

Abstract
Toll-like receptor (TLR) signaling plays an important role in cerebral ischemia, but downstream signaling events, which can be organ-specific, are incompletely understood. We thereby investigated involvement of the MyD88-dependent (MyD88) and MyD88-independent (TRIF) TLR signaling pathways in 2 in vitro and 2 in vivo models of cerebral ischemia. For in vitro studies, we used a model of oxygen-glucose deprivation (OGD) followed by flow cytometric analysis to determine:1) viability of PC12 cells following knock-down with MyD88 siRNA compared to negative control siRNA and 2) viability, apoptosis and necrosis of cortical neurons from MyD88 null (-/-), TRIF mutant, and wild type (WT) mice. In addition, in vivo, 1) We examined CA1 neuronal survival 7 days after global forebrain ischemia and 2) infarct volumes 24h after Middle Cerebral Artery Occlusion (MCAO) in all 3 types of mice. OGD: 1) There were no differences in either percent viability of PC12 cells transfected with MyD88 compared to negative control siRNA or 2) in percent viability, apoptosis and necrosis of cortical neurons from MyD88-/-,TRIF mutant and WT mice. Global ischemia: neuronal survival was similar in all 3 groups of mice. Finally, MCAO: infarct volumes were not statistically different among all 3 groups of mice: MyD88 -/-, 23.9 ± 9.9 mm3, TRIF mutant, 26.7 ± 5.8mm3 and WT, 17.9 ± 8.4mm3. These findings show that disruption of MyD88 or TRIF signaling does not confer protection in brain ischemia and suggests the possibility of additional or alternate downstream adaptors during TLR signaling in cerebral ischemia.
Keywords: Toll-like receptor (TLR), Focal ischemia, MCAO, Global ischemia, MyD88, TRIF
1. Introduction
Stroke remains the third leading cause of death and the leading cause of long-term serious disability in the United States and new approaches for treatment are desperately needed. One such approach involves understanding the role of pathways leading to inflammation such as the Toll-like receptor (TLR) signaling pathway in controlling the brain's response to ischemia.
Toll-like receptors are evolutionarily conserved pattern-recognition receptors important for initiation of innate immune responses to pathogen-associated molecular patterns (PAMPs). However, the “danger model” of immunity, suggests that “danger signals” that activate the innate immune response to infection are not restricted to PAMPs such as lipopolysaccharide (LPS;TLR4 ligand) (Matzinger, 2002a; Matzinger, 2002b), but can include endogenous danger-associated molecular patterns (DAMPs) released during ischemic stress or during the subsequent restoration of blood flow (reperfusion) (Medzhitov, 2001).
Evidence supporting the involvement of the TLR signaling pathway in the innate immune response to brain ischemia includes elevated TLR4 message levels in blood cells of patients with acute cerebral ischemia compared to controls (Yang et al., 2008) and protection against cerebral ischemia in mice with Toll-like receptor deletions/mutations (Caso et al., 2007), (Ziegler et al., 2007) (Tang et al., 2007). While these studies focus on upstream TLR signaling events, very little is known about the molecules that propagate TLR signals downstream during cerebral ischemia.
Understanding these downstream signaling events is important as it may lead to the identification of specific points along the pathway that can be modulated during cerebral ischemia. Also, there is evidence that downstream TLR signaling pathways may be organ-specific. For instance in cardiac ischemia, disruption of Myeloid Differentiation factor 88 (MyD88) signaling is protective (Hua et al., 2005). In hepatic ischemia-reperfusion, however, downstream TLR signaling seems to occur via the Toll/IL-1 Receptor-containing adapter inducing IFN-β (TRIF) as opposed to the MyD88 pathway (Tsung et al., 2006). In addition, in renal ischemia, downstream TLR 2 signaling seems to be mediated via a MyD88-independent pathway, as mice with deletions of TLR2 are better protected from renal ischemia compared to mice with disruptions of the currently known TLR2 downstream adaptor, MyD88 (Shigeoka et al., 2007).
Currently, downstream TLR signaling is known to occur via two major pathways, the MyD88-dependent pathway and the MyD88-independent pathway or TRIF pathway (Kumar et al., 2009).
In light of the fact that the Toll-like receptors previously shown to be important in cerebral ischemia both use the MyD88 and TRIF adaptors for downstream signaling, we sought to systematically determine whether one, both, or neither, of these two known TLR adaptors, MyD88 and TRIF, were important or preferentially used in downstream TLR signaling during cerebral ischemia. We used two in vitro and two in vivo models of cerebral ischemia in our study.
2. Results
2.1 Confirmation of MyD88-/- and TRIF mutant genotypes
Polymerase Chain Reaction (PCR)
PCR of tail DNA, from all 3 types of mice, using MyD88 primers, to confirm the genotype of MyD88-/- mice compared to TRIF mutant and WT mice, yielded the expected DNA size fragments. (Data not shown).
2.1.1 DNA Sequencing
Similarly, DNA sequencing of DNA fragments, from PCR above, confirmed deletion of exons IV and V of the MyD88 gene in MyD88-/- mice. In TRIF mutant mice, DNA sequencing confirmed deletion of a single guanine from position 6734 of the TRIF gene.
In vitro
2.2 MyD88 knock-down does not affect PC12 cell viability in vitro
PC12 cells transfected with MyD88 siRNA showed significant knock-down of MyD88 protein compared to PC12 cells transfected with negative control siRNA 96h following Amaxa nucleofection of respective siRNA into cells; (p=0.0004); Figures 1a&b. Results represent average values from 7 independent transfection experiments. There were no differences in PC12 cell viability, as determined by flow cytometric analysis using Hoechst 33342 (Hoechst) and Propidium Iodide (PI) staining, following 15h oxygen-glucose deprivation (OGD) in vitro, in cells transfected with negative control siRNA compared to MyD88 siRNA. Cell viability was 53.1 ± 13.4% in PC12 cells transfected with negative control siRNA compared to 53.4 ±17.4 % in PC12 cells transfected with MyD88 siRNA (p=0.743). Viability of untreated PC12 cells did not differ from that of negative control treated cells in preliminary studies. For control PC12 cells, not exposed to OGD, cell viability was 73.9% ± 9.4% in PC12 cells transfected with negative control siRNA compared to 70.9±11.4% in PC12 cells transfected with MyD88 siRNA; Figure 1c. Results represent averages from 3 independent experiments.
Figure 1.
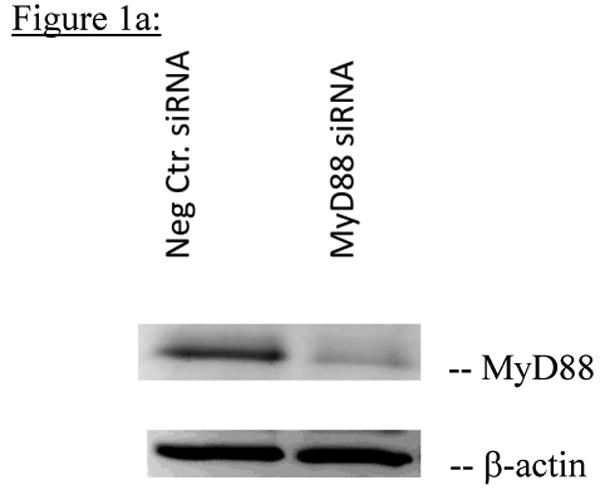


Figure 1a: Representative Western blot of 7 independent transfections of total PC12 cell lysates incubated with anti-MyD88 antibody showing decreased expression of MyD88 protein in PC12 cells transfected with MyD88 siRNA for 96h compared to PC12 cells transfected with negative control siRNA. β-actin loading controls are shown below each band.
Figure 1b: Average quantitation of MyD88 protein expression in PC12 cells transfected with negative control siRNA compared to PC12 cells transfected with MyD88 siRNA from 7 independent experiments. Quantitation of MyD88 protein levels 96h after transfection with respective siRNAs was done by densitometry using Alpha Ease software, version 4.0 (Alpha Innotech). MyD88 protein expression was significantly decreased in PC12 cells transfected with MyD88 siRNA compared to PC12 cells transfected with negative control siRNA (p=0.0004).
Figure 1c: Average viability of PC12 cells following OGD after knock-down of MyD88 expression. PC12 cells were transfected with MyD88 or negative control siRNA for 96h, and cells exposed to OGD for 15h. Data represent averages from 3 independent experiments. Percent cell viability was determined after OGD by means of flow cytometric analysis using Hoechst 33342 and PI staining. Percent cell viability following OGD was not statistically different in PC12 cells transfected with MyD88 siRNA compared to cells transfected with negative control siRNA as shown above.
2.3 Disruption of MyD88 or TRIF has no effect on cortical neuron viability or percent apoptosis and necrosis of cell death in vitro
Cell viability, apoptosis and necrosis following 5-hour OGD in vitro was assayed by flow cytometric analysis using Hoechst and PI staining. Percent cell viability was WT, 69.6 ± 13.8%, MyD88 -/-, 72.2 ± 4.4% and TRIF mutant, 67.4 ± 7.9%; and these differences were not statistically different among cortical neurons from any of the groups of mice based on averages from 4 independent experiments; (p=0.69) (Figure 2a).
Figure 2.


Figure 2a: Embryonic cortical neurons were isolated from WT, MyD88-/- and TRIF mutant mice and subjected to 5h OGD. Cell viability following OGD was determined by flow cytometric analysis. Percent neuron survival for each group was determined by comparing cell viability after OGD to cell viability of control cortical neurons that did not undergo OGD within each group. Data shown represent average percent neuron survival from 4 independent experiments for each group. Average percent neuron survival following 5h OGD was similar for cortical neurons from all 3 groups of mice.
Figure 2b: The percent of cell death due to apoptosis (A) and necrosis (N) was determined for each experimental group by flow cytometric analysis using Hoechst 33342 and Propidium Iodide staining. Results represent data from 4 independent experiments. Percentages of apoptotic and necrotic cells were not significantly different for cortical neurons from all 3 groups of mice following 5h OGD.
As with the percentage cell viability, the percentage of cortical neurons in different stages of apoptotic (A) and necrotic (N) cell death as determined by means of flow cytometric analysis using Hoechst and PI staining was WT – (A); 41.9 ± 12.9%, (N); 58.0 ± 12.9%, MyD- (A); 46.0± 8.0%, (N) 53.9 ± 8.0%, TRIF- (A) - 43.3 ± 8.3%, (N); 56.7± 8.2%. Neither percent apoptosis nor percent necrosis of cell death differed significantly among cortical neurons from any of the 3 groups of mice using averages from 4 independent experiments; (p=0.47) (Figure 2b).
BCCAO
2.4 Neurological Score
Mice from all 3 groups had no obvious neurological impairment as determined by neurological scoring for the 7 days that mice survived following BCCAO. Neurological score was 0 (Normal moves freely) for mice from all 3 groups, every day, as assessed daily for the7 days following BCCAO.
CA1 neuronal survival is unaffected by MyD88 or TRIF disruption in global ischemia
There was no intra-op or post-op mortality among the 6 MyD88 mice or 6 TRIF mutant mice, but two of eight WT mice died during 6 min BCCAO surgery with one minute left to go in both cases. Mice from all 3 groups had a similar post-op course. Neurological score for all mice for all 7 days after BCCAO was the same in all animals as determined by gait and posture scores of 0 for mice in all 3 groups. Fig 3a shows representative hippocampal CA1 sections from all 3 groups of mice following 6 minutes of BCCAO and 7 days of reperfusion. There were no obvious differences in the gross morphology of CA1 hippocampal neurons among the groups of mice. The percentages of neurons that survived in a region of CA1 hippocampus were: WT = 77.4 ±25.3 %, MyD88-/- = 76.3 ± 20.3%, and TRIF mutant = 81.2 ±17.4 % (Figure 3b) and these differences were not statistically different among mice from all 3 groups (p=0.95).
Figure 3.
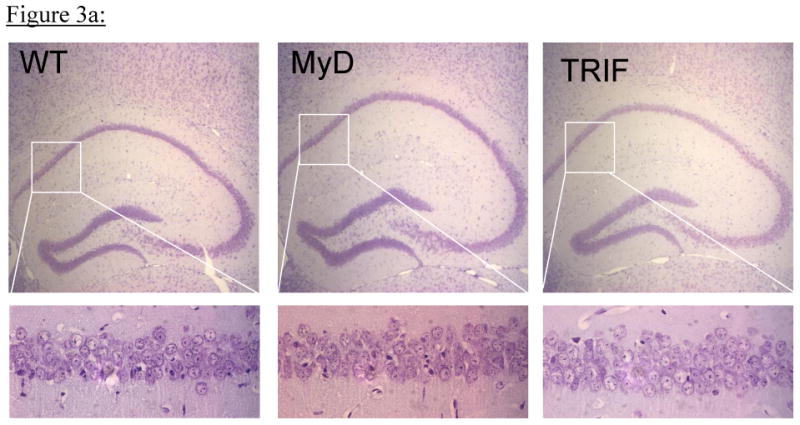

Figure 3a: Mice were subjected to global forebrain ischemia via 6-minute bilateral common carotid occlusion and hippocampal CA1 neuron survival was determined 7 days later (total N= 30; n= 12, 9 and 9 per group for WT, MyD88-/- and TRIF mutant mice respectively). Representative hippocampal sections (5X) from all 3 groups of mice are shown in the upper panels and in the lower panels are representative CA1 sections (40 X) from the same hippocampi.
Figure 3b: The percentage of CA1 neurons that survived 7 days following 6-minute BCCAO, as determined by light microscopy, from WT, MyD88-/- and TRIF mutant mice are shown. There were no differences in percent CA1 neuronal survival following BCCAO among the 3 groups of mice.
2.5 MCAO
No differences in infarct sizes in WT, MyD88 and TRIF mutant mice after pMCAO
No animals died intra or post-op in any group. The 24-hr post-op course was similar for mice in all 3 groups. Permanent MCAO resulted in obvious infarction in all 3 groups of mice. Representative Cresyl Violet-stained brain sections from all 3 groups of mice are shown in Fig 4a. Absolute infarct volumes were: WT 17.9 ± 8.4mm3, MyD88 -/- 23.9 ± 9.9 mm3 and TRIF mutant 26.7 ± 5.8mm3 (Fig 4b). There was a trend towards larger infarct volumes in MyD88-/- and TRIF mutant mice compared to WT mice (Fig 4b). However, these differences were not statistically significant (p=0.16).
Figure 4.
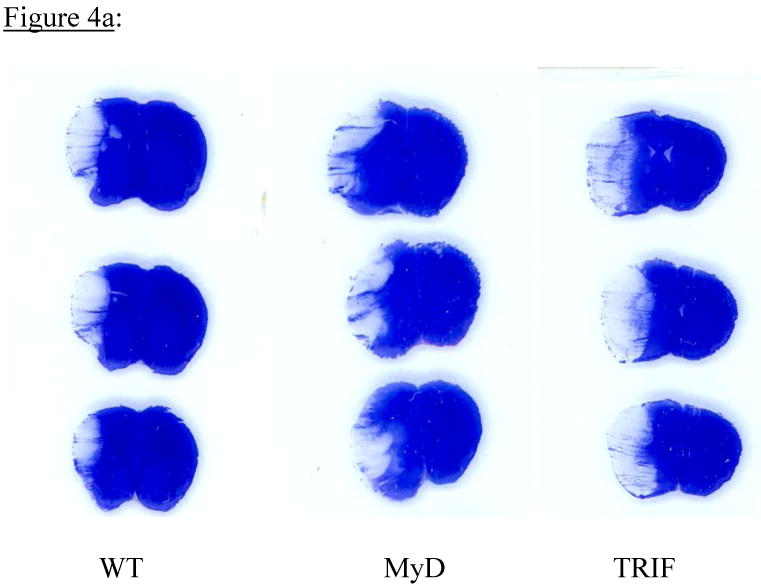

Figure 4a: Mice were subjected to permanent MCAO) and infarct volumes determined 24 h later (total=21; n=7 per group). Representative coronal brain sections, stained with Cresyl violet show infarct sizes from WT, MyD88-/- and TRIF mutant mice. Infarct sizes were similar in all 3 groups of mice.
Figure 4b: Comparison of infarct volumes from WT, MyD88-/- and TRIF mutant mice following permanent MCAO (n=7 per group). Infarct areas from sections as shown in Fig 4a were determined using NIH image J software and absolute infarct volumes were calculated as described in the methods section. There was a trend towards increased infarct volumes in MyD88-/- and TRIF mutant mice compared to WT mice, but these differences were not statistically significant, (p=0.16).
3. Discussion
Our results showed that neither deletion of MyD88 nor disruption of TRIF had any clear effect on 1) cell viability in in vitro ischemia, 2) CA1 neuron survival in global forebrain ischemia or 3) on infarct size in a model of permanent focal ischemia in contrast to the previously reported roles of MyD88 and TRIF in downstream TLR signaling in cardiac and hepatic ischemia respectively. (Hua et al., 2005), (Tsung et al., 2006). However, there was an unexpected trend towards larger infarct volumes in MyD88-/- mice compared to WT controls. This finding may suggest that in addition to the predominant inflammatory role of the MyD88 pathway, there may be some role for MyD88, in the acute stress response during the early innate response to cerebral ischemia. Overall, our in vivo results, using focal and global ischemia, confirm results from our in vitro models of ischemia in PC12 and cortical neurons from all 3 types of mice demonstrating that neither disruption of MyD88 nor TRIF downstream signaling had any effect on cortical neuron viability or apoptosis following OGD.
Cell viability in our models of in vitro ischemia using PC12 cells and cortical neurons and in global forebrain ischemia was not affected by disruption of MyD88 or TRIF. One limitation of this model is that our siRNA studies did not result in complete knock-down of the MyD88 protein. However, the results of the siRNA studies are consistent with results from the other in vitro model and with the results from the other 2 in vivo models of ischemia. To the best of our knowledge, this is the first study evaluating these downstream TLR signaling pathways in in vitro ischemia and in global ischemia demonstrating that disruption of either of these pathways may not play a major role in cerebral ischemia. This is in contrast to previous studies focusing on proximal events at the receptor showing the importance of TLR4 in global ischemia (Hua et al., 2007) and the importance of TLR2 and TLR4 in in vitro ischemia using cortical embryonic neurons, as used in our study(Tang et al., 2007). In one other study of downstream TLR signaling in in vitro ischemia, treatment of WT cortical neurons with agonists of the TRIF pathway was shown to be protective (Marsh et al., 2009). These studies, however, differ from our studies because the TRIF pathway was activated after OGD in WT cortical neurons with intact TRIF and MyD88 pathways as opposed to assessing the viability of cortical neurons with disruptions of either pathway after OGD as done in our study.
Furthermore, results of our focal ischemia model are consistent with clinical studies showing no changes in peripheral MyD88 message levels in patients with acute cerebral ischemia, but increased TLR 4 levels in these same patients. (Yang et al., 2008). While peripheral changes in MyD88 may not reflect cerebral events, the results from experimental studies accompanying these clinical studies, exploring the mechanism of increased TLR4 message levels in patients with acute cerebral ischemia, also support our findings by showing that MyD88-/- mice were not protected against cerebral ischemia in a model of 6h of focal cerebral ischemia followed by 24h reperfusion (Yang et al., 2008).
Interestingly, in a few reported studies, that emerged during the preparation of this manuscript, involving downstream TRIF signaling, in different models of focal cerebral ischemia than used in our study, mice with deletion of IRF3, a transcription factor downstream from TRIF, were not protected against cerebral ischemia (Marsh et al., 2009), and neither were mice with non-functional TRIF (Hua et al., 2009),(Yang et al., 2010).
To authenticate our results, we confirmed the genotypes of the MyD88 -/- and TRIF mutant mice. In addition, preliminary data from follow-up studies to explain our current results show significantly decreased expression of chemokines/cytokines specific to the MyD88 and TRIF pathways following permanent MCAO. These findings confirm that the deletion/mutations interfered with signal transduction in either pathway (unpublished results). Thereby, compensation by the dominant pathway when the other pathway is non-functional is an unlikely explanation for our current results.
Another consideration that addresses our current results, especially with regards to the MyD88-/- mice used in this study, involves the fact that the MyD88 TIR domain required for downstream signaling was replaced with a neomycin resistance gene in these mice and hence downstream TLR signaling via MyD88 was completely abolished (Adachi et al., 1998). However, the 5′ end of the gene, as well as exons 1-3 and the non-coding exon 6, were left in place. Since it is known that the 5′ end of the gene contains IL-6 and IFN-gamma-activated sequences (GAS), (Adachi et al., 1998),(Strengeil et al., 2002), the role of these sequences in the portion of the gene that were left in place is not known in light of the cerebral response to ischemia by MyD88. Also, the death domain (exons 1 and 2) through which MyD88 dimerizes with other death domain-containing proteins, as well as the intermediary domain, remain in place in the MyD88-/- animals (Adachi et al., 1998). Alternatively, other forms of MyD88 which predominate in the brain may be more important in the cerebral response to ischemia (Kim et al., 2007).
Other considerations include the involvement of MyD88 in other signaling pathways such as IL-1 and IL-18 (Adachi et al., 1998). However, our current results with regards to MyD88 can't be explained by the involvement of this adaptor in these signaling pathways since deletion of IL-18 is not protective against cerebral ischemia (Wheeler et al., 2003). On the other hand, deletion of IL-1, similar to deletion of some TLRs, is protective in cerebral ischemia and agents that block the IL-1 receptor are being studied as potential therapeutic agents for acute stroke (Emsley et al., 2005). However, deletion of MyD88, the downstream adaptor through which IL-1 and these TLRs are thought to signal, is not protective according to our current studies and other reports. Thereby, IL-1 and these TLRs, shown to be important in cerebral ischemia, may signal via a downstream adaptor other than the presumed downstream MyD88, during cerebral ischemia.
The possibility of alternate downstream TLR signaling pathway in ischemia exists in the kidney wherein TLR2 -/- mice were better protected from renal ischemia compared to MyD88 -/- mice and compared to combined MyD88-/- X TRIF knock-out mice (Shigeoka et al., 2007).These studies suggested the presence of a TLR 2 mediated MyD88-independent pathway. It remains to be determined whether such a pathway is present in the brain.
Furthermore, there is emerging evidence supporting the probability of different downstream adaptors in cerebral ischemia showing that TLR signaling can occur via PI3 kinase in cortical neurons (Peltier et al., 2010). However, while TLR signaling occurs via PI-3 kinase in neurons, the involvement of this pathway and the production of any harmful downstream effectors during cerebral ischemia remains to be explored.
In summary, we describe, systematically, in the same study, using 2 different in vitro and in vivo models, evaluation of the roles of downstream TLR signaling via MyD88 and TRIF signaling in cerebral ischemia. Our findings as well as other reports, strongly suggest that MyD88 and TRIF do not play a major role in downstream TLR signaling during cerebral ischemia. These findings, in light of previous reports of protection against cerebral ischemia in mice with receptor deletions (Caso et al., 2007), (Ziegler et al., 2007), (Tang et al., 2007), fit into a model whereby these receptors might use different downstream adaptors/adaptor during TLR signaling. Our results also suggest that there may be organ-specificity in spite of the heterogeneity of downstream TLR adaptors during ischemia.
Conclusions
Finally, these findings highlight the complexities and incomplete state of knowledge with regards to downstream TLR signaling during cerebral ischemia. Therefore therapies limited to blocking MyD88 or TRIF are not likely to be protective against acute brain ischemia.
4. Experimental Procedures
4.1 Animals
We used 12-16 week old, male, WT C57BL6/J and TRIF mutant mice (Jackson Laboratories) and MyD88 knock-out mice (breeder pairs from Institute for Systems Biology, Seattle, WA). Genotypes of both MyD88-/- and TRIF mutant mice were confirmed by Polymerase Chain Reaction (PCR) and DNA sequencing. WT C57BL6/J mice served as controls since both TRIF mutant and MyD88 knock-out mice were on a C57BL6/J background. All mice were housed and bred together in the institutional animal facility. Mice had free access to food and water pre and post-surgery. All animal procedures were approved by the National Institute of Neurological Disorders and Stroke (NINDS) Animal Care and Use Committee.
Experimental design
In vitro
For in vitro studies, PC12 cells and cortical neurons from MyD88, TRIF mutant and WT mice were used in OGD studies.
In vivo
Global and focal ischemia were carried out on MyD88, TRIF mutant and WT mice. For MCAO studies, the average pre-op weights of mice were: WT 28.3±2.1g, MyD88-/- 29.18 ±2.6g and TRIF mutant 28.4±2.2g. Infarct volumes were determined 24h after focal ischemia. While for Bilateral Common Carotid Artery Occlusion (BCCAO) studies, the average pre-op weights were: WT 26.9±2.5g, MyD88-/-, 27.0 ±2.2g and TRIF, 29.0 ±3.4g. CA1 neuronal survival was determined 7 days after global ischemia.
4.1.1 Genotype confirmation
Genotypes of MyD88-/- and TRIF mutant mice were verified by means of PCR of tail DNA followed by DNA sequencing of PCR products.
PCR
MyD88 primers flanking exons IV and V of the MyD88 gene:
5′ GGC AAC TAG AAC AGA CAG AC-3′
3′ CAA ATG CTG AAA CTA TAG G-5′.
TRIF primers flanking the single base-pair deletion in the TRIF gene:
5′ CAC AGT CCC AAT CCT TTC-3′
3′ TCA CTC TGG AGT CTC AAG- 5′
PCR conditions: The PCR protocol consisted of an initial denaturing step at 95°C for 5 min, then 30 cycles of 95°C for 30 sec, 55°C for 30 sec, 72°C for 1 min, followed by final elongation step of 72°C for 7 min using Platinum Blue Super mix (Invitrogen) on an I-Cycler (Bio-Rad). Primers were synthesized by Sigma-Genosys.
4.1.2 DNA sequencing
Purified DNA fragments from PCR amplification were sequenced using the above MyD88 and TRIF primers. Sequencing was carried out by the NINDS Core DNA sequencing facility.
In vitro
4.2 MyD88 Knock-down in PC12 cells
Undifferentiated PC12 cells rat pheochromocytoma cells (ATCC, Manassas,VA, Cat # CRL 1721) were transfected with 120nM of MyD88 (Qiagen, SI02018317) or 120nM of scrambled negative control siRNA (Qiagen,1027310), using Amaxa Nuclefector (cell line Nucleofector kit V, PC12 (ATCC), U-029 program) according to manufacturer's instructions. Transfected PC12 cells were seeded at ˜ 700×103 cells per well in poly-D lysine coated 6-well plates and incubated with 1 ml RPMI supplemented with 10% horse serum (HS) (ATCC) and 5% Fetal Bovine Serum (FBS) (Atlanta Biologicals) at 37°C for 96h. This transfection protocol gave the most reproducible MyD88 knock-down compared to lower siRNA concentrations and shorter transfection time frames. The day after transfection, medium in each well was replaced with 2mls of fresh RPMI with 10% HS and 5% FBS.
4.2.1 PC12 cell viability following MyD88 knock-down and OGD
Following 96h of transfection with MyD88 or negative control siRNA, cells were washed with Phosphate Buffered Saline (PBS) and prepared for OGD by incubating with Roswell Park Memorial Institute (RPMI) media without glucose (Invitrogen) under anaerobic conditions. Briefly, cells were placed in sealed modular chambers as previously described (Hillion et al., 2005) and flushed with 95% nitrogen/5% CO2 at 4 liters/minute at room temperature for 30 minutes prior to being incubated at 37°C for 15h. Following OGD, cells were harvested using papain dissociation method and cell viability determined by means of flow cytometric analysis using Hoechst 33342 (Hoechst) and propidium iodide (PI) staining as previously described (Hillion et al., 2005).
4.2.2 Western Blotting
MyD88 protein knock-down with MyD88 siRNA compared to knock-down with negative control siRNA was determined 96 h following transfection with the respective siRNAs. Cells were washed with PBS and cells lysates prepared. Protein concentrations were determined by the Bradford protein assay using a spectrophotometer (Molecular Devices). Equal amounts of protein were separated electrophoretically on 4-20% Tris-glycine, SDS Polyacrylamide gels (Invitrogen) and proteins transferred to a PolyVinyliDene Fluoride (PVDF) membrane (Invitrogen). After blocking PVDF membranes with 5% non-fat dry milk at room temperature for 1 hour, membranes were incubated with primary antibodies followed by incubation with peroxidase-linked secondary antibodies. Incubation of the same membranes with anti-β-actin was used as a loading control. Protein bands were detected by chemiluminescence using an Alpha Innotech FluorChem 8900 imaging system (Merced, CA). Densitometric quantitation of immunoreactive protein bands was done from Integrated Density Values (IDV) of bands using the Alpha Ease Stand Alone software version 4.0. Primary antibodies used included anti-MyD88 antibodies (Imgenex- IMG 5499) and anti-β -actin (Sigma). Secondary antibodies were anti-rabbit and anti-mouse (Cell signaling).
4.3 Cortical neuron isolation and culture
Cortical neurons were isolated from embryonic cortices from all 3 groups of mice and cultured in Neuro Basal A medium (Gibco) as previously described (Lee et al., 2009) for 4-5 days at 106 cells per well in six-well plates.
4.3.1 OGD
All cultures were prepared for OGD as previously described (Lee et al., 2009) except that chambers were flushed with 95% nitrogen/5% CO2 at 4 liters/minute at room temperature for 30 minutes and incubated at 37°C for 5 hours with anaerobic colorimetric indicator strips that detected a 0.2% oxygen threshold (Becton Dickinson).Control plates were incubated with 1 ml glucose-containing Neuro basal A medium under normoxic conditions. After OGD, medium in all cultures was replaced with 2 ml glucose-containing Neurobasal A medium and incubated at 37°C, under normoxic conditions for 18-24hr.
4.3.2 Flow cytometric analysis of cell viability and cell death
Following reperfusion, cells were harvested using a standard papain dissociation protocol, and stained with Hoechst and PI to determine the percentage of viable, apoptotic and necrotic cells, as previously described (Hillion et al., 2005). In brief, decreased intensities of Hoechst staining were used to identify decreased DNA content of cells undergoing progressively advanced stages of apoptosis. In contrast, increased PI staining was used to detect increased cell membrane permeability of cytotoxic/necrotic cell populations and apoptotic cell populations with increased cell permeability due to advanced apoptosis.
Cell Quest acquisition and analysis software (BD Biosciences) was used to quantify fluorescence intensities from Hoechst and PI staining as previously described (Hillion et al., 2005). The percentages of viable, apoptotic and necrotic cortical neurons cultured under normoxic conditions with glucose were compared to those analyzed within each group after OGD.
In vivo
4.4 Anesthesia and euthanasia
Anesthesia for MCAO and BCCAO surgeries was induced with 5% and maintained with 1-1.5% isoflurane in 70% oxygen / 30% nitrous oxide mixture via face mask. Mice were euthanized with a lethal dose of isoflurane and nitrous oxide followed by bilateral pneumothorax.
BCCAO: a total of 30 mice were used; 9 mice each for MyD88-/- and TRIF mutant groups; 6 experimental and 3 control mice per group. The control WT group had 6 experimental and 6 control mice.
MCAO: a total of 21 mice used for MCAO studies; 7 per group for MyD88, TRIF and WT mice.
4.4.1 BCCAO
Following anesthesia induction and ventral mid-line incision, both common carotid arteries were separated from the vagus nerve and occluded with microaneurysm clips for 6 minutes. We used 6 minute BCCAO for reproducible CA1 damage in all groups of mice. Rectal temperatures were maintained at 37°C using a heating lamp and warming pad (Physitemp). Restoration of blood flow was visually confirmed following BCCAO.
4.4.2 Neurological score determination
Mice were scored for gait and posture every day for 7 days following BCCAO as follows: 0 - Normal moves freely, 1 - Mild incoordination when stimulated, 2 - Hunched posture, 3 - Obvious ataxia or head tilt; drags one or both limbs.
4.4.3 Determination of delayed neuronal death
Mice were euthanized seven days following BCCAO, perfused transcardially with PBS, followed by 4% paraformaldehyde, before overnight immersion in 4% paraformaldehyde. Fixed brains were paraffin-embedded, cut into 6-μm sections and stained with 0.5% Cresyl Violet. Normal pyramidal neurons were defined as large, pale and > 10μm diameter in the short axis. Cells were counted, in a blinded fashion, under light microscopy on 3 consecutive 300μm lengths of CA1 from a stereotactically determined level (bregma P1.80 mm-P2.20 mm). The number of normal neurons in both hippocampi were totaled for each animal and averaged for each group.
The percentage of normal neurons in each group was determined by comparing the number of normal neurons, 7 days after BCCAO, in a region of CA1 hippocampus, to the baseline CA1 neuronal density in that region in control mice from each group that had not undergone surgery. The percentage of surviving CA1 neurons in MyD88-/- and TRIF mutant mice were then compared to the percentage of surviving neurons in WT mice.
4.5 Permanent MCAO
After anesthesia induction, the distal left middle cerebral artery, lateral to the olfactory tract and inferior to the inferior cerebral vein, was exposed at the skull base, with a micro drill and cauterized with a bipolar electrocoagulator (ICC 200, EBRE Inc.). Rectal temperatures were maintained at 37°C using a heating lamp and warming pad (Physitemp). Mice recovered in warm boxes before being returned to the Animal facility.
4.5.1 Infarct volume determination
Twenty-four hours after MCAO, mice were euthanized, their brains removed and quickly placed on dry ice, and stored at -80°C. Frozen brains were cut coronally into 20-μm sections, starting from the anterior pole of the caudate nucleus. After the 1st slice which included the infarct area, every 17th serially cut slice within the infarct area, was placed on a slide and stained with Cresyl Violet. The infarct areas (mm2) were determined using NIH image J software. Absolute infarct volumes were calculated as follows: sum of infarct areas (mm2) per brain X 17 X slice thickness (20μm). Correction for edema was done by dividing the infarct volume by the ratio of the volume of the ischemic hemisphere compared to the volume of the non-ischemic hemisphere (edema index) as previously described (Leach et al., 1993). Infarct volumes from MyD88-/- and TRIF mutant mice were compared to infarct volumes from WT mice.
4.6. Statistical analysis
Comparisons of mean values among experimental groups were carried out with SAS, version 9.1.3, (Cary, N.C), using the non-parametric Kruskal-Wallis test for 3-group comparisons of means. For comparison of means between 2 groups, two-sample t-test was used after checking for normality and equality of variances. Differences among groups were expressed as mean ± standard deviation and considered significant at p < 0.05.
Acknowledgments
Dr Maria Spatz for editorial and scientific suggestions and Dr. Sungyoung Auh for help with statistical analysis.
Footnotes
Disclosures: We have no conflict of interest and nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–72. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion JA, Takahashi K, Maric D, Ruetzler C, Barker JL, Hallenbeck JM. Development of an ischemic tolerance model in a PC12 cell line. Journal of Cerebral Blood Flow & Metabolism. 2005;25:154–62. doi: 10.1038/sj.jcbfm.9600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Commun. 2005;338:1118–25. doi: 10.1016/j.bbrc.2005.10.068. [DOI] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. Journal of Neuroimmunology. 2007;190:101–11. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Wang J, Sayeed I, Ishrat T, Atif F, Stein DG. The TRIF-dependent signaling pathway is not required for acute cerebral ischemia/reperfusion injury in mice. Biochem Biophys Res Commun. 2009;390:678–83. doi: 10.1016/j.bbrc.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Zhou P, Qian LP, Chuang JZ, Lee J, Li CJ, Iadecola C, Nathan C, Ding AH. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. Journal of Experimental Medicine. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochemical and Biophysical Research Communications. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- Leach MJ, Swan JH, Eisenthal D, Dopson M, Nobbs M. BW619C89, a glutamate release inhibitor, protects against focal cerebral ischemic damage. Stroke. 1993;24:1063–7. doi: 10.1161/01.str.24.7.1063. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Castri P, Bembry J, Maric D, Auh S, Hallenbeck JM. SUMOylation participates in induction of ischemic tolerance. J Neurochem. 2009;109:257–67. doi: 10.1111/j.1471-4159.2009.05957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AEB, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. Journal of Neuroscience. 2009;29:9839–49. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P. An innate sense of danger. Annals of the New York Academy of Sciences. 2002a;961:341–2. doi: 10.1111/j.1749-6632.2002.tb03118.x. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002b;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nature Reviews Immunology. 2001;1:135–45. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Peltier DC, Simms A, Farmer JR, Miller DJ. Human Neuronal Cells Possess Functional Cytoplasmic and TLR-Mediated Innate Immune Pathways Influenced by Phosphatidylinositol-3 Kinase Signaling. Journal of Immunology. 2010;184:7010–7021. doi: 10.4049/jimmunol.0904133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. Journal of Immunology. 2007;178:6252–8. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]
- Strengeil M, Sareneva T, Foster D, Julkunen I, Matikainen S. IL-21 UP-Regulates the expression of genes associated with innate immunity and Th1 response. Journal of Immunology. 2002;169:3600–3605. doi: 10.4049/jimmunol.169.7.3600. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, Chan MH, Jeyabalan G, Yim JH, Geller DA. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1261–8. doi: 10.1152/ajpgi.00460.2005. [DOI] [PubMed] [Google Scholar]
- Wheeler RD, Boutin H, Touzani O, Luheshi GN, Takeda K, Rothwell NJ. No role for interleukin-18 in acute murine stroke-induced brain injury. J Cereb Blood Flow Metab. 2003;23:531–5. doi: 10.1097/01.WCB.0000059587.71206.BA. [DOI] [PubMed] [Google Scholar]
- Yang Qw, Li Jc, Lu Fl, Wen Aq, Xiang J, Zhang Ll, Huang Zy, Wang Jz. Upregulated expression of Toll-like receptor 4 in monocytes correlates with severity of acute cerebral infarction. Journal of Cerebral Blood Flow & Metabolism. 2008;28:1588–96. doi: 10.1038/jcbfm.2008.50. [DOI] [PubMed] [Google Scholar]
- Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC, Fang CQ, Wang JZ. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2010 doi: 10.1038/jcbfm.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochemical & Biophysical Research Communications. 2007;359:574–9. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]