

Abstract
Rationale
Post-translational phosphorylation of connexin43 (Cx43) has been proposed as a key regulatory event in normal cardiac gap junction expression and pathologic gap junction remodeling. Nonetheless, the role of Cx43 phosphorylation in the context of the intact organism is poorly understood.
Objective
To establish whether specific connexin43 phosphorylation events influence gap junction expression and pathologic remodeling.
Methods and Results
We generated Cx43 germline knock-in mice in which serines 325/328/330 were replaced with phosphomimetic glutamic acids (S3E) or non-phosphorylatable alanines (S3A). The S3E mice were resistant to acute and chronic pathologic gap junction remodeling (GJR) and displayed diminished susceptibility to the induction of ventricular arrhythmias. Conversely, the S3A mice showed deleterious effects on cardiac gap junction formation and function, developed electrical remodeling and were highly susceptible to inducible arrhythmias.
Conclusions
These data demonstrate a mechanistic link between post-translational phosphorylation of Cx43 and gap junction formation, remodeling and arrhythmic susceptibility.
Keywords: gap junction, casein kinase, arrhythmias, optical mapping, mouse, connexin
INTRODUCTION
Gap junctional communication is essential for normal cardiac impulse propagation. A growing body of literature provides compelling evidence supporting the concept that dysregulation of cell-cell coupling in the heart contributes to a pro-arrhythmic substrate.1 The proximate pathologic stimuli that result in aberrant cardiac gap junction expression and function are diverse. They include common forms of acquired heart disease including hypertrophy and ischemic heart disease1, as well as rare genetic causes such as the somatic mutations in connexin 40 associated with sporadic cases of atrial fibrillation2 or the inherited mutations in Cx43 responsible for oculodentodigital dysplasia.3
The cardiac connexins (i.e. connexins 43, 40 and 45) are all phosphoproteins. Post-translational phosphorylation is thought to affect multiple aspects of the connexin lifecycle including transport to the plasma and junctional membrane, oligomerization into connexons (hemi-channels), gap junction assembly, gating of assembled gap junction channels, as well as degradative processes involving lysosomal and proteasomal pathways.4, 5 Gap junction remodeling (GJR) is likely to reflect perturbations at any number of these steps in the lifecycle of connexins.
Cx43, the major ventricular gap junction protein, undergoes post-translational phosphorylation in its carboxy-terminus at as many as a dozen amino acids. Significant progress has been made in the identification of specific kinases and phosphatases that act upon Cx43 as well as their specific target sites. Protein kinase A (PKA)-dependent phosphorylation of Cx43 results in increased trafficking to the plasma membrane;6, 7 phosphorylation at CK1δ sites on Cx43 plays a significant role in gap junction assembly;8-10 Cx43 interaction with Src kinase has the dual effect of destabilizing Cx43 interaction with the scaffolding protein ZO-1 leading to Cx43 internalization/degradation;11-13 as well as down regulation of gap junctional cell-cell communication;14-16 MAPKs also inhibit intercellular communication by reduced channel open probability;17-19 while protein kinase C (PKC)-dependent phosphorylation of Cx43 influences unitary conductance of assembled channels 20-22 and may be further involved in cell-cycle progression.23 The protein phosphatases PP1 and PP2A co-localize with Cx43 and were found to be upregulated in heart failure models, suggesting a role in GJR.24, 25 Much of these data were gleaned from experiments using pharmacological approaches as well as in vitro studies of site-specific connexin mutants. While these studies have been informative, the relevance of these approaches to in vivo physiology and pathophysiology is uncertain.
CK1δ phosphorylation of Cx43 occurs at serines 325/328/330. Cell culture studies suggest that these sites play an integral role in gap junction assembly as well as the formation of the slower migrating phospho-isoforms of Cx43 observed when subjected to SDS-PAGE.8, 9 Interestingly, recent data indicate that phosphorylation at these sites is markedly reduced in response to cardiac ischemia, and the loss of phosphorylation is associated with relocalization of the protein from gap junctions to the lateral borders of myocytes.9 Moreover, our laboratory demonstrated that not only acute stressors such as ischemia, but also the imposition of chronic pressure-overload hypertrophy resulted in early and progressive dephosphorylation of Cx43 at these same residues, in association with GJR and slowing of impulse propagation.10 These changes were blunted by mineralocorticoid receptor blockade, a therapy that has been shown to diminish sudden cardiac deaths in patients with heart failure.26
In the present study, we tested the hypothesis that modulation of post-translational phosphorylation of Cx43 at this triplet of serines would influence gap junction remodeling and alter arrhythmic susceptibility. We generated two new strains of genetically engineered knock-in mice. Compared to wild type mice, cardiac gap junctions in mice harboring phosphatase-resistant phosphomimetic glutamic acids at residues 325, 328 and 330 (S3E), were resistant to pathologic remodeling and to the induction of ventricular arrhythmias. In contrast, introduction of non-phosphorylatable alanines into the carboxyl terminus (S3A) interfered with gap junction formation and function and promoted a proarrhythmic phenotype. Our data provide the first in vivo evidence to date demonstrating a mechanistic link between Cx43 phosphorylation status, electrophysiological substrate and arrhythmic susceptibility.
MATERIALS AND METHODS
An expanded Materials and Methods Section is found in the online supplement
Generation of Cx43 Phospho-mutant Constructs and Mutant Mice
Site-directed mutagenesis was performed to introduce the S3E and S3A (residues 325,328,330) into the same gene-targeting vector previously used to conditionally inactivate Cx43 in the heart and other lineages.27-29 Cx43 phospho-mutant targeting vectors were introduced into the 129/Sv-derived ES cell line R127, 30 and correctly targeted ES clones were injected into C57BL/6 blastocysts. Highly chimeric male mice were crossed with wild-type (WT) CD1 females to generate F1 Cx43S3A/WT and Cx43S3E/WT heterozygous mutant mice, which were then bred to generate homozygous Cx43S3A/S3A and Cx43S3E/S3E mice. Both heterozygous and homozygous intercrosses were maintained and for all experiments, WT littermates were used as controls.
Echocardiography
Left ventricular dimensions and function were assessed by echocardiography as previously described using an ATL 5000CV Ultrasound System (Philips Medical, Bothell, WA).27
Western Blot Analysis
Total protein lysates and Triton X-100 insoluble pellet fractions were prepared from the apical two-thirds of the ventricle as previously described.31 Signals were visualized and quantified using the Odyssey Imaging System (Li-Cor, Lincoln, NE).
Immunofluorescence, Confocal Microscopy, and Cx43 Quantification
Immunofluorescent staining was performed on formalin-fixed, paraffin embedded sections using rabbit polyclonal anti-Cx43 antibody (Sigma c6219) and mouse anti-N-cadherin antibody (Invitrogen). Gap junction remodeling was quantified by determining the extent of colocalization of Cx43 and N-cadherin at the intercalated discs (CoLocalizer Pro software, CoLocalization Research Software).
Heart Isolation and Optical Mapping
High-resolution optical mapping experiments were performed as previously described, using a CMOS video camera (Ultima-L; SciMedia, Inc).27, 32 Epicardial conduction velocity (CV) and optical action potential duration and APD dispersion measurements were obtained from the left ventricular surface at a basic cycle length (BCL) of 100 ms.
Electrophysiology Studies (EPS)
EPS was performed as previously described using a 2-French octapolar intracardiac catheter.33 Standard pacing protocols (single, double and triple extrastimulation and burst pacing were performed to test for ventricular tachycardia (VT) inducibility. VT was defined as ≥ 4 consecutive beats.
Transverse aortic constriction
Transverse aortic constriction was performed on male mice at 3-4 months of age as previously described.34
No-Flow Ischemia Protocol
A Langendorff global ischemia model was performed as previously described.35
Statistical Analysis
Data are presented as mean ± SEM and analyzed by Student’s t-test. For multiple groups, one way ANOVA was performed with post-hoc Newman-Keuls test. Arrhythmia inducibility was analyzed by Fisher’s exact test. P values less than 0.05 were considered statistically significant.
RESULTS
S3A and S3E Mice Develop Normally
To evaluate the effects of Cx43 phosphorylation on gap junction expression and function in vivo, we generated site-specific knock-in mice expressing either the S3A or S3E mutations, using a strategy identical to that previously utilized to generate Cx43 oculodentodigital dysplasia (ODDD) mutant mice.36 Homozygous S3A and S3E mice were grossly indistinguishable from WT controls at birth and throughout development into adulthood (> 3 months). Histological examination of S3A and S3E adult hearts appeared entirely normal without any evidence of fibrosis, hypertrophy, or myofibrillar disarray (data not shown). There was no statistically significant difference between the three genotypes with respect to contractile performance, left ventricular chamber sizes, wall thicknesses, or heart weight to body weight ratio (Supplemental Table I).
Altered Cx43 Phosphorylation and Gap Junction Formation in S3A and S3E Mice
Immunoblotting studies were performed to determine whether the site-specific mutations were associated with changes in Cx43 electrophoretic fractionation. Prior reports have shown that Cx43 resolves into multiple electrophoretic isoforms when subjected to SDS/PAGE, including a fast migrating non-phosphorylated band termed P0 and slower migrating phosphorylated bands referred to as P1, P2 and higher.4 Consistent with this notion, immunoblotting of highly resolved total protein lysates from mice of each genotype revealed distinctive banding patterns consistent with altered phosphorylation. WT hearts with polyclonal panCx43 antibodies revealed a faint P0 band as well as more prominent P1, P2, and P3 bands. In contrast, homozygous S3A mice demonstrated faint amounts of P0 and an accumulation of the P1 isoform, while total protein lysates from the hearts of homozygous S3E mice displayed a near absence of all Cx43 isoforms faster than P2 (Figure 1A upper panel). Heterozygous mice showed intermediate phenotypes. We also determined the banding pattern of Triton X-100 insoluble protein extracts, which are enriched in junctional membrane proteins.37 This fractionation resulted in the preferential recovery of more slowly migrating forms of Cx43 for all genotypes (Figure 1A lower panel).
Figure 1. Electrophoretic mobility of Cx43 mutants.
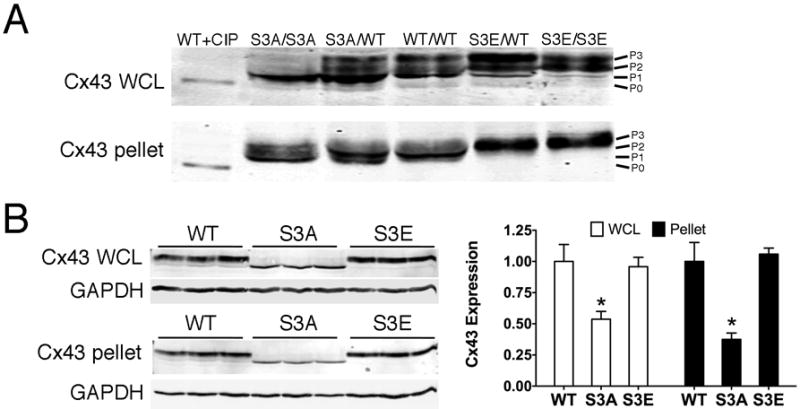
(A) High resolution Western blot analysis of whole cell lysates (WCL) or Triton X-100 insoluble pellets (pellet) prepared from ventricles of mice with the indicated genotypes, probed with polyclonal panCx43 antisera. Wild type Cx43 lysate treated with calf intestine phosphatase (CIP) migrates at P0 and is shown for comparison to various major phosphorylated forms of Cx43 (P1, P2, P3). (B) Western blot analysis of whole cell lysates (upper) or Triton X-100 insoluble pellets (lower). N=3 for each genotype. Quantification is shown to the right. *P<0.05 compared to WT.
To quantitatively determine whether the site-specific mutants affected the steady-state accumulation of Cx43, we performed Western blot analysis of whole cell lysates as well as Triton X-100 insoluble pellet fractions prepared from the hearts of a cohort of wild type, homozygous S3A and S3E mutant mice. Compared to hearts expressing wild type Cx43, the amount of total Cx43-S3A protein found in whole cell lysates as well as amounts within the Triton X-100 insoluble fraction was significantly reduced to less than 50% of control levels, indicative of less accumulation of Cx43 into gap junctions (Figure 1B). In contrast, the abundance of the phosphomimetic S3E mutant protein was indistiguishable from wild type levels in both whole cell lysates and Triton X-100 insoluble fractions (Figure 1B).
To directly visualize gap junctional plaques, we next performed immunofluorescent staining of hearts from each of the three strains of mice. Hearts of S3A mice showed significantly less total Cx43 co-localized with N-cadherin at intercalated discs compared to WT controls or S3E mutant mice (Figure 2A upper images and Figure 2B). Taken together, these findings are consistent with prior in vitro data suggesting that phosphorylation of Cx43 at serine residues 325/328/330 leads to P2 formation, and also suggest that inhibition of this phosphorylation event in vivo interferes with normal Cx43 trafficking and/or stability.9
Figure 2. Cx43-S3E phospho-mimetic mutation prevents pathologic gap junction remodeling.
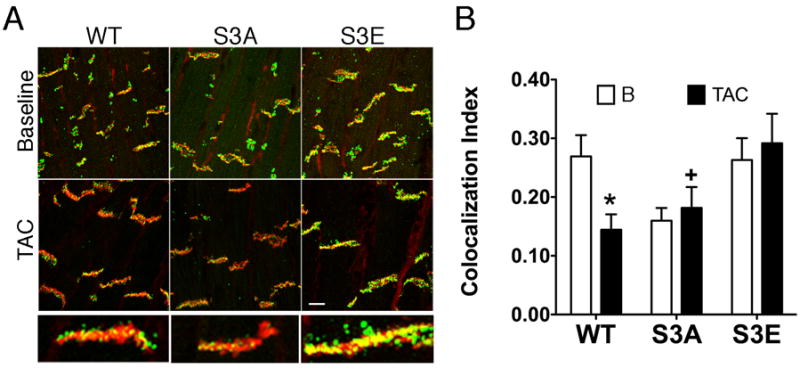
(A) Representative immunofluorescent staining with panCx43 (Cx43, green) and N-cadherin (N-cad, red) antibodies at baseline (B) and four weeks following transverse aortic constriction (TAC). Scale bar = 10 μm. Magnified views of individual gap junction plaques for each genotype after TAC are shown below. (B) Quantitative analysis of Cx43 and N-cadherin colocalization at baseline (□) and following TAC (■). *P<0.05 compared to S3E, Post-TAC; +P<0.05 compared to WT and S3E.
S3E Mice are Resistant to Structural GJR
We previously demonstrated that chronic pressure overload hypertrophy secondary to transverse aortic constriction (TAC) results in dephosphorylation of Cx43 at serines 325/328/330, as well as structural GJR and slowing of impulse propagation.10 To determine whether modification of these sites affected the heart’s response to chronic pathologic stress, wild type and homozygous S3A and S3E mutant mice were subjected to TAC and followed for up to 4 weeks. Mice of all three genotypes exhibited comparable structural remodeling, with deterioration of contractile function following imposition of pressure overload (Supplemental Table I). As anticipated from our previous work10, analysis of whole cell lysates from wild type hearts subjected to TAC showed molecular evidence of GJR, with an obvious shift toward higher mobility/hypophosphorylated forms of Cx43 compared to sham-operated mice (Figure 3A). In contrast, the more rapid electrophoretic mobility of Cx43-S3A and the retarded mobility of Cx43-S3E seen at baseline was largely unaffected by imposition of TAC, consistent with the phosphatase-resistant design of both mutant proteins. However, analysis of Triton X-100 insoluble fractions, enriched in gap junctions, revealed a significant reduction in abundance of Cx43 in WT hearts and particularly in S3A mutant hearts after TAC, whereas the S3E hearts were resistant to molecular GJR (Figure 3B). In agreement with these data, we found by confocal immunofluorescence that while TAC led to significant GJR in WT and S3A mice, the S3E mice were resistant to pathologic remodeling, with preservation of Cx43 at the intercalated discs, largely colocalizing with N-cadherin (Figure 2A, compare upper and lower panels and quantification in Figure 2B).
Figure 3. S3E mice are resistant to structural GJR after chronic pathologic stimuli.
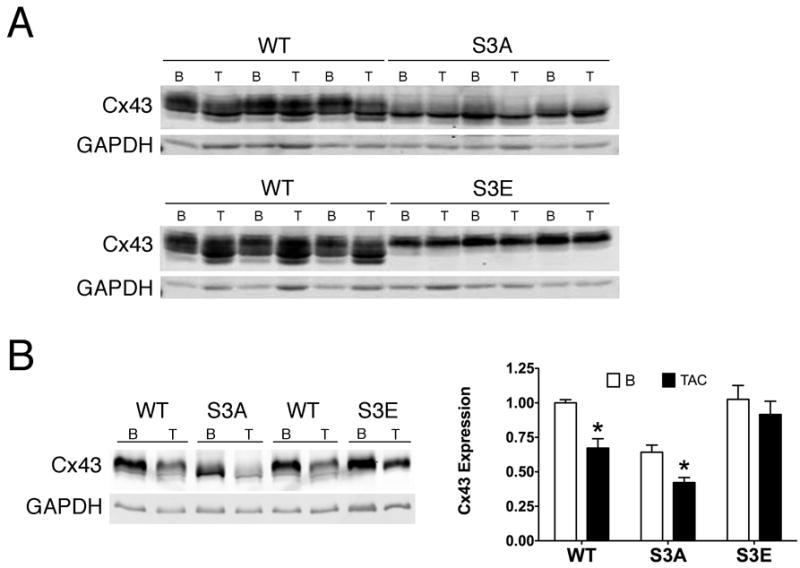
(A) Representative immunoblots of whole cell lysates at baseline (B) and after TAC (T), probed with polyclonal panCx43 and GAPDH antibodies. (B) Quantitative analysis of Cx43 in gap junction enriched Triton X-100 insoluble fractions, at baseline and after TAC, normalized to GAPDH. N=3 hearts for each group. *P<0.05, TAC vs same genotype at baseline.
To further examine the relevance of post-translation phosphorylation of Cx43 at serines 325/328/330, we subjected isolated hearts from WT, S3A and S3E mice to acute global ischemia, as previously described.35 Confirming prior studies, there was a significant increase in the amount of P0 Cx43 in WT hearts post-ischemia. Post-ischemia S3A hearts also showed an accumulation in the non-phosphorylated Cx43 band; in contrast, the S3E post-ischemia hearts displayed significantly less dephosphorylation (Figure 4).
Figure 4. S3E mice are resistant to structural GJR after acute pathologic stimuli.
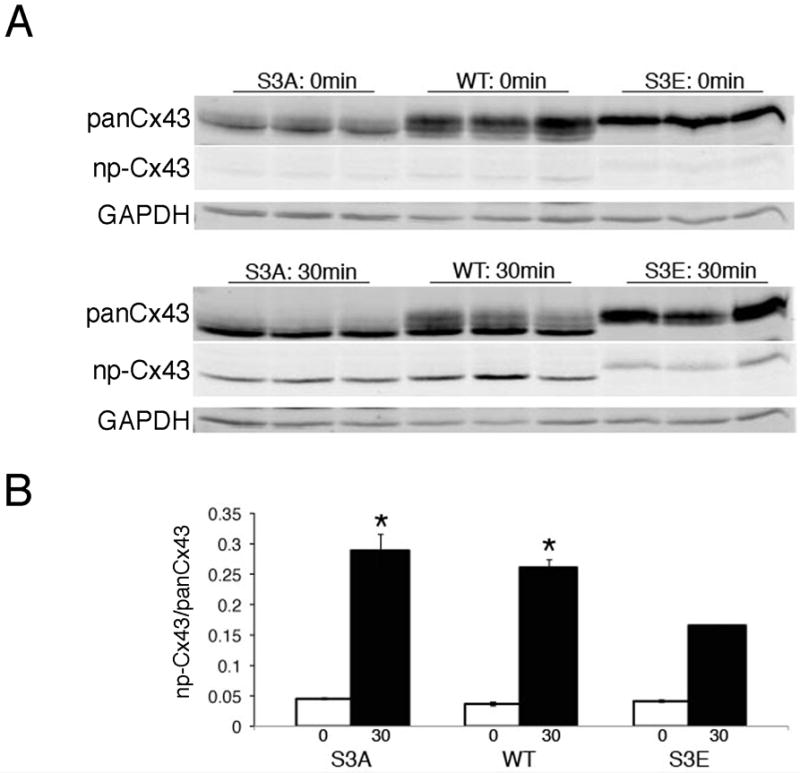
(A) Representative immunoblots of samples probed with polyclonal panCx43, monoclonal non-phosphorylated Cx43 (np-Cx43) and GAPDH antibodies prior to (0 min) and 30 minutes (30min) after imposition of global ischemia. (B) Quantitative analysis of np-Cx43 signal relative to panCx43 signal. N=3 hearts for each group. *P<0.05 compared to S3E at 30 minutes global ischemia.
S3E Mice are Resistant to Conduction Slowing
To determine whether these affects on gap junctional remodeling were associated with functional sequelae, we performed optical mapping to quantify conduction velocities in WT and mutant mice, both at baseline and following TAC. As shown in Figure 5, optical mapping of WT and S3A hearts revealed significantly diminished CVmax and CVmin post-TAC, but S3E conduction velocities were preserved, consistent with the lack of structural GJR.
Figure 5. Functional analysis of isolated-perfused hearts.
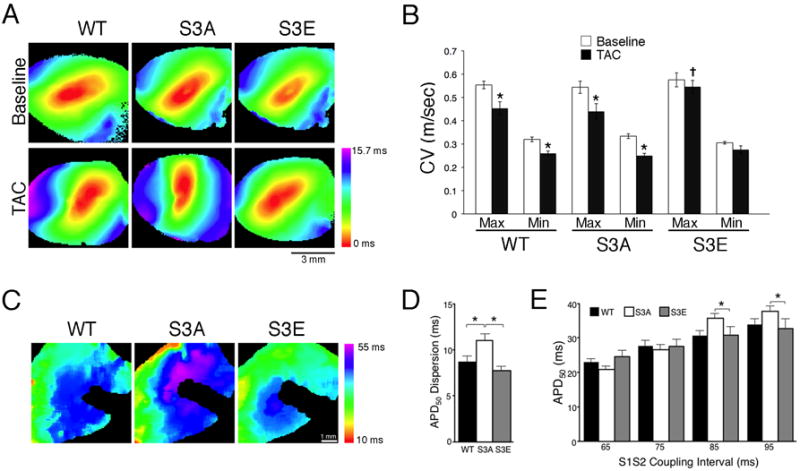
(A) Representative activation maps and (B) quantification of conduction velocity at baseline and after TAC. *P<0.05 compared to baseline for each genotype; +P<0.05 compared to TAC WT and S3A. (C) Representative APD50 maps. The shadow in each image represents a non-pacing positioning. (D) Quantification of averages values of APD50 dispersion at an S1S2 coupling interval of 95 ms. (E) Average values of APD50 taken at a range of coupling intervals. CV, conduction velocity. *P<0.05. N=6 hearts for each genotype.
Connexin Mutations Differentially Influence Electrical Remodeling
Recent data suggest that the intercalated disc may behave as a functional unit and changes in gap junction expression may influence other ionic currents.32, 38, 39 Therefore, we also measured action potential parameters in isolated-perfused hearts. Interestingly, hearts from S3A mice showed a significant increase in APD dispersion compared to WT and S3E mutants (Figure 5A-D). Moreover, the restitution properties of the S3A hearts also differed from the other two genotypes; the relationship between coupling interval and APD was steeper in these mice, with a trend toward shorter APD50 values at short S1S2 coupling intervals and greater APD50 values at longer coupling intervals (Figure 5E).
S3E Mice Are Less Susceptible to Induction of Ventricular Arrhythmias
A key goal of strategies to modify GJR is amelioration of arrhythmic activity in the intact organism. Accordingly, using two different provocative methods, we tested whether the changes in electrophysiological properties identified above were associated with alterations in arrhythmic propensity. Baseline WT and mutant mice were subjected to PES with premature extrastimuli. Using this protocol, 40% of S3A mice and 33% of WT mice developed ventricular tachyarrhythmias, but none of the S3E mice were inducible. Using the more aggressive burst pacing protocol, 70% of S3A mice and 42% of WT mice developed VT, but only 18% of S3E mice were inducible, as shown in Figure 6.
Figure 6. S3E mice are resistant to inducible ventricular arrhythmias.
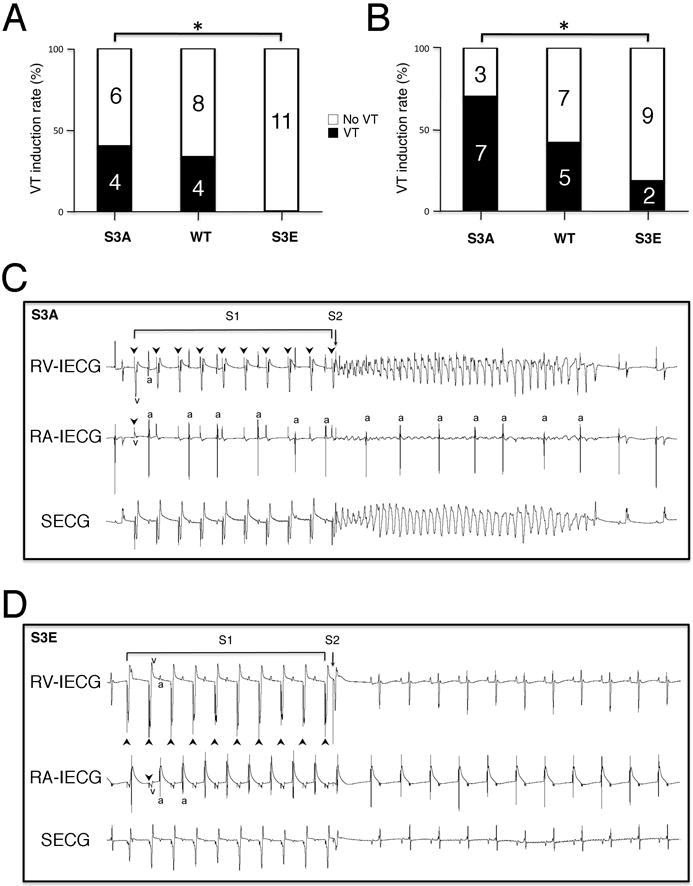
Percentage of mice of each genotype with ventricular tachycardia (VT) induced by premature extrastimuli (A) or burst pacing (B). (C) Representative intracardiac and surface electrocardiograms of an S3A mouse during an episode of VT induced by a single extrastimulus. AV dissociation is noted after onset of VT. (D) Representative intracardiac and surface electrocardiograms of an S3E mouse, showing resumption of sinus rhythm after the premature beat. RV-IECG, right ventricular intracardiac electrocardiogram; RA-IECG, right atrial intracardiac electrocardiogram; SECG, surface electrocardiogram; Arrowheads, stimulus artifact; v, ventricular electrogram; a, atrial electrogram. *P<0.05.
DISCUSSION
Sudden cardiac death from ventricular tachyarrhythmias results in as many as 462,000 deaths annually in the United States alone.40 Prior studies suggest that GJR plays an important role in the pathophysiology of these lethal cardiac arrhythmias. Cx43 is the major cardiac gap junction protein, and we have previously shown that the elimination of Cx43-dependent intercellular coupling between cardiomyocytes results in a slowing of ventricular conduction velocity and uniform sudden arrhythmic death in mice.27 These results illustrate the importance of Cx43 to normal cardiac function, but do not address the underlying mechanism and functional consequences of pathologic GJR that occur in response to myriad myopathic stimuli associated with acquired forms of heart disease.
This study was performed to further explore the significance of post-translational phosphorylation of Cx43 at serines 325/328/330 and the role of this regulatory event in impulse propagation and arrhythmogenicity. Our focus on these particular residues derives from in vitro studies demonstrating the importance of these sites for gap junction formation, as well as our recent observation that dephosphorylation at these residues is an early response to pressure-overload hypertrophy, a clinically important pathologic stimulus associated with increased mortality and sudden cardiac death.41 We established strains of mutant knock-in mice to determine the relevance of these sites in vivo. The results of our analysis indicate that pseudo-phosphorylation of Cx43 at serines 325/328/330 (S3E mutant mice) leads to gap junctions that appear resistant to pathologic remodeling associated with both acute (no-flow ischemia) and chronic (TAC) pathologic stimuli. Conversely, phosphorylation-deficient S3A mutant mice display aberrant gap junction expression even at baseline. Importantly, while WT mice have a similar incidence of VT induction by PES as previously reported33, the S3E mice are highly resistant to the induction of life-threatening ventricular arrhythmias, and the S3A mice display increased arrhythmic susceptibility.
The results of this study are consistent with prior in vitro studies using specific CK1δ inhibitors that caused decreased gap junction formation 8, as well as in vivo work using phosphospecific antibodies that found decreased expression of Cx43 phosphorylated at residues 325/328/330 in gap junctions after ischemia and TAC.9, 10 Our data expand on these findings by showing a link between phosphorylation of Cx43, GJR, and susceptibility to arrhythmia induction in vivo.
It is important to point out that although S3A mutants are non-phosphorylated at serines 325/328/330, P2 and slower bands were present on immunoblotting of the Triton X-100 insoluble fraction. This may indicate that CK1δ-dependent phosphorylation is electrophoretically silent, but facilitates an additional phosphorylation event, which in turn is responsible for the formation of “P2”. In this scenario, the second phosphorylation event responsible for P2 formation may be less efficient in the S3A mutants but not completely abrogated. Alternatively, it may simply be the case that with so many potential phosphorylatable residues within the carboxy-terminus of this gap junctional protein, the various “P”-forms represent heterogeneous populations of Cx43 phospho-isoforms. Indeed, it has previously been shown that certain Cx43 phospho-isoforms can even migrate at P0.8, 9, 42
Interestingly, the Cx43 phosphorylation-site mutations appear to differentially influence action potential properties. Mice expressing the S3A mutation displayed significantly longer APD50 values as well as an increase in the dispersion of repolarization. Moreover, hearts from the S3A mice showed steeper restitution curves. Taken together, these properties may play an unexpectedly significant role in conferring the pro-arrhythmic phenotype observed in these mutant mice. Indeed we previously reported significant electrical remodeling in Cx43 deficient hearts.38 These observations reinforce recent suggestions that the intercalated disc may act as a functional unit, with crosstalk between connexins, scaffolding proteins such as ankyrins, and various sodium and potassium channel subunits.39
There are several limitations to our study. While these experiments provide definitive evidence linking Cx43 phosphorylation state to gap junction stability and resistance to GJR, additional work will be required to more precisely identify what step(s) in the connexin life-cycle are primarily responsible for this salutary behavior. One hypothesis for the increased stability seen with CK1δ phosphorylated Cx43 is that phosphorylation at these sites confers protection from degradative pathways. Gap junctions are dynamic structures whose degradation involves internalization and eventual breakdown by both lysosomal43-45 and proteasomal pathways.45-48 Ubiquitin conjugation to gap junctional Cx43 is thought to initiate endocytosis. The ubiquitin protein ligase Nedd4 is known to bind the carboxyl terminus of Cx43, and the phosphorylation status of Cx43 may modulate this interaction.49 Previous studies have shown that epidermal growth factor and 12-O-tetradecanoylphorbol-13-acetate treatment of rat epithelial cells induces ubiquitination of Cx43 and this process is associated with hyperphosphorylation of Cx43 by MAPK and PKC.50 These findings illustrate the close relationship between Cx43 phosphorylation status and ubiquitination, as well as the importance of specific target residues. It is conceivable that phosphorylation at serines 325/328/330 interferes with endocytosis and subsequent degradation of gap junctions, while dephosphorylation at these sites enhances degradation. Our studies using heterologous expression systems have suggested the half-live of the various mutant Cx43 proteins differ from WT Cx43, however attempts to perform comparable studies in intact hearts using translational inhibitors have not been revealing. Alternatively, the major effect of Cx43 phosphorylation could involve trafficking. The S3E modification might augment (and conversely, the S3A mutation inhibit) the direct targeting of connexons to the adherens junction.51 Additional studies will be required to distinguish among these potential mechanisms.
Inasmuch as the phosphorylation status of Cx43 reflects the balance of various kinases and phosphatases such as CK1δ and PP2A, we measured the abundance of these two enzymes. Given the recent report of increased expression of PP2A and enhanced association of PP2A with Cx43 in a model of non-ischemic heart failure, we anticipated a similar finding in the hypertrophied hearts. However, we found no change in the abundance of either of CK1δ or PP2A in hearts with pressure-overload hypertrophy and no evidence of increased interaction between PP2A and Cx43 (data not shown). While this does not exclude changes in enzymatic activity, additional studies will be required to clarify the mechanisms responsible for the altered phosphorylation state of Cx43 we observed in hypertrophied hearts.
In summary, these data provide the first in vivo evidence to date of a mechanistic link between Cx43 phosphorylation status, gap junction expression and arrhythmic susceptibility. Our results provide additional evidence indicating that modulation of Cx43 phosphorylation status may be a rational anti-arrhythmic strategy.52
Supplementary Material
Novelty and Significance.
What is known?
Gap junction comprise intercellular channels that electrotonically couple cardiomyocytes with one another and facilitate impulse propagation and normal rhythmicity in the heart.
Diverse disease-causing stimuli promote abnormal expression of gap junctions, i.e., pathologic gap junction remodeling (GJR), and various lines of experimental evidence indicate that GJR contributes to increased arrhythmic propensity.
Connexin43 (Cx43), the major cardiac gap junction protein, is post-translationally phosphorylated by numerous kinases and these post-translational events are thought to regulate channel assembly, membrane trafficking, gating and turnover.
In vitro studies indicate that phosphorylation of Cx43 by casein kinase 1δ (CK1δ) at serines 325, 328 and 330 may promote gap junction assembly, suggesting this event may be an important regulator of intercellular coupling and cardiac rhythmicity.
What new information does this article contribute?
Using genetically engineered knock-in mice with site-specific mutations introduced into the Cx43 gene, we demonstrate that phospho-mimetic mutants of Cx43 at CK1δ-dependent target sites enhance gap junction formation and confer resistance both to pathologic GJR and the induction of ventricular arrhythmias.
Conversely, inhibition of phosphorylation at these same target sites diminishes gap junction formation and confers enhanced arrhythmic susceptibility.
Histologic pathologic studies have demonstrated abnormal expression of gap junctions in diverse cardiac disease states, including most acquired and a number of inherited cardiomyopathies. At the molecular level, this process of GJR is associated with hypophosphorylation of Cx43, the major gap junctional protein. While expression and function of various kinases and phosphatases that act upon Cx43 have been examined in vitro, the precise role of these regulatory pathways in the context of the intact organism remain unknown. Using genetically engineered mice with site-specific mutations introduced into the Cx43 protein that either mimic (glutamic acid) or prevent (alanine) phosphorylation, we tested the role of CK1δ-dependent phosphorylation of Cx43 on gap junction expression and function. The phospho-mimetic mutant mice were resistant to GJR and induction of ventricular arrhythmias, whereas the phospho-resistant mutant mice showed the opposite phenotype. These data, demonstrate for the first time a mechanistic link between post-translational phosphorylation of Cx43 and gap junction formation, remodeling, and arrhythmic susceptibility in vivo
Acknowledgments
SOURCES OF FUNDING This study was supported by National Institutes of Health grants HL64757, HL82727, HL81336 and 1S10RR026881 (GIF) and HL076751 (GEM), Glorney-Raisbeck Cardiovascular Fellowships (BR and JML), and a Sarnoff Cardiovascular Research Fellowship (FMV).
Non-standard Abbreviations and Acronyms
- Cx43
Connexin43
- CK1δ
Casein kinase 1δ
- CV
Conduction velocity
- GJR
Gap junction remodeling
- TAC
Transverse aortic constriction
- EPS
Electrophysiology study
Footnotes
DISCLOSURES None
References
- 1.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 3.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreno AP, Lau AF. Gap junction channel gating modulated through protein phosphorylation. Prog Biophys Mol Biol. 2007;94:107–119. doi: 10.1016/j.pbiomolbio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, Johnson RG. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113(Pt 17):3037–3049. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- 7.TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem. 2002;277:44962–44968. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- 9.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–3442. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, Lampe PD, Fishman GI. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365–371. doi: 10.1161/CIRCRESAHA.108.184044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilleron J, Carette D, Fiorini C, Benkdane M, Segretain D, Pointis G. Connexin 43 gap junction plaque endocytosis implies molecular remodelling of ZO-1 and c-Src partners. Commun Integr Biol. 2009;2:104–106. doi: 10.4161/cib.7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilleron J, Fiorini C, Carette D, Avondet C, Falk MM, Segretain D, Pointis G. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008;121:4069–4078. doi: 10.1242/jcs.033373. [DOI] [PubMed] [Google Scholar]
- 13.Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 14.Duffy HS, Ashton AW, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–222. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- 15.Postma FR, Hengeveld T, Alblas J, Giepmans BN, Zondag GC, Jalink K, Moolenaar WH. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 17.Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee JD, Abe J, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase1/ERK5 but not ERK1/2 kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 18.Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol. 2003;284:C511–520. doi: 10.1152/ajpcell.00214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warn-Cramer BJ, Lampe PD, Kurata WE, Kanemitsu MY, Loo LW, Eckhart W, Lau AF. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- 20.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreno AP, Fishman GI, Spray DC. Phosphorylation shifts unitary conductance and modifies voltage dependent kinetics of human connexin43 gap junction channels. Biophys J. 1992;62:51–53. doi: 10.1016/S0006-3495(92)81775-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno AP, Saez JC, Fishman GI, Spray DC. Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ Res. 1994;74:1050–1057. doi: 10.1161/01.res.74.6.1050. [DOI] [PubMed] [Google Scholar]
- 23.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 24.Duthe F, Plaisance I, Sarrouilhe D, Herve JC. Endogenous protein phosphatase 1 runs down gap junctional communication of rat ventricular myocytes. Am J Physiol Cell Physiol. 2001;281:C1648–1656. doi: 10.1152/ajpcell.2001.281.5.C1648. [DOI] [PubMed] [Google Scholar]
- 25.Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. 2005;96:54–63. doi: 10.1161/01.RES.0000152325.07495.5a. [DOI] [PubMed] [Google Scholar]
- 26.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 27.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Presley CA, Lee AW, Kastl B, Igbinosa I, Yamada Y, Fishman GI, Gutstein DE, Cancelas JA. Bone marrow connexin-43 expression is critical for hematopoietic regeneration after chemotherapy. Cell Commun Adhes. 2005;12:307–317. doi: 10.1080/15419060500514200. [DOI] [PubMed] [Google Scholar]
- 29.Sridharan S, Simon L, Meling DD, Cyr DG, Gutstein DE, Fishman GI, Guillou F, Cooke PS. Proliferation of adult sertoli cells following conditional knockout of the Gap junctional protein GJA1 (connexin 43) in mice. Biol Reprod. 2007;76:804–812. doi: 10.1095/biolreprod.106.059212. [DOI] [PubMed] [Google Scholar]
- 30.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagy JI, Li WE, Roy C, Doble BW, Gilchrist JS, Kardami E, Hertzberg EL. Selective monoclonal antibody recognition and cellular localization of an unphosphorylated form of connexin43. Exp Cell Res. 1997;236:127–136. doi: 10.1006/excr.1997.3716. [DOI] [PubMed] [Google Scholar]
- 32.Leaf DE, Feig JE, Vasquez C, Riva PL, Yu C, Lader JM, Kontogeorgis A, Baron EL, Peters NS, Fisher EA, Gutstein DE, Morley GE. Connexin40 imparts conduction heterogeneity to atrial tissue. Circ Res. 2008;103:1001–1008. doi: 10.1161/CIRCRESAHA.107.168997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maguire CT, Wakimoto H, Patel VV, Hammer PE, Gauvreau K, Berul CI. Implications of ventricular arrhythmia vulnerability during murine electrophysiology studies. Physiol Genomics. 2003;15:84–91. doi: 10.1152/physiolgenomics.00034.2003. [DOI] [PubMed] [Google Scholar]
- 34.Rockman HA, Wachhorst SP, Mao L, Ross J., Jr ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol. 1994;266:H2468–2475. doi: 10.1152/ajpheart.1994.266.6.H2468. [DOI] [PubMed] [Google Scholar]
- 35.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 36.Kalcheva N, Qu J, Sandeep N, Garcia L, Zhang J, Wang Z, Lampe PD, Suadicani SO, Spray DC, Fishman GI. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci U S A. 2007;104:20512–20516. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danik SB, Rosner G, Lader J, Gutstein DE, Fishman GI, Morley GE. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. Faseb J. 2008;22:1204–1212. doi: 10.1096/fj.07-8974com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 40.State-specific mortality from sudden cardiac death--United States, 1999. MMWR Morb Mortal Wkly Rep. 2002;51:123–126. [PubMed] [Google Scholar]
- 41.Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- 42.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- 43.Larsen WJ, Hai N. Origin and fate of cytoplasmic gap junctional vesicles in rabbit granulosa cells. Tissue Cell. 1978;10:585–598. doi: 10.1016/s0040-8166(16)30351-2. [DOI] [PubMed] [Google Scholar]
- 44.Naus CC, Hearn S, Zhu D, Nicholson BJ, Shivers RR. Ultrastructural analysis of gap junctions in C6 glioma cells transfected with connexin43 cDNA. Exp Cell Res. 1993;206:72–84. doi: 10.1006/excr.1993.1122. [DOI] [PubMed] [Google Scholar]
- 45.Laing JG, Tadros PN, Westphale EM, Beyer EC. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- 46.Laing JG, Beyer EC. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem. 1995;270:26399–26403. doi: 10.1074/jbc.270.44.26399. [DOI] [PubMed] [Google Scholar]
- 47.Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- 48.Leithe E, Cruciani V, Sanner T, Mikalsen SO, Rivedal E. Recovery of gap junctional intercellular communication after phorbol ester treatment requires proteasomal degradation of protein kinase C. Carcinogenesis. 2003;24:1239–1245. doi: 10.1093/carcin/bgg066. [DOI] [PubMed] [Google Scholar]
- 49.Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci. 2006;119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- 50.Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem. 2004;279:50089–50096. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- 51.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kjolbye AL, Haugan K, Hennan JK, Petersen JS. Pharmacological modulation of gap junction function with the novel compound rotigaptide: a promising new principle for prevention of arrhythmias. Basic Clin Pharmacol Toxicol. 2007;101:215–230. doi: 10.1111/j.1742-7843.2007.00123.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.