.94 | 1.19 | 1.37 | 1.13 | 0.57-2.77 |
| B-cells | 75 | 422 | 169 | 410 | 150 | 210 | 268 | 317 | 562 | 304 | 200-400 * 106/L |
| NK-cells | 23 | 50 | 39 | 35 | 48 | 21 | 21 | 55 | 42 | 32 | 200-400 * 106/L |
| IgA | 0.56 | 1.32 | 1.06 | 1.8 | 0.92 | 0.59 | 0.53 | 1.49 | 0.89 | 0.29 | 0.80-2.80 g/L |
data represent single random values, obtained on the same day for all cases. Values outside the normal range (2.5-97.5 % percentile) are bolded.
For the 2 paediatric cases, follow-up analyses are available in Supplementary Table 1 and 2.
Table II.
Haematological & immunological parameters in female carriers
| Case | I.1 | I.2 | I.3 | I.7 | II.5 | II.9 | II.25 | III.17b | Normal adult range |
|---|---|---|---|---|---|---|---|---|---|
| Age (y) | 61 | 59 | 56 | 36 | 31 | 27 | 27 | 8 | |
| Leucocytes a | 3.6 | 4.1 | 3.7 | 4.8 | 6 | 4 | 4.5 | 2.5 | 4.0-11 * 109/L |
| Neutrophils | 1.5 | 1.8 | 1.9 | 1.5 | 3.9 | 2.1 | 2.4 | 1.7 | 2.0-7.0 * 109/L |
| Monocytes | 0.2 | 0.2 | 0.2 | 0.3 | 0.5 | 0.2 | 0.4 | 0.1 | 0.2-1.8 * 109/L |
| Eosinophils | 0.0 | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.2 | ND | 0.02-0.5 * 109/L |
| Platelets | 135 | 144 | 136 | 121 | 193 | 168 | 158 | 215 | 150-400 * 109/L |
| Lymphocytes | 1.9 | 2.1 | 1.5 | 2.9 | 1.5 | 1.5 | 1.5 | 1.4 | 1.0-3.0 * 109/L |
| T-cells (CD3+) | 1486 | 1473 | 1351 | 2378 | 1114 | 1160 | 1112 | 511 | 900-1700 * 106/L |
| CD4+ | 904 | 973 | 763 | 1636 | 694 | 551 | 571 | 291 | 700-1100 * 106/L |
| CD8+ | 591 | 536 | 606 | 779 | 399 | 578 | 522 | 185 | 500-900 * 106/L |
| CD4/CD8 ratio | 1.53 | 1.81 | 1.26 | 2.1 | 1.74 | 0.95 | 1.09 | 1.57 | 0.57-2.77 |
| B-cells | 255 | 318 | 238 | 419 | 363 | 301 | 335 | 137 | 200-400 * 106/L |
| NK cells | 118 | 173 | 65 | 63 | 153 | 122 | 94 | 94 | 200-400 * 106/L |
| IgA | 1.45 | 1.54 | 1.49 | 0.77 | 1.35 | 1.72 | 1.27 | 0.38 | 0.80-2.80 g/L |
| X-chromosome inactivation | NS c | NS | NS | NS | skewed | skewed | NS | NS |
data represent single random values. Values outside the normal range (2.5-97.5% percentile) are bolded.
the normal range for these paediatric cases was obtained from (Bain 1995)
NS: not skewed
Figure 1.
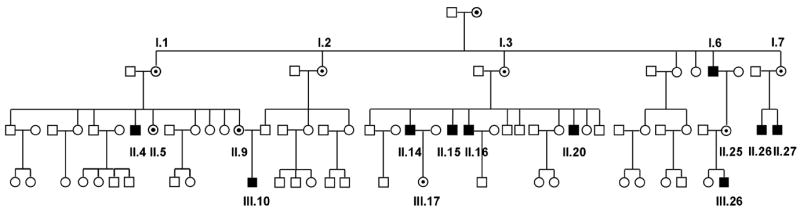
Pedigree showing all 60 screened individuals, from I.1 till III.26: affected males (black squares), carrier females (filled circles) and unaffected family members (unfilled symbols).
Five of ten affected males in the I294T family had monocytopenia. I294T mutant cases also had low or low-normal numbers of lymphocytes. Among the lymphocyte subsets, the CD3-/CD16+56+ NK cell subset was most strikingly decreased, consistent with the report of Ancliff et al (Ancliff, et al 2006). This feature, although not reported originally (Devriendt, et al 2001), is also present in the original L270P family (unpublished data). Absolute B-lymphocyte counts are decreased in all ten affected males. The absolute numbers of CD4+ T-helper cells and of CD8+ cytotoxic T-cells were decreased in seven out of ten and five out of ten patients respectively. In contrast to the original XLN family and two cases recently described (Ancliff, et al 2006), an inverted CD4/CD8 ratio was not a feature in this family. The cytopenias within the lymphocyte subsets result in decreased absolute lymphocyte numbers in five out of ten I294T cases and low-normal lymphocyte numbers in the remaining ones.
Mildly reduced platelet counts (seven males) and low-normal platelet counts (two males) were seen in nine of the ten affected males. The mean platelet volume was normal. Finally, the mean IgA level in affected adult males was 1.034 g/L, significantly lower than in adult unmutated members (2.013 g/L) (p = 0.0020) (Figure 2), although for most of them the serum IgA remains within the lower end of the normal range (0.80-2.8g/L). IgA levels in 2 of 3 tested XLN patients with the L270P mutation were also decreased (unpublished data). Therefore, low-normal IgA levels also appear a feature of XLN.
Figure 2.
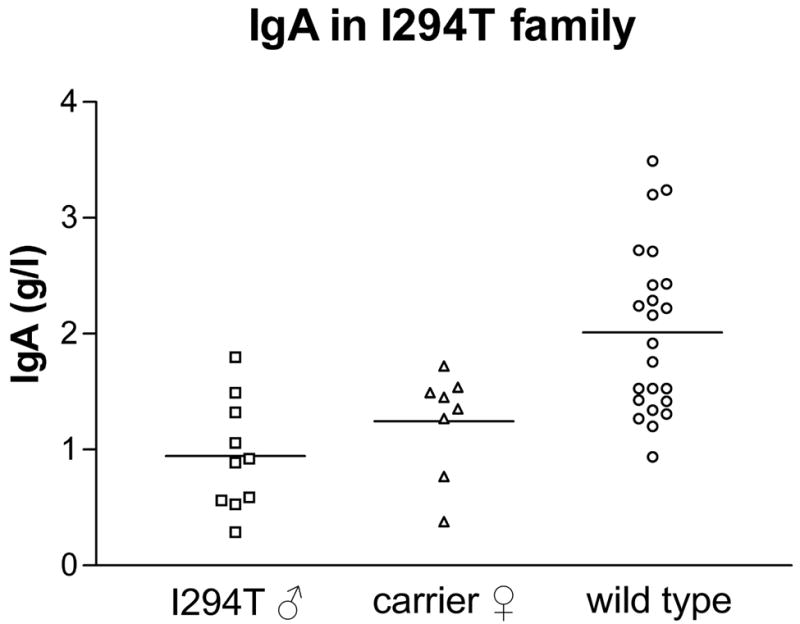
Mean IgA levels in g/L for male XLN patients (n=10), I294T carriers (n=8) and unaffected family members (n=23). P-value for IgA is 0.002 as calculated by the Mann-Whitney test comparing adult affected cases with unaffected family members.
Bone marrow aspirates, marrow biopsies and cytogenetics that had been done on five of the affected males, reportedly did not reveal evidence of or evidence of cytogenetic abnormalities.
Of note, female carriers showed intermediate findings with low-normal neutrophil and platelet counts (Table II). NK cell counts were higher than in affected males, but still below the normal range. Individual II.25 required frequent antibiotics as a child and has been using G-CSF intermittently in the past. Individual I.1 suffers from recurrent mouth ulcers and individual II.5 has frequent herpes labialis infections. Case I.7 was frequently admitted for URTI’s as a child. Female carriers showed intermediate values for IgA (Figure 2).
Biochemical findings
The 882T>C mutation in exon 9 of the WAS--gene results in an I294T mutation. In order to examine the functional effects of the I294T mutation, the I294 mutation was incorporated into a GBD-VCA construct. Similar to an L270P GBD-VCA construct, the I294T GBD-VCA construct has a reduced melting temperature (36°C), as measured by circular dichroism spectroscopy (78°C for wild-type) (Figure 3). Finally, we investigated the activity of the I294T GBD-VCA construct towards actin polymerisation. As shown in figure 4, the I294T GBD-VCA construct was nearly completely active towards the Arp2/3 complex (comparable to free VCA domain), in the absence of Cdc42, the Rho-GTPase that is the physiological activator of WASP.
Figure 3.
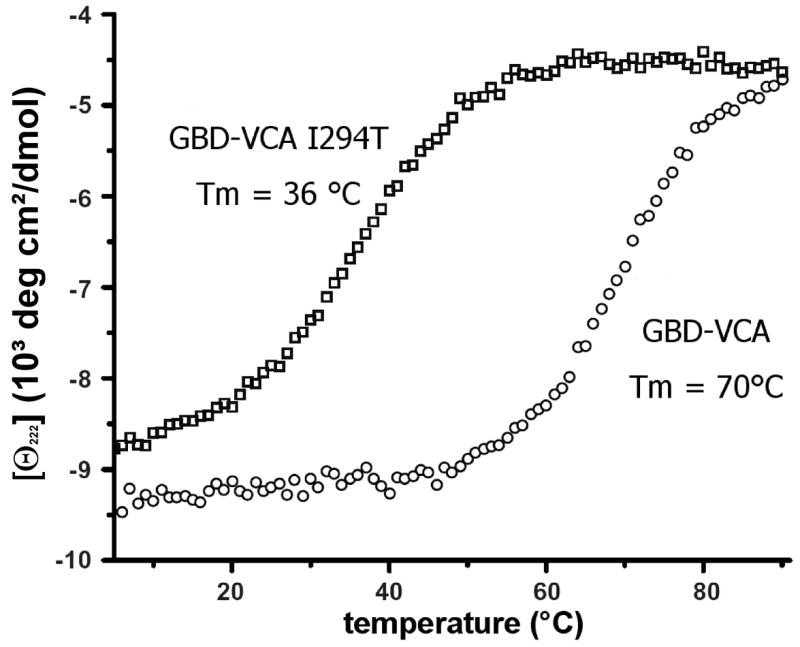
Thermal denaturation of wild type GBD-VCA and GBD-VCA I294T by circular dichroism (CD) spectroscopy. CD measurements (222 nm) were obtained on 10 μM samples in 25 mM phosphate (pH 7), 150 mM NaCl, and 1 mM DTT, as described in Materials and Methods.
Figure 4.
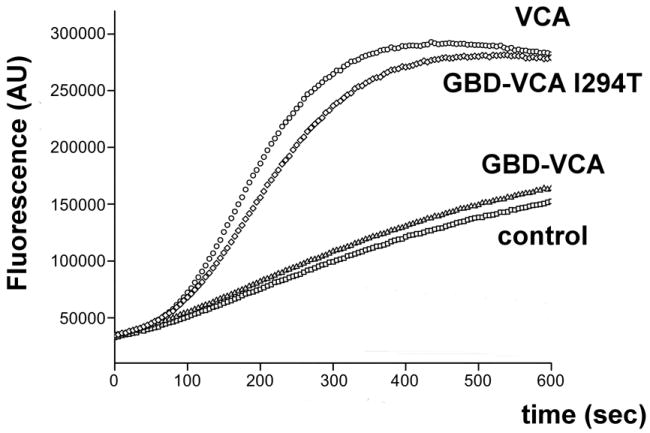
Pyrene actin polymerization assays show that GBD-VCA I294T display Cdc42-independent activation. Pyrene fluorescence at 407 nm (excitation wavelength 365 nm) was monitored over time, as described in Materials and Methods. Control : 4 μM actin (6% pyrene), 10 nM bovine Arp2/3 complex. 500 nM VCA, 500nM GBD-VCA or 500 nM GBD-VCA I294T were added as indicated.
X-chromosome inactivation studies in carrier females
The Human Androgen Receptor Assay (HUMARA) was used to investigate X-chromosome inactivation patterns in peripheral blood leucocyte subsets in female carriers. This method is based on methylation sensitive digestion of the polymorphic Human Androgen Receptor gene on Xcen-q13. Epithelial cells from mouth swabs were used to obtain the constitutional lyonisation pattern in tissues not expressing WASP.
In the L270P family, asymmetrical X-chromosome inactivation of unfractionated peripheral blood leucocytes was previously found in female carriers, supporting selection against L270P WASP in hematopoietic development. In addition, Ancliff et al. also reported asymmetrical X-chromosome inactivation in carriers of I294T and S272P WASP. However, in the present I294T family, asymmetrical X-inactivation could not be demonstrated in haematopoietic cells of 4 out of 6 tested carriers. In case II.9, the X-inactivation pattern was skewed with preferential inactivation of the mutant WAS allele in PMN cells, but not in the other fractions. In case II.5, PMN, monocytes and T-cells, but not the buccal swabs, exhibited asymmetrical X-inactivation leading to inactivation of the chromosome with wild type WAS. Thus, no consistent pattern of asymmetrical X-inactivation was identified.
Discussion
In 2001 we described X-linked neutropenia as a new syndrome of neutropenia caused by a gain-of-function type mutation of the WAS gene (Devriendt, et al 2001). Since, two additional isolated cases have been described, one with an I294T mutation and a second with an S272P mutation (Ancliff, et al 2006). We here report a large family with XLN, in which the I294T mutation was identified. This family had previously been described in the literature as “a large kindred with congenital neutropenia and low serum immunoglobulin A” (Cryan, et al 1988). This largest XLN kindred reported to date allowed us to describe the clinical, hematological and immunological phenotype of XLN in more detail.
Chronic neutropenia and monocytopenia are confirmed as prominent features of XLN, although some cases have values that fall within the low-normal range. This leads to a variable infectious phenotype which is not distinctive and overall clinically mild in view of the depth of neutropenia. The rate of infections does not correlate closely with the neutrophil count. Therefore, it seems reasonable to reserve the use of G-CSF for severe infections, so as to avoid the long-term risks of chronic G-CSF administration (Germeshausen, et al 2007). The large number of affected males in this three-generation family allowed us to identify several, more subtle features of the phenotype of XLN: For instance, NK cell numbers, B-lymphocyte counts, platelet counts and levels of serum IgA were all subnormal or in the low-normal range. Importantly, the normal mean platelet volume allows to distinguish between X-linked neutropenia and X-linked thrombocytopenia, the attenuated phenotype of Wiskott-Aldrich syndrome with microplatelets. In contrast with 3/3 evaluated cases with L270P XLN, and two recently described cases with I294T and S272P XLN, an inverted CD4/CD8 ratio or elevated peripheral CD3+/CD8+ cell counts were not found in this family. Additional recent testing of cases IV.1 and IV.3 with L270P XLN did not reveal an inverted CD4/CD8 cell ratio either (unpublished data). Therefore, an inverted CD4/CD8 ratio does not seem an essential presenting feature of XLN. However, there was a tendency for decreased numbers of CD3+CD4+ helper T-cells, while the levels of CD3+CD8+ cytotoxic T-cells were better preserved.
The infectious pattern in XLN patients is not suggestive of a deficiency in T-cell mediated immunity. An elaborate immunological study was not performed in this family, although we attempted T-cell stimulation assays on frozen samples of 6 XLN patients (data not shown). Only in patient II.4, the recovery and condition of thawed cells was satisfactory for further testing. There was no clear deficit in T-cell production of cytokines in this patient, consistent with our previous findings in the L270P family. A possible explanation for the difference with classic Wiskott-Aldrich syndrome could be that only a truncated or absent WASP protein leads to deficient T-cell immunity. In contrast, full length WASP albeit mutated as in XLN or XLT remains compatible with preservation of a largely normal B-and T-cell function.
The thermal dissociation and actin-polymerisation studies allow to conclude that the I294T WAS mutation affects the normal function of WASP by a mechanism similar to L270P: that is, substitution of the non-polar isoleucine in position 294 by a hydrophilic threonine leads to disruption of the hydrophobic fold, loss of the auto-inhibited structure of inactive WASP and constitutive availability of the carboxy-terminal VCA-segment for actin-polymerisation.
Recent studies have partly elucidated the pathogenesis of neutropenia in XLN: I294T WASP causes hyperactivation and delocalisation of actin polymerisation, genomic instability and increased apoptosis (Moulding, et al 2007). This could fit with the diagnosis of myelodysplastic syndrome (MDS) in one case (Ancliff, et al 2006), and an eventual evolution towards MDS and acute myeloid leukaemia in two cases in the L270P family (Beel, et al 2006). However, in the present kindred, no cases of MDS or leukaemia have occurred nor did available bone marrow and cytogenetic examinations reveal evidence for myelodysplasia or other myeloid malignancies.
X-chromosome inactivation studies, performed on sorted leukocyte populations from female I294T carriers of this family, did not reveal a consistent asymmetrical X-inactivation pattern. This was unexpected as previous studies on unfractionated leucocytes in the L270P family had shown asymmetrical X-inactivation in 2/2 tested carrier females (Devriendt, et al 2001). In addition, three carriers with S272P and one carrier with I294T WAS also displayed asymmetrical X-inactivation in haematological cells. (Ancliff, et al 2006). These seemingly conflicting results could reflect that activating mutations in the GBD of WASP do not provide a sufficiently strong signal to consistently result in skewed X-chromosome inactivation. Indeed, carriers of XLT may have random X-inactivation in haematological cells, while carriers of inactivating WAS mutations, leading to Wiskott-Aldrich Syndrome, uniformly show non-random inactivation of the X-chromosome harbouring mutant WAS (Puck, et al 1990; De Saint-Basile, et al 1991; De Saint-Basile, et al 1996). Alternatively, Carrel and Willard have recently shown that WAS belongs to the small proportion of genes that can be variably expressed from the inactivated X-chromosome (Carrel, et al 2005), which could also explain the lack of skewing in hematopoietic cells of this family and the mild neutropenia that is observed in carrier females. Finally, other background genetic factors may contribute to determine the ratio of X-chromosome inactivation (Puck, et al 1998).
In conclusion, the data from this large pedigree with the I294T WAS mutation provide important, new and independent genetic evidence that mutations disrupting the auto-inhibitory GBD domain of WASP are the cause of XLN. Clinically, the severity of the neutropenia in I294T XLN does not seem to translate into an extreme susceptibility to infections. NK cell numbers, platelets and IgA levels in XLN are consistently below normal or in the low-normal range. There is a tendency for decreased numbers of CD4, but reversed CD4/CD8 ratio’s, as previously described by us and others (Ancliff, et al 2006, Devriendt, et al 2001), do not seem to be invariably present. Unexpectedly for an X-linked disease, female carriers of XLN have intermediate neutrophil, NK cell and platelet numbers and intermediate levels of IgA. To date, no affected members of this family have developed malignancies of myelopoiesis. However, as most affected males are younger than in the Belgian family, it is advisable that they are followed up closely. In particular long term G-CSF usage may predispose to CSF3R mutations, and this is highly predictive for malignant transformation (Germeshausen, et al 2007). Therefore, it might be reasonable to limit the use of G-CSF to episodes of severe infection and chronic use to merely correct neutrophil count should be discouraged.
Finally, further investigation is needed to reveal how L270P and I294T WASP mutations lead to XLN.
Supplementary Material
Acknowledgments
We thank the patients and their families for participating in this study. Particular thanks go to Amanda Kiely in OLCH Dublin for assisting with the documentation and administration; to Liz O’Sullivan AMNCH Dublin for pulling charts and supplying laboratory records, and to Diarmaid O’Donghaile, Amy Lee Chong and Raveen Shahdadpuri for phlebotomy assistance. We thank Prof.dr. J. Ceuppens and L. Coorevits for T-cell activation assays.
This study was supported by a research grant form FWO Vlaanderen to P.V. KB is an aspirant researcher and PV is a senior clinical investigator of FWO Vlaanderen. This text presents research results of the Belgian programme of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister’s Office, Science Policy Programming. The scientific responsibility is assumed by the authors.
Footnotes
Authors contributions: KB designed and performed the experimental work, analysed and interpreted the data and wrote the article, MC discovered the family and contributed clinical and haematological data, JB, JB, GL, SR, OPS and FG contributed haematological and laboratory data. VV offered expertise in flow cytometry, MKR and DWL designed and performed WASP activation studies, Peter Vandenberghe designed the study, analysed and interpreted the data and wrote the paper.
References
- Ancliff PJ, Blundell MP, Cory GO, Calle Y, Worth A, Kempski H, Burns S, Jones GE, Sinclair J, Kinnon C, Hann IM, Gale RE, Linch DC, Thrasher AJ. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108:2182–2189. doi: 10.1182/blood-2006-01-010249. [DOI] [PubMed] [Google Scholar]
- Bain B. Blood cells, a practical guide. Blackwell Science Ltd; London: 1995. [Google Scholar]
- Beel K, Schollen E, Uyttebroeck A, Verhoef G, Demuynck H, Devriendt K, Vandenberghe P. Gain-of-Function WASP Mutations in Pediatric and Adult Patients with Myelodysplasia or AML. Blood (ASH Annual Meeting Abstracts) 2006;108:4516. [Google Scholar]
- Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, Maragh M, Gilliland DG. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Cryan EF, Deasy PF, Buckley RJ, Greally JF. Congenital neutropenia and low serum immunoglobulin A: description and investigation of a large kindred. Thymus. 1988;11:185–199. [PubMed] [Google Scholar]
- De Saint-Basile G, Schlegel N, Caniglia M, Le Deist F, Kaplan C, Lecompte T, Piller F, Fischer A, Griscelli C. X-linked thrombocytopenia and Wiskott-Aldrich syndrome: similar regional assignment but distinct X-inactivation pattern in carriers. Annals of Hematology. 1991;63:107–110. doi: 10.1007/BF01707282. [DOI] [PubMed] [Google Scholar]
- De Saint Basile G, Lagelouse RD, Lambert N, Schwarz K, Le Mareck B, Odent S, Schlegel N, Fischer A. Isolated X-linked thrombocytopenia in two unrelated families is associated with point mutations in the Wiskott-Aldrich syndrome protein gene. Journal of Pediatrics. 1996;129:56–62. doi: 10.1016/s0022-3476(96)70190-7. [DOI] [PubMed] [Google Scholar]
- Delforge M, Demuynck H, Verhoef G, Vandenberghe P, Zachee P, Maertens J, Van Duppen V, Boogaerts MA. Patients with high-risk myelodysplastic syndrome can have polyclonal or clonal haemopoiesis in complete haematological remission. British Journal of Haematology. 1998;102:486–494. doi: 10.1046/j.1365-2141.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Human Mutation. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D, Rosen MK, Vandenberghe P. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nature Genetics. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007;109:93–99. doi: 10.1182/blood-2006-02-004275. [DOI] [PubMed] [Google Scholar]
- Moulding DA, Blundell MP, Spiller DG, White MR, Cory GO, Calle Y, Kempski H, Sinclair J, Ancliff PJ, Kinnon C, Jones GE, Thrasher AJ. Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. Journal of Experimental Medicine. 2007;204:2213–2224. doi: 10.1084/jem.20062324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puck JM, Siminovitch KA, Poncz M, Greenberg CR, Rottem M, Conley ME. Atypical presentation of Wiskott-Aldrich syndrome: diagnosis in two unrelated males based on studies of maternal T cell X chromosome inactivation. Blood. 1990;75:2369–2374. [PubMed] [Google Scholar]
- Puck JM, Willard HF. X inactivation in females with X-linked disease. New England Journal of Medicine. 1998;338:325–328. doi: 10.1056/NEJM199801293380611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.