

Abstract
Mutant src−/− mice have osteopetrosis resulting from defective osteoclasts, the cells that resorb bone. However, signaling pathways involving Src family members in osteoclasts remain unclear. We demonstrate that expression of a truncated Src molecule, Src251, lacking the kinase domain, induces osteopetrosis in wild-type and src+/− mice and worsens osteopetrosis in src−/− mice by a novel mechanism, increased osteoclast apoptosis. Induction of apoptosis by Src251 requires a functional SH2, but not an SH3, domain and is associated with reduced AKT kinase activity. Expression of Src251 dramatically reduces osteoclast survival in response to RANKL/TRANCE/OPGL, providing evidence that Src family kinases are required in vivo for survival signaling pathways downstream from TNF family receptors.
Keywords: Src, RANK, osteoclast, apoptosis
The c-Src tyrosine kinase has been implicated in multiple signaling pathways that regulate cellular growth, migration, and survival (Brown and Cooper 1996; Thomas and Brugge 1997). First defined as the normal cellular counterpart of the transforming gene of Rous Sarcoma Virus, c-src is overexpressed in a variety of human tumors, and an activating mutation has recently been found in a subpopulation of colon carcinomas (Bolen et al. 1987; Cartwright et al. 1989; Irby et al. 1999). Src kinase activity increases in response to a number of signaling pathways, most notably those downstream from tyrosine kinase growth factor receptors, G-protein coupled receptors, and integrin cell surface adhesion molecules (Brown and Cooper 1996; Thomas and Brugge 1997). Recent data suggest that Src can be activated downstream from RANK (receptor activator of NF-κB/osteoprotogerin-ligand-receptor/TRANCE-receptor) (Wong et al. 1999a), a member of the tumor necrosis factor (TNF) receptor superfamily that is involved in osteoclast formation and survival, as well as in osteoclast and dendritic cell function (Fuller et al. 1998; Dougall et al. 1999; Wong et al. 1999b; Li et al. 2000). The association of Src with these multiple signaling pathways makes it a potential therapeutic target for several diseases, including osteoporosis and cancer.
Src is a member of a family of nonreceptor tyrosine kinases defined by a common structure (see Fig. 1) that includes a myristoylated unique amino-terminal domain followed by the Src homology (SH) protein interaction domains: the SH3 domain interacts with proline-rich regions, and the SH2 domain interacts with phosphorylated tyrosine residues (Pawson and Gish 1992; Brown and Cooper 1996). The tyrosine kinase catalytic domain comprises the second half of the molecule and is followed by a short carboxy-terminal tail containing a negative regulatory tyrosine (Y527). This tyrosine is normally phosphorylated by the CSK kinase, causing an intramolecular interaction with Src's own SH2 domain that leads to a closed conformation and inactivation of the kinase (see Fig. 7C below; Brown and Cooper 1996). Mutational and crystallographic data suggest that a second intramolecular interaction, between the SH3 domain and the region that links the SH2 domain to the kinase, is also important for kinase regulation (Jove and Hanafusa 1987; Parsons and Weber 1989; Yamaguchi and Hendrickson 1996; Sicheri et al. 1997; Xu et al. 1997). Together, these intramolecular associations closely control Src kinase activity, but they also regulate the availability of the SH2 and SH3 domains for interactions with other molecules and influence the subcellular localization of Src, so that activated Src becomes detergent-insoluble.
Figure 1.



Src251 induces osteopetrosis. (A) Structure of TRAPSrc transgenic constructs. The TRAP promoter, first exon, and intron were fused to the ATG of wild-type (WT) and mutant versions of the chicken c-src gene. (U) Unique region; (SH3) Src homology 3 region; (SH2) Src homology 2 region; (Kinase) kinase domain. Crosses overlay domains containing inactivating mutations. (B) Radiographic analyses of Src251 mice. X-rays, frontal view. Brackets indicate regions that demonstrate increased bone density. (C) Histological analyses of tibia. Sections were stained both with Hematoxylin and Eosin (H&E) and for TRAP activity. (Upper panel) low power (4× magnification). (Lower panel) intermediate power (20×), showing TRAP-positive osteoclasts. (D) Histomorphometric analyses of mice. Genotype is indicated on the X axis. Percentage bone volume (amount of bone matrix as a percentage of the cancellous bone) is indicated on the Y axis. Osteoclasts were counted as TRAP-positive cells.
Figure 7.
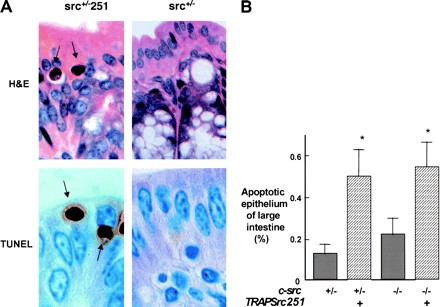

Src251 induces apoptosis of intestinal epithelial cells. (A) Histological analyses of large intestine (100×). Arrows indicate apoptotic epithelial cells. (Upper panels) H&E-stained sections; (lower panels) TUNEL staining showing apoptotic cells. (B) Percentage apoptotic large-intestine epithelial cells. Numbers were obtained from 3000 epithelial cells counted in each of two sections. (C) Model for action of Src251. Src251 is a particularly effective dominant-negative molecule because it efficiently blocks interactions of the Src homology protein interaction domains.
The widespread expression of c-src, its exquisite regulation, and the many pathways that activate this kinase, suggest that Src and Src-family kinases are critical for normal cellular function.
Nonetheless, targeted disruption of the c-src gene in mice leads to only one predominant phenotype, osteopetrosis, or a failure to break down bone, resulting from an intrinsic functional defect in osteoclasts, a cell type that expresses high levels of Src (Soriano et al. 1991; Horne et al. 1992; Lowe et al. 1993; Lowell et al. 1996). The src−/− mice develop osteoclasts, but the cells are morphologically abnormal, having aberrant cytoskeletal organization and lacking ruffled borders, the polarized part of the cell responsible for bone resorption (Boyce et al. 1992; Insogna et al. 1997; Schwartzberg et al. 1997). Accordingly, src−/− osteoclasts fail to resorb bone effectively and show defective phosphorylation of cellular proteins in response to extracellular stimuli, such as M-CSF and engagement of integrin receptors (Tanaka et al. 1996; Insogna et al. 1997; Duong et al. 1998; Nakamura et al. 1998). Recent in vitro data suggest that Src may be activated by a RANK-mediated cell-survival pathway in osteoclasts (Wong et al. 1999a). However, to date, there is no evidence of decreased osteoclast survival in src−/− mice in vivo. Indeed, src−/− mice have normal to elevated numbers of osteoclasts (Boyce et al. 1992).
Although the phenotype associated with Src deficiency may appear relatively limited, osteoclasts present a unique, complex cell type in which to examine Src function in a physiological setting. To investigate the multiple possible functions of Src in the osteoclast, we have undertaken a genetic dissection of Src function by reexpressing wild-type (wt) and mutated versions of c-src in osteoclasts of Src-deficient mice under the control of the promoter for tartrate-resistant acid phosphatase (TRAP), a gene that is highly expressed in osteoclasts. Using this system, we have shown previously that expression of a wild-type chicken c-src cDNA can fully complement src−/− osteopetrosis (Schwartzberg et al. 1997). Surprisingly, expression of a kinase-defective point mutant of Src (SrcK295M) also rescued many aspects of the osteopetrosis resulting from Src deficiency. Combined with experiments in src−/− fibroblasts indicating that Src kinase activity may not be strictly required for signaling from integrin receptors (Kaplan et al. 1995; Schlaepfer et al. 1998; P.L. Schwartzberg, unpubl.), these results pose the possibility that in certain pathways Src may function, in part, as an adaptor molecule.
To further examine the requirements for Src function in the osteoclast, we constructed a transgene, Src251, which drives expression of the amino-terminal 251 amino acids of Src, lacking the entire kinase domain and the regulatory tyrosine 527. Surprisingly, the Src251 transgene induced osteopetrosis in wild-type and src+/− mice and worsened osteopetrosis in src−/− mice. Osteopetrosis in Src251 mice was associated with a novel phenotype not observed in src−/− mice: increased osteoclast apoptosis. We present genetic and biochemical evidence indicating that this apoptosis may be caused by misregulation of the Src251 protein interaction domains. Expression of Src251 was associated with decreased AKT kinase activity and decreased osteoclast survival in response to RANKL, providing in vivo evidence for regulation by Src family kinases of cell-survival pathways involving TNF family receptors and AKT. These results further indicate that proper regulation of Src protein interaction domains is a critical element of Src function in vivo and may be a particularly effective target for Src-based therapeutics.
Results
Src251 induces osteopetrosis
We have demonstrated previously that expression of either wild-type or a kinase-defective point mutant of Src in osteoclasts of transgenic mice improved bone histology in many src−/− animals (Schwartzberg et al. 1997). To further examine the requirement for the kinase domain in osteoclast function, we constructed a transgene in which the first 251 amino acids of chicken c-Src, Src251, including the unique region and SH2 and SH3 domains, but lacking the kinase domain and regulatory tyrosine (Y527), was expressed under the control of the TRAP promoter (Fig. 1A).
In contrast to the full-length kinase-defective Src mutant (SrcK295M), which has no effect in a wild-type background, expression of the Src251 transgene caused a failure of tooth eruption. This is one of the most obvious manifestations of osteopetrosis, resulting from a failure of osteoclasts to erode a channel for erupting teeth (Soriano et al. 1991). Progeny from several founders revealed varying degrees of penetrance of this dental phenotype, with multiple transgenic lines showing a lack of tooth eruption in both wild-type and heterozygous backgrounds (Table 1). An additional line in which tooth eruption was normal did not express the transgene, confirming that the phenotype correlated with expression of Src251. Interbreeding with c-src heterozygous mice to generate src−/−TRAPsrc251 mice further demonstrated that this transgene was unable to complement osteopetrosis in src−/− mice.
Table 1.
Induction of osteopetrosis by TRAPSrc251transgene
| Transgenic line
|
c-src
|
% with tooth eruption
|
|---|---|---|
| — | +/− | 100% |
| — | +/− | 100% |
| — | −/− | 0% |
| Src251V | +/+ | 18% |
| Src251V | +/− | 0% |
| Src251V | −/− | 0% |
| Src251Y | +/+ | 70% |
| Src251Y | +/− | 43% |
| Src251Y | −/− | 0% |
| Src251Z | +/+ | 96% |
| Src251Z | +/− | 2% |
| Src251Z | −/− | 0% |
Radiological examination confirmed increased bone density typical of osteopetrosis in src+/+ and src+/− mice expressing the Src251 transgene (Fig. 1B). Surprisingly, however, osteopetrosis in src−/− mice was also exacerbated significantly by expression of Src251. Histologically, variable degrees of osteopetrosis were observed both in long bones and vertebrae of Src251 mice, with significant increases in bone volume in all c-src genotypes (Fig. 1C,D; data not shown). The increased osteopetrosis in Src-deficient animals indicated that expression of the Src251 transgene caused a more profound phenotype than Src deficiency alone. Thus, Src251 may interfere with more pathways than does the lack of Src, perhaps by interfering with the actions of other Src family kinases expressed in osteoclasts.
Src251 induces apoptosis in osteoclasts
Several mouse models of osteopetrosis, including mice with mutations in the genes encoding c-Fos, PU.1, CSF-1, RANKL (osteoprotogerin ligand/TRANCE), RANK, and the NF-κB subunits p50 and p52, result from a lack of osteoclast development (Wiktor-Jedrzejczak et al. 1990; Yoshida et al. 1990; Wang et al. 1992; Franzoso et al. 1997; Tondravi et al. 1997; Dougall et al. 1999; Kong et al. 1999b; Li et al. 2000). In contrast, src−/− mice develop normal to increased numbers of osteoclasts, but their function is defective (Boyce et al. 1992).
Src251 transgenic mice, however, had reduced numbers of osteoclasts (Fig. 1C,D), and many of these cells exhibited the classic morphological features of apoptosis, including pyknotic, fragmented nuclei and condensed cytoplasm. These features were readily visible in TRAP-stained sections (Fig. 2A) and were confirmed by TUNEL staining (data not shown).
Figure 2.

TRAPSrc251 transgenic mice show increased osteoclast apoptosis. (A) Histological analyses, high power (100×), showing TRAP-positive osteoclasts. Osteoclasts in Src251 mice are intensely staining with pyknotic nuclei characteristic of apoptotic cells (arrows). (B) Percentage of osteoclasts that were apoptotic.
Significantly, the percentage of apoptotic osteoclasts correlated with the degree of osteopetrosis and was inversely correlated with the levels of endogenous Src present in the wild-type and heterozygous mice (Fig. 2B). Culture of bone marrow or splenocytes from Src251 mice in vitro resulted in generation of osteoclasts, unlike the results from osteopetrotic strains that lack osteoclast development. However, cultures from Src251 mice yielded significantly fewer osteoclasts than those from either wild-type or src−/− mice, and significantly more of the osteoclasts were apoptotic (data not shown; see Fig. 6 below). Thus, Src251 may cause osteopetrosis by a novel mechanism, that of inducing osteoclast apoptosis. These data suggest that Src and/or other Src family members may be involved in cell signaling pathways that are required for cell survival in the osteoclast.
Figure 6.





Src251 blocks osteoclast responses to RANKL but not to M-CSF. (A) Osteoclasts were differentiated in culture, then washed twice with PBS and left in either complete media or media plus RANKL overnight. Wells (in quadruplicate) were stained for TRAP activity either before (pre) or after treatment (media or RANKL), and TRAP-positive multinucleated cells were counted. (B) AKT activity in response to RANKL. Osteoclast cultures were serum starved, treated with RANKL for 10 min, lysed, and AKT-immunoprecipitated; and AKT kinase activity was evaluated using GST–GSK3α as a substrate. (C) WT, Src251, Src251-ΔSH3, and Src251-R175L osteoclasts were treated with RANKL overnight, as in A. Src251-ΔSH3 blocks cell survival in response to RANKL, paralleling its induction of osteopetrosis. (D) Osteoclasts from src−/− and Src251 mice both respond poorly to RANKL, but respond normally to M-CSF. Osteoclasts differentiated in culture were treated as above or cultured in media with M-CSF overnight. (E) Src251 does not block AKT activation in response to M-CSF. Osteoclast cultures were serum starved, then treated with M-CSF for 10 min, lysed, and immunoblotted with anti-pS473-AKT to monitor AKT activation.
Abnormal localization of Src251 in osteoclasts
Src251 lacks kinase activity as a result of the absence of the kinase domain. However, Src251 appears to elicit a quite different phenotype from the full-length kinase-defective point-mutant, SrcK295M, which can partially rescue certain of the defects associated with Src deficiency, allowing increased resorption of bone by osteoclasts in many src−/−TRAPsrcK295M mice (Schwartzberg et al. 1997). Since Src251 differs from SrcK295M by the deletion of the kinase domain, one difference between these two molecules is the lack of regulation of the SH2 and SH3 protein interaction domains, which would not be masked by the kinase domain in Src251.
To examine whether misregulated domains of Src251 can affect Src function, we analyzed the subcellular partitioning of Src251 into detergent-soluble and insoluble fractions within osteoclasts. In unstimulated fibroblasts, wild-type Src is mostly in an inactive closed conformation that is detergent-soluble and associated with endosomes (Hamaguchi and Hanafusa 1987; Kaplan et al. 1992). Activation of Src is accompanied by translocation to a detergent-insoluble fraction (see Fig. 7C below; Clark and Brugge 1993; Walker et al. 1993; Kaplan et al. 1994); this subcellular change requires the protein interaction domains, but is independent of kinase activity, for inactivating the kinase by the K295M mutation does not prevent translocation. In contrast, Src251, with its available SH2 and SH3 domains, has been found to be constitutively detergent-insoluble in fibroblasts, where it is located in focal adhesions, the usual site of Src in an activated open conformation (Kaplan et al. 1994). Similarly, in osteoclasts, we found that Src251 resides predominantly in the detergent-insoluble fraction (Fig. 3A, lane 2). In osteoclasts, we also find that wild-type Src is more prevalent in the detergent-insoluble fraction, perhaps reflecting its high expression and activity in this cell type (Fig. 3A, lane 4; Horne et al. 1992; Lowell et al. 1996). Significantly, however, when Src251 was expressed in osteoclasts, we observed a smaller proportion of the endogenous c-Src in the detergent-insoluble fraction than seen typically in wild-type osteoclasts (Fig. 3A, cf. lanes 2 and 4), suggesting that the presence of Src251 prevents wild-type Src from residing in the detergent-insoluble fraction. In contrast, we have found that expression of Src251 did not appear to alter significantly the kinase activity of c-Src in cultured osteoclasts (data not shown). Furthermore, Src251 did not appear to alter the subcellular localization of several other signaling molecules within the cell, including the tyrosine kinase Pyk2, the p85 subunit of Phosphatidylinositide 3-kinase (PI3K), and the serine-threonine kinase AKT (Fig. 3B; data not shown). These results suggest that Src251 may block cell-survival pathways by specifically competing with Src and perhaps other Src family kinases, preventing them from trafficking and interacting with other molecules upon activation.
Figure 3.


Src251 is detergent-insoluble in osteoclasts. (A) Osteoclasts were differentiated from bone marrow in culture, then lysed gently in 0.5% Triton X-100 for varying times (2-min lysis is shown here; similar results were obtained at other time points) and the remaining detergent-insoluble fraction then lysed with RIPA. Src was immunoprecipitated from detergent-soluble and -insoluble fractions and analyzed by immunoblotting (Kaplan et al. 1994). (B) Detergent solubility of Pyk2 and AKT are not altered by expression of Src251. Osteoclasts were lysed as above, and lysates were immunoblotted for Pyk2 and AKT. Similar results were seen at 1-, 2-, and 4-min detergent solubility lysis conditions (1- and 4-min conditions are shown here). (S) Detergent-soluble fraction; (In) detergent-insoluble fraction.
Other Src mutants also induce apoptosis of osteoclasts
If misregulation of the protein interaction domains of Src251 causes apoptosis of osteoclasts in Src251 transgenic mice, it might be expected that other Src mutants would also induce osteoclast apoptosis. In particular, high levels of expression of the SrcK295M mutant may not be properly regulated by Csk and, thus, might induce a similar phenotype. Although we did not see evidence of increased osteoclast apoptosis in our initial analyses of TRAPSrcK295M mice, overexpression of SrcK295M has been found to block signaling in a number of systems, including those activated by growth-factor receptors and G-protein-coupled receptors (Courtneidge et al. 1993; Twamley-Stein et al. 1993; Luttrell et al. 1996). To determine if increased levels of K295M also affect osteoclast survival, we examined bones from TRAPSrcK295M transgenic mice that were bred to homozygosity to obtain higher levels of transgene expression. Surprisingly, we noted that the TRAPSrcK295M/K295M homozygotes were less likely than hemizygotes to rescue the src−/− phenotype (data not shown).
Significantly, we observed increased bone volume, more severe osteopetrosis, and increased osteoclast apoptosis in the nonrescued src−/−TRAPSrcK295M/K295M homozygous mice compared to src−/− mice (Fig. 4A,B). Interestingly, when we compared levels of protein expression from the two series of transgenic mice, we found that the SrcK295M mutant is expressed at higher levels than Src251 (Fig. 4C). Thus, SrcK295M appears to be less potent, but may still act similarly to Src251 when expressed at high levels, perhaps because of misregulation of the protein interaction domains.
Figure 4.


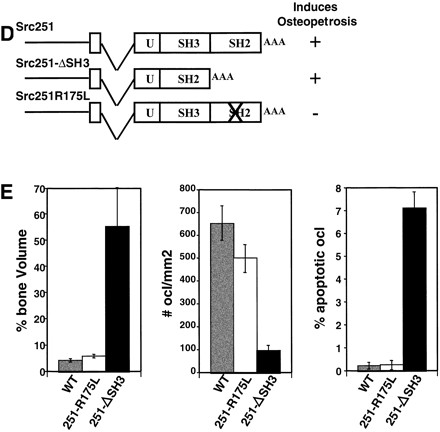
Other Src mutants induce apoptosis of osteoclasts. (A) Histological analyses of src−/− mice expressing the K295M kinase-inactive mutant that were not rescued. Arrows indicate apoptotic osteoclasts. (B) Bone volume and percentage apoptotic osteoclasts in wild-type (WT), src−/−, and src−/− mice that showed rescue or did not show rescue by TRAPSrcK295M. (C) Relative levels of expression of Src251 and SrcK295M transgenes. Lysates from osteoclast cultures of Line V Src251 and Line NN SrcK295M transgenics were normalized for TRAP levels, Src-immunoprecipitated with monclonal 327, and immunoblotted with anti-avian specific Src monoclonal EC10, which only reacts with the transgenic Src (left panel), or monoclonal 327, (right panel) which reacts only with both endogenous and transgenic Src. Similar results were seen with other transgenic lines. (D) Phenotype of mutants affecting the SH3 and SH2 domains of Src251. (E) Bone histomorphometry, osteoclast numbers, and percent apoptotic osteoclasts from mutants affecting the SH3 and SH2 domains of Src251.
To further define the role of the SH2 and SH3 protein interaction domains in the induction of osteoclast apoptosis, we generated two additional transgenic lines that either deleted the region encoding the SH3 domain (Src251-ΔSH3) or changed a highly conserved arginine in the FLVRES sequence of the SH2 domain that is required for SH2 binding to phosphotyrosine residues (Src251-R175L) (Fig. 4D,E). The TRAPSrc251-ΔSH3 transgene also induced osteopetrosis that was associated with increased osteoclast apoptosis. However, mutation of the SH2 domain prevented the induction of apoptosis and osteopetrosis by Src251. Thus, induction of apoptosis by Src251 does not require the SH3 domain of Src251 but may require a functional SH2 domain.
Src251 affects AKT function
In several signaling pathways, protection from apoptosis involves the PI3K-mediated activation of the serine–threonine kinase AKT (Datta et al. 1997). Src family kinases have been implicated in the activation of PI3K through interactions with the PI3K p85 regulatory subunit (Fukui and Hanafusa 1991; Pleiman et al. 1994). Furthermore, pharmacological inhibition of PI3K activity can induce apoptosis of cultured osteoclasts (S. Kadapakkam and B.F. Boyce, unpubl.), suggesting that these signaling intermediates are important for osteoclast survival. To determine if Src251 may interfere with PI3K-mediated pathways, we examined AKT kinase activity in cultured osteoclasts. Osteoclasts cultured from Src251 mice had reduced AKT kinase activity compared to their wild-type counterparts (Fig. 5A). In contrast, the activation status of ERK1 and ERK2, other kinases involved in cell survival, did not appear to differ significantly between wild-type and Src251 cells (Fig. 5B). Thus, at least one pathway that may be altered by expression of Src251 is the activation of AKT.
Figure 5.

Src251 osteoclasts have reduced AKT kinase activity. (A) Osteoclasts were differentiated from bone marrow in culture, then lysed, immunoprecipitated with anti-AKT serum, and in vitro kinase assays were performed using GST–GSK3α as a substrate. Products were detected using an anti-phospho GSK-3 antibody by immunoblotting. After normalization for AKT protein levels, AKT activity was observed to be decreased twofold. In this experiment, twofold lower levels of AKT protein were also observed. (Lower panel) Lysates were normalized by immunoblotting for TRAP protein. (B) Osteoclasts were lysed as above and TRAP-normalized lysates were immunoblotted with anti-active ERK. Equivalent levels of total ERK were observed in these samples.
Src251 decreases osteoclast survival in response to RANKL
Multiple pathways involved in cell survival have been shown to activate AKT, including those downstream from tyrosine kinase growth factor receptors, integrins, and TNF family receptors. Significantly, Src has been implicated in signaling from all these receptors (Thomas and Brugge 1997). In particular, recent data have suggested that Src is activated by the TNF family member RANKL via a TRAF6-mediated pathway that activates AKT and may be involved in osteoclast survival (Wong et al. 1999a). Nonetheless, src−/− mice have high numbers of osteoclasts and do not appear to have decreased osteoclast survival in vivo. Indeed, we find that the percentage of apoptotic osteoclasts in bone sections from src−/− mice (0.7% ± 0.1) is similar to that in wild-type mice (0.5% ± 0.2), making the importance of Src in RANKL-mediated survival pathways unclear.
To examine whether Src251 interferes with this survival pathway in osteoclasts, we examined osteoclast survival in the presence or absence of RANKL. Wild-type osteoclasts rapidly lose viability in the absence of RANKL, whereas addition of RANKL promotes their survival (see Fig. 6A; Fuller et al. 1998). In contrast, osteoclasts cultured from Src251 mice failed to respond to RANKL, with little or no increase in viability (Fig. 6A). Additionally, whereas we observe only slight increases in AKT activity in response to RANKL, these responses are reduced in osteoclasts from Src251 mice (Fig. 6B). Osteoclasts expressing the Src251-ΔSH3 mutant also failed to respond to RANKL, consistent with the ability of Src251-ΔSH3 to induce osteoclast apoptosis and osteopetrosis (Fig. 6C). Src251-R175L, in contrast, did not block osteoclast responses to RANKL, just as it did not induce osteopetrosis. Furthermore, as reported previously, src−/− osteoclasts also responded poorly to RANKL in culture (Fig. 6D; Wong et al. 1999a). The observation that Src deficiency prevents cell-survival responses to RANKL in vitro, as does expression of the Src251 and Src251-ΔSH3 mutants, strongly suggests that these mutants are blocking Src specific pathways in a dominant-negative manner. Thus, expression of these mutants may reveal a role for Src in RANKL-mediated cell survival in vivo that is not readily apparent from the phenotype of the Src-deficient mice alone.
Finally, another major pathway affecting osteoclast survival is that activated by the macrophage colony stimulating factor (M-CSF), which also rescues osteoclasts from apoptosis (Fuller et al. 1993). Experiments both in osteoclasts and in heterologous cells have implicated Src in M-CSF signaling (Courtneidge et al. 1993; Insogna et al. 1997). However, in contrast to the results with RANKL, src−/− osteoclasts and osteoclasts expressing Src251 were able to survive normally in response to M-CSF in culture (Fig. 6D). Furthermore, AKT activity in response to M-CSF appeared normal in Src251 osteoclasts (Fig. 6E). Thus, the increased apoptosis in cells expressing Src251 may result from a specific defect in response to RANKL.
Src251 can induce apoptosis in other cell types
To determine if the induction of apoptosis by Src251 may be a more generalized phenomenon shared with other cell types, we examined other cells that express this transgene. We have shown previously that virtually all mice carrying TRAPSrc transgenes express them in the intestine (Schwartzberg et al. 1997). Consistent with these patterns of expression, we found evidence of increased apoptosis in large-intestinal epithelial cells of Src251 transgenic mice, suggesting that Src251 may inhibit cell-survival pathways in multiple cell types (Fig. 7A,B). Although we do not yet know the pathway(s) disrupted by Src251 in these cells, these results suggest that regulation of cell survival by Src-family kinases is not restricted to the osteoclast.
Discussion
Apoptosis, or programmed cell death, is an important regulatory process for many cells during embryonic development and normal function. For osteoclasts, as for many other hematopoietic cells, apoptosis is a normal part of their life cycle. The factors that contribute to osteoclast survival are therefore critical elements of bone homeostasis and development, which requires an active balance between the bone-generating activity of osteoblasts and the bone resorption of osteoclasts (Roodman 1999). Many factors have been found that influence osteoclast survival, including M-CSF and the recently discovered RANKL (Fuller et al. 1993, 1998). For example, estrogens induce osteoclast apoptosis via a TGF-β-mediated pathway that is associated with increased expression of the soluble decoy receptor osteoprotegerin, which binds and blocks RANKL activity (Hughes et al. 1996; Hofbauer et al. 2000). Thus, after menopause, as endogenous estrogens are reduced, not only is osteoclast formation increased, but osteoclast apoptosis is also decreased. The resulting increase in bone resorption is a major contributor to postmenopausal osteoporosis. Similarly, bisphosphonates, the first synthetic drugs found to reduce bone fractures in the elderly, increase osteoclast apoptosis (Hughes et al. 1995).
We demonstrate here that expression of a truncated form of Src with unregulated SH2 and SH3 protein interaction domains dramatically increases osteoclast apoptosis. Induction of apoptosis depends on a functional SH2 domain and is accompanied by decreased AKT activity and decreased responsiveness to RANKL. Recently, Wong and co-workers have implicated Src in RANKL-mediated activation of AKT via a TRAF6–Src interaction (Wong et al. 1999a). Our results suggest that Src family kinases are critical in vivo for the signaling pathways required for cell survival downstream from this TNF family receptor. Of note, although the in vitro data of Wong et al. implicated the Src SH3 domain in an interaction with TRAF6, our data indicate that this domain is not required for blocking this pathway. Rather, our data suggest that other parts of Src such as the SH2 and perhaps the unique domain may be critical for interactions with molecules in this pathway.
The implication of Src family kinases and tyrosine phosphorylation signaling in cell-survival pathways downstream from a TNF family receptor reveals a novel previously unappreciated cell survival signaling pathway from these receptors. Apoptosis has been associated with two distinct types of pathways that differ in their dependence on mitochondrial cytochrome C release and their ability to be blocked by expression of Bcl-2 family members. The availability of transgenic mice that express BclXL in osteoclasts may allow further dissection of the RANK-induced signaling pathways involved in cell survival and the induction of apoptosis by Src251. Additionally, the use of Src mutants may permit further dissection of these cell-survival pathways.
Although RANKL is implicated in both osteoclast formation and survival, the effects of Src251 are most pronounced on osteoclast survival. It should be noted that the osteoclast survival experiments performed here used osteoclast cultures that had been differentiated in the presence of RANKL. Thus, neither Src deficiency nor expression of Src251 prevent osteoclast differentiation but, rather, they interfere specifically with osteoclast survival in response to RANKL. One interpretation of this data is that Src-family kinases may be less important for the signals downstream from RANK required for osteoclast formation. NFκB is also activated downstream from RANK via TRAF6 (Wong et al. 1998). Combined deficiency of the NFκB p50 and p52 subunits or deficiency of RANK cause osteopetrosis as a result of defective osteoclast formation (Franzoso et al. 1997), indicating that distinct signaling pathways downstream from RANK may be involved in osteoclast formation and survival. Alternatively, it is possible that Src-family kinases may be involved in this NFκB activation pathway (Fig. 7D). Our results may then be secondary to relative amounts of Src-family kinases expressed in preosteoclasts and to the temporal patterns of expression of the Src251 transgene driven from the TRAP promoter, which may not be efficiently expressed in osteoclast precursors. RANKL has also been shown to activate osteoclasts to increase bone resorption (Fuller et al. 1998). Although it remains to be determined if defects in signaling from RANK contribute to the defects in bone resorption in src−/− osteoclasts, the recent report of osteopetrosis associated with defective ruffled borders in TRAF6−/− mice (Lomaga et al. 1999), similar to the src−/− phenotype, suggests that this is likely. Indeed, our results raise the possibility that phenotypes associated with deficiency of other Src-family kinases in mice may result from defects associated with TNF-family receptor signaling.
Although src−/− osteoclasts in culture also have reduced responses to RANKL in vitro, osteoclasts in Src-deficient mice do not show increased apoptosis, suggesting that other factors or pathways that are not present in vitro can compensate for the lack of Src in vivo. These could include other Src-family kinases or interacting molecules with which Src251 may interfere. Indeed, there is evidence that TRAF6 can coimmunoprecipitate with multiple Src-family kinases (Wong et al. 1999a; A. Venegas and P. Schwartzberg, unpubl.). Although nonspecific interactions or toxicity of Src251 cannot be ruled out in these experiments, the degree of apoptosis induced in osteoclasts appears to depend on a balance between the levels of endogenous Src and Src251. The titration of this phenotype against the level of endogenous Src protein suggests that Src251 acts by specifically blocking interactions of Src or other Src-family kinases with other molecules. Thus, Src251 may inhibit cell-survival pathways by specifically competing with Src, perhaps by constitutively associating with proteins that normally interact with Src and other Src-family kinases (Fig. 7C). Indeed, preliminary experiments suggest that although Src251 does not alter the subcellular localization of Pyk2 and AKT, it may alter the detergent solubility of TRAF6 (P. Schwartzberg, unpubl.), providing further evidence for Src's connection to this signaling pathway in osteoclasts.
The different phenotypes elicited from expression of different Src mutants in the osteoclast, or even from the same mutant (K295M) under different levels of expression, highlights the many roles for Src and Src-family kinases in signal transduction. Although eliminating kinase activity may not affect certain pathways such as integrin signaling, it may severely impair others.
Our data with Src251 argue that inference with the regulation of protein interaction domains may be a particularly effective means of blocking Src-family function and again emphasize the importance of these domains in Src function. Expression of Src251 can also act as a potent block to VEGF-induced angiogenesis in a chicken chorioallantoic membrane model system (Eliceiri et al. 1999) and can induce apoptosis in intestinal epithelial cells (Fig. 7A,B). Thus, as in the osteoclast, expression of the dominant-negative Src251 may reveal roles for Src-family kinases in pathways that are not obvious from the gross phenotype of src−/− mice.
It is therefore important to note that whereas our results highlight the potential of Src inhibition as a drug-targeting strategy, they also underscore the potential hazards of Src inhibition that may not be discerned in studies using isolated tissue culture systems. Osteoclasts provide a model system for understanding Src function in a physiologic setting and help provide information for therapeutic development based on Src family members. In bone, this may be particularly useful for the treatment both of osteoporosis and of rheumatoid arthritis, where blocking RANKL activity has been found to decrease bone destruction in animal models (Kong et al. 1999a). However, such therapeutics may also be useful for cancers and other pathophysiologic conditions in which Src or other Src-family kinases have been implicated.
Material and methods
Transgenic mice
Generation of the TRAPSrcWT and TRAPSrcK295M transgenic constucts and transgenic mice were described previously. TRAPSrc251 was constructed by PCR amplification of the sequence encoding the first 251 amino acids of the chicken c-src gene, and a BstY1–Xba1 fragment was used to replace a portion of the wild-type chicken src gene in pBSTRAPSrc. To generate TRAPSrc251-R175L, TRAPSrc251 was mutagenized using the Quickchange kit (Stratagene) using the following primer and its reverse complement: (GGA ACC TTC TTG GTC CTC GAG AGC GAG ACG). To generate TRAPSrc251-ΔSH3, a fragment was subcloned into TRAPSrc251 from Src251-D11 (Kaplan et al. 1994). Transgenic mice were generated via standard technology. Transgenic founders were bred to src+/− mice and then interbred to generate TRAPSrc transgenic mice in wild type, src+/−, and src−/− backgrounds. Genotyping was performed as described previously (Schwartzberg et al. 1997).
Antibodies and reagents
The following antibodies were used in this study: Monoclonal anti-c-Src 327 (Oncogene Science and gift of Joan Brugge, Harvard Medical School, Boston, MA); Src2 polyclonal, Pyk2 polyclonal, p85 PI3K polyclonal, and anti-active ERK monoclonal E4 (Santa Cruz), EC10 anti-chicken Src specific monoclonal (UBI), anti-TRAcP monoclonal (Zymed), and anti-GSK3 polyclonal, anti-AKT polyclonal, and anti-pS473-AKT polyclonal (New England Biolabs). RANKL was a generous gift from Dr. Dirk Anderson (Immunex Corporation, Seatttle, WA) and Dr. Mehrdad Tondravi (Redcross, Rockville, MD).
Histologic and radiographic analyses
Preparation and analyses of bones were performed as described previously (Schwartzberg et al. 1997). Briefly, bones were fixed in 10% buffered formalin, decalcified in 10% EDTA, and embedded in Paraffin. Sections were stained with Hematoxylin and Eosin and for TRAP activity. Bone volume (amount of bone matrix as a percentage of cancellous space) and osteoclast numbers (per millimeter of the cancellous bone surface) in the cancellous space of the long bones were measured as described previously (Boyce et al. 1992). For histology, samples were examined from a minimum of four mice each. Apoptotic osteoclasts were determined by TRAP staining or TUNEL analyses (Hughes et al. 1996). The assessment of colonic enterocytes apoptosis was done in the upper half of the crypts of sections of the ascending colon. Three thousand enterocytes were counted in each of two sections, and apoptotic cells were identified using standard morphologic criteria on H&E stained sections. Radiographs were performed on a Faxatron X-ray machine.
Osteoclast cultures
Osteoclasts were differentiated from bone marrow in the presence of Vitamin D3 (10−9 M) with or without Dexamethasone (5 nm/mL) as described previously (Takahashi et al. 1988b). Alternatively, bone marrow and splenocytes were cultured in the presence of osteoblasts derived from calvaria from newborn mice (Takahashi et al. 1988a). Cultures were enriched for osteoclasts by removing osteoblast feeder layers by rinsing with phospho buffered saline (PBS) without Ca2+ and Mg2+.
For cell-survival studies, osteoclasts were differentiated from CFU-GM cultures (Uy et al. 1995). Briefly, splenocytes were cultured in methylcellulose in the presence of recombinant human GM-CSF (20 ng/mL) and M-CSF (30 ng/mL) for 11–14 d. Pools of five colonies were plated in α-MEM with 10% fetal calf serum, glutamine, penicillin, and streptomycin in the presence of 80 ng/mL RANKL and 30 ng/mL M-CSF. Under these conditions, osteoclasts had differentiated by 3–5 d.
Survival assays
Differentiated osteoclasts were washed three times with PBS, then either stained for TRAP activity or incubated overnight in complete α-MEM alone or supplemented with 100 ng/mL RANKL or 30 ng/mL M-CSF. After 24 h, cells were stained for TRAP activity. TRAP-positive cells containing three or more nuclei were scored as osteoclasts.
Immunoprecipitations and Western analyses
For detergent-insolubility assays, osteoclasts differentiated in six well plates were lysed by gently pipetting 400 μL of 0.5% Triton X-100 buffer for varying times. Soluble material was removed by pipetting, then cells were further lysed to obtain detergent-insoluble material by the addition of RIPA (Radio-immunoprecipitation buffer: 50 mM Hepes at pH 7.0, 1% Triton X-100, 1 mM EDTA, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 150 mM NaCl with protease inhibitors and sodium orthovanadate) for 10 min at 4°C (Kaplan et al. 1994). Detergent-soluble and -insoluble fractions were normalized by addition of the opposite lysis buffer, and lysates were clarified by centrifugation at 14,000g for 10 min. Src was immunoprecipitated from cell lysates using anti-Src monoclonal 327; immunoprecipitates were resolved on a 10% SDS–polyacrylamide gel and immunoblotted with a combination of EC10 and Src-2 antibodies. Alternatively, lysates were resolved by SDS-PAGE and immunoblotted for Pyk2, p85, or AKT.
To evaluate transgenic protein levels, osteoclast cultures were lysed in RIPA, Src-immunoprecipitated with monoclonal 327, and immunoblotted with either monoclonal EC10, which only recognizes the chicken c-Src transgenic protein, or 327, which recognizes both endogenous and transgenic Src.
AKT kinase assays
Osteoclasts were differentiated from bone marrow as above, osteoblasts were washed away with PBS, and the cells were lysed in 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton-X-100, 2.5 mM sodium pyrophosphate, 1 mM beta glycerophosphate, 1 mM sodium orthovanadate, 1 μg/mL leupeptin, 1 mM PMSF, plus Protease Inhibitor Mini-Tablet/10 mL (Roche Molecular Biochemicals). AKT was immunoprecipitated, and kinase activity was assayed with the New England Biolabs AKT kinase assay kit (NEB 9840) using GST–GSK-3α as a substrate. Products were detected using an anti-phospho-GSK3 antibody as per the manufacturer's recommendation.
To examine responses to RANKL or M-CSF, differentiated osteoclast cultures were serum starved in 0.5% serum for 6 h, then treated with RANKL (1 μg/mL) or M-CSF (300 ng/mL) for 10 min and lysed as above. Lysates were resolved by SDS-PAGE and immunoblotted with anti-pS473-AKT or anti-AKT sera for M-CSF stimulated cultures, or analyzed for AKT kinase activity as above (RANKL-treated cultures).
Acknowledgments
This work was initiated in the laboratory of H.E. Varmus. The authors thank Phillipe Soriano, Clifford Lowell, Ken Kaplan, Alan Kimmel, James Dodson, and members of the Schwartzberg, Boyce, and Varmus laboratories for mice, reagents, advice, and discussions; Beryl Story, Arlene Farias, Jennifer Harvey, and Zhenxing Tai for excellent technical assistance; and Gene Elliot, Theresa Hernandez, and Madeva Chetana for assistance with mouse husbandry. P.L. Schwartzberg is supported in part with funds from the Searle Scholar's Program/Chicago Community Trust. This work was also supported by grant AR-43510 from National Institutes of Health to B.F. Boyce.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pams@nhgri.nih.gov; FAX (301) 402-2170.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.840301.
References
- Bolen JB, Veillette A, Schwartz AM, DeSeau V, Rosen N. Activation of pp60c-src protein kinase activity in human colon carcinoma. Proc Natl Acad Sci. 1987;84:2251–2255. doi: 10.1073/pnas.84.8.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce B, Yoneda T, Lowe C, Soriano P, Mundy G. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and to resorb bone in mice. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Cooper JA. Regulation, substrates, and functions of Src. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- Cartwright CA, Kamps MP, Meisler AI, Pipas JM, Eckhart W. pp60c-src activation in human colon carcinoma. J Clin Invest. 1989;83:2025–2033. doi: 10.1172/JCI114113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Redistribution of activated pp60c-src to integrin-dependent cytoskeletal complexes in thrombin-stimulated platelets. Mol Cell Biol. 1993;13:1863–1871. doi: 10.1128/mcb.13.3.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtneidge SA, Dhand R, Pilat D, Twamley GM, Waterfield MD, Roussel MF. Activation of Src-family kinases by colony stimulating factor-1, and their association with its receptor. EMBO J. 1993;12:943–950. doi: 10.1002/j.1460-2075.1993.tb05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al. RANK is essential for osteoclast and lymph node development. Genes & Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong LT, Lakkakorpi PT, Nakamura I, Machwate M, Nagy RM, Rodan GA. PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of α(v)β3 integrin, and phosphorylated by src kinase. J Clin Invest. 1998;102:881–892. doi: 10.1172/JCI3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-κB in osteoclast and B-cell development. Genes & Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, Hanafusa H. Requirement of phosphatidylinositol-3 kinase modification for its association with p60src. Mol Cell Biol. 1991;11:1972–1979. doi: 10.1128/mcb.11.4.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ. Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med. 1993;178:1733–1744. doi: 10.1084/jem.178.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller K, Wong B, Fox S, Choi Y, Chambers TJ. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. 1998;188:997–1001. doi: 10.1084/jem.188.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi M, Hanafusa H. Association of p60src with Triton X-100-resistant cellular structure correlates with morphological transformation. Proc Natl Acad Sci. 1987;84:2312–2316. doi: 10.1073/pnas.84.8.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- Horne W, Neff L, Chatterjee D, Lomri A, Levy J, Baron R. Osteoclasts express high levels of pp60c-src in association with intracellular membranes. J Cell Biol. 1992;119:1003–1013. doi: 10.1083/jcb.119.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DE, Wright KR, Uy HL, Sasaki A, Yoneda T, Roodman GD, Mundy GR, Boyce BF. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J Bone Miner Res. 1995;10:1478–1487. doi: 10.1002/jbmr.5650101008. [DOI] [PubMed] [Google Scholar]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-β. Nat Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- Insogna KL, Sahni M, Grey AB, Tanaka S, Horne WC, Neff L, Mitnick M, Levy JB, Baron R. Colony-stimulating factor-1 induces cytoskeletal reorganization and c-src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest. 1997;100:2476–2485. doi: 10.1172/JCI119790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu Rev Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- Kaplan KB, Swedlow JR, Varmus HE, Morgan DO. Association of p60c-src with endosomal membranes in mammalian fibroblasts. J Cell Biol. 1992;118:321–333. doi: 10.1083/jcb.118.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 1994;13:4745–4756. doi: 10.1002/j.1460-2075.1994.tb06800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes & Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999a;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999b;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci. 2000;97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes & Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C, Yoneda T, Boyce BF, Chen H, Mundy GR, Soriano P. Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proc Natl Acad Sci. 1993;90:4485–4489. doi: 10.1073/pnas.90.10.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA, Niwa M, Soriano P, Varmus HE. Deficiency of the Hck and Src tyrosine kinases results in extreme levels of extramedullary hematopoiesis. Blood. 1996;87:1780–1792. [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- Nakamura I, Jimi E, Duong LT, Sasaki T, Takahashi N, Rodan GA, Suda T. Tyrosine phosphorylation of p130Cas is involved in actin organization in osteoclasts. J Biol Chem. 1998;273:11144–11149. doi: 10.1074/jbc.273.18.11144. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Weber MJ. Genetics of src: Structural and functional organization of a protein tyrosine kinase. Curr Top Microbiol Immunol. 1989;147:79–127. doi: 10.1007/978-3-642-74697-0_3. [DOI] [PubMed] [Google Scholar]
- Pawson T, Gish GD. SH2 and SH3 domains: From structure to function. Cell. 1992;71:359–362. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- Pleiman CM, Hertz WM, Cambier JC. Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 1994;263:1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Cell biology of the osteoclast. Exp Hematol. 1999;27:1229–1241. doi: 10.1016/s0301-472x(99)00061-2. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: Summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzberg PL, Xing L, Hoffmann O, Lowell CA, Garrett L, Boyce BF, Varmus HE. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes & Dev. 1997;11:2835–2844. doi: 10.1101/gad.11.21.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley J, Martin TJ, Suda T. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988a;123:2600–2602. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Yamana H, Yoshiki S, Roodman GD, Mundy GR, Jones SJ, Boyde A, Suda T. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988b;122:1373–1382. doi: 10.1210/endo-122-4-1373. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Amling M, Neff L, Peyman A, Uhlmann E, Levy JB, Baron R. c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature. 1996;383:528–531. doi: 10.1038/383528a0. [DOI] [PubMed] [Google Scholar]
- Thomas S, Brugge J. Cellular functions regulated by Src family kinases. Ann Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- Twamley-Stein GM, Pepperkok R, Ansorge W, Courtneidge SA. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc Natl Acad Sci. 1993;90:7696–7700. doi: 10.1073/pnas.90.16.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uy HL, Dallas M, Calland JW, Boyce BF, Mundy GR, Roodman GD. Use of an in vivo model to determine the effects of interleukin-1 on cells at different stages in the osteoclast lineage. J Bone Miner Res. 1995;10:295–301. doi: 10.1002/jbmr.5650100217. [DOI] [PubMed] [Google Scholar]
- Walker F, deBlaquiere J, Burgess AW. Translocation of pp60c-src from the plasma membrane to the cytosol after stimulation by platelet-derived growth factor. J Biol Chem. 1993;268:19552–19558. [PubMed] [Google Scholar]
- Wang Z-Q, Ovitt C, Grigoriadis AE, Möhle-Steinlein U, Rüther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Wiktor-Jedrzejczak W, Bartocci A, Ferrante A, Ahmed-Ansari A, Sell K, Pollard J, Stanley E. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci. 1990;87:4828–4832. doi: 10.1073/pnas.87.12.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–28359. doi: 10.1074/jbc.273.43.28355. [DOI] [PubMed] [Google Scholar]
- Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol Cell. 1999a;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- Wong BR, Josien R, Choi Y. TRANCE is a TNF family member that regulates dendritic cell and osteoclast function. J Leukoc Biol. 1999b;65:715–724. doi: 10.1002/jlb.65.6.715. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S-I, Kunisada T, Ogawa M, Nishikawa S, Okumura H, Sudo T, Shults LD, Nishikawa S-I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]