

Abstract
Henipaviruses encode several proteins from the P gene, of which V and W have been demonstrated by gene-based transfection studies to antagonize the innate immune response, blocking both type I interferon production and signaling. This study examines the effects of henipavirus infection on the innate immune response in human cell lines. We report that henipavirus infection does not result in interferon production, with the virus antagonizing this response. In contrast to published transfection studies, our study found that the interferon signaling pathways are only partially blocked by henipavirus infection of human cell lines.
Zoonotic henipaviruses naturally infect flying foxes, the reservoir host, without causing any clinical symptoms but have caused serious diseases of humans and livestock in Australia, Malaysia, Singapore, India, and Bangladesh. Hendra virus (HeV) was identified in September 1994 in the Brisbane suburb of Hendra, Australia, after an outbreak of acute respiratory disease in thoroughbred horses (17, 18, 23). Numerous outbreaks of HeV infection (between 1994 and 2010) have resulted in 44 infected horses and seven cases of human disease, four of which were fatal (16). Nipah virus (NiV) emerged in Malaysia during 1998 and 1999 as the causative agent of disease in humans and pigs. More than 1 million pigs were culled to prevent the spread of the virus, and by May 1999 NiV had caused 265 cases of acute encephalitis in humans resulting in 105 deaths (3, 4). There was a reemergence of NiV encephalitis in humans in Bangladesh in 2001 (11), and there have been fatalities in that country almost every year since its emergence (8, 9, 14, 15).
The innate immune system is the host's first line of defense against foreign pathogens, such as invading viruses. A major component of the innate immune response is the production and signaling pathways of type I interferons (alpha/beta interferon [IFN-α/β]), which act to establish antiviral states within both infected and neighboring cells. The production of interferon leads to signaling cascades, which results in the transcriptional upregulation of interferon-stimulated genes (ISGs). Many viruses, including most paramyxoviruses, encode proteins which counteract the innate immune system by targeting different parts of the interferon production and signaling pathways, leading to evasion of the interferon-induced antiviral state of the host cell (6, 7). Previous studies based on transient expression of individual gene products have demonstrated that the henipavirus P gene products, the P, V, W, and C proteins, could exert antagonistic effects on both the interferon production and interferon signaling pathways by interaction with the cellular MDA5 and STAT proteins (10, 20–22, 24, 25).
To examine whether the results from single gene transfection studies are reflected in the response following virus infection, 293T cells were transfected with an interferon-stimulated response element (ISRE) luciferase plasmid (pISRE-Luc) (PathDetect pISRE-Luc cis-reporter plasmid; Stratagene) and then infected at a multiplicity of infection (MOI) of 10 with individual henipaviruses, Hendra virus (HeV; Hendra virus/Australia/Horse/1994/Hendra), a Nipah virus Malaysian isolate (NiV-M; Nipah virus/Malaysia/Human/1999/PKL), and a Nipah virus Bangladesh isolate (NiV-B; Nipah virus/Bangladesh/Human/2004/Rajbari R1). Cells were lysed at 24 h postinfection, and the luciferase activities of lysates were measured using a Dual-Luciferase reporter assay system (Promega). Similar levels of luciferase activity were observed in both infected and mock-infected cell lysates (Fig. 1 a). Since only basal levels of luciferase were induced by infection, it can be concluded that all three henipaviruses tested were antagonizing one or more components of the interferon production or signaling pathway.
Fig. 1.
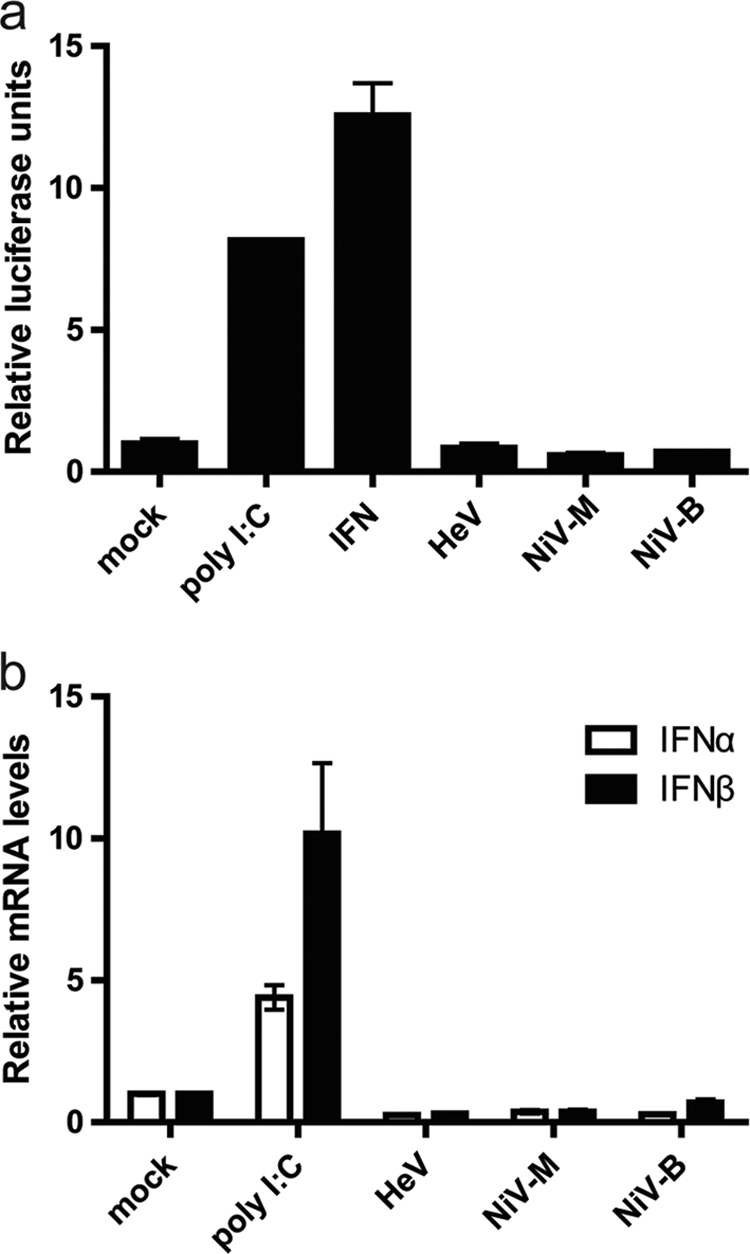
Production of type I interferons following henipavirus infection. (a) 293T cells were transfected with 500 ng of pISRE-Luc and 500 ng Renilla luciferase reporter plasmid (pRL-tK; Promega) for normalization. After 24 h, cells were infected with each virus (HeV, NiV-M, and NiV-B) at an MOI of 10 for 3 h and then assayed for luciferase activity. n = 2, with error bars indicating standard errors of the means (SEM). Cells treated with human IFN-β (1,000 U; Sigma-Aldrich) and poly(I:C) (10 μg; cotransfected with pISRE-Luc and pRL-tK) were used as positive controls. (b) HEp-2 cells were infected at an MOI of 10 for 3 h. Total RNA was isolated, and quantitative real-time PCR was performed using SYBR green, with primer sequences from IFN-α (26), IFN-β (24), and 18s rRNA (19). A total of 10 μg poly(I:C) was used as a positive control. n = 2, with error bars indicating SEM.
To confirm that the luciferase results are not cell specific, HEp-2 cells were subsequently utilized for real-time PCR assays to measure transcriptional upregulation of interferon genes following virus infection. HEp-2 cells infected at an MOI of 10 for 3 h produced both IFN-α and IFN-β mRNA transcripts at levels comparable to those observed in mock-infected cells (Fig. 1b). This result demonstrates that the interferon production pathway is antagonized by each of the three tested henipaviruses. Similar results were obtained in 293T cells infected with NiV-M (data not shown).
Both the luciferase assays and the real-time PCR assays failed to demonstrate an interferon-mediated effect following infection with henipaviruses, which is consistent with previously published transfection studies investigating the function of P gene products in modulating an interferon response (2, 24). Importantly, despite apparent differences in disease patterns, transmission, and pathogenicity between the viruses, there appears to be little or no difference in the antagonistic effects of the interferon production pathway during HeV, NiV-M, and NiV-B cell-based infection studies.
To examine the effect of viral infection on the interferon signaling pathway, 293T cells were transfected with pISRE-Luc and infected 6 h later with henipaviruses at an MOI of 1. To confirm that transfection did not inhibit virus replication, immunofluorescence was performed, demonstrating that more than 90% of cells were infected (data not shown). The cells were treated with exogenous human interferon for 3 h. Cells were then lysed and assayed for luciferase activity (Fig. 2 a). The interferon signaling pathway appeared to be only partially blocked in infected cells following treatment with exogenous interferon since luciferase activity was only slightly reduced compared to that in mock infection (Fig. 2a). The finding that henipavirus infection does not block interferon signaling contrasts with results from transfection studies in which a complete block of ISG expression was observed (Fig. 2b) (5, 13, 24). To confirm that the results were not due to mutant viruses, all P genes were sequenced and their sequences were demonstrated to be identical to reference sequences. During this study, an additional study looking at the interferon response following infection with henipaviruses was published (27). In that study by Yoneda et al., a block in interferon signaling was observed in Vero cells after NiV-M infection (27). Differences in the results reported may be accounted for by differences in the cell types and levels of infection. We optimized our experiment so that more than 90% of attached cells were infected at the time of interferon treatment, which is allowed to proceed for 3 h only. Figure 2a demonstrates that this is sufficient time to observe a considerable ISRE-luciferase response.
Fig. 2.

Henipavirus infection and antagonism of the interferon signaling pathway following treatment with exogenous human interferon. (a) 293T cells were transfected with 500 ng pISRE-Luc and pRL-tk and after 6 h infected at an MOI of 1. At 24 h postinfection, cells were treated with 1,000 U of human IFN-β for 3 h and then assayed for luciferase activity. The error bars indicate standard deviations for three independent experiments. (b) 293T cells were transfected with pISRE-Luc and pRL-tK concurrently with pCAGGS expression plasmids (HeV P, V, and W; 1 μg in 2 × 105 cells) using Lipofectamine 2000. At 24 h posttransfection, cells were treated with 1,000 U of human IFN-β for 3 h and then assayed for luciferase activity. Error bars indicate SEM. (c) HEp-2 cells were infected at an MOI of 1 at 24 h postinfection, followed by IFN-β (1,000 U) treatment for 3 h. Real-time PCR was then performed for ISG54, ISG56 (24), and 18s rRNA (19). The error bars indicate standard deviations for three independent experiments.
To look at the levels of transcriptional activation, RNA was extracted from HEp-2 cells following infection and interferon treatment. Cells were infected separately with each of the three viruses (MOI of 1), and real-time PCR was used to assess the expression levels of ISG54 and ISG56 (Fig. 2c). The mRNA expression levels of both ISG54 and ISG56 in infected cells prior to interferon treatment paralleled those observed in mock-infected cells. Following treatment with exogenous interferon for 3 h, only a slight reduction in ISG transcription was observed compared to that in mock-infected cells (Fig. 2c). This partial blocking effect supports the results obtained with luciferase reporter assays with transfected 293T cells. It is apparent that the results observed for cells transfected with individual henipavirus genes are not representative of what occurs in virus-infected cells. Greater amounts of protein are expected to be present in transfected cells than in infected cells due to the use of high-efficiency promoters to drive overexpression. We hypothesize that the complete block in interferon signaling observed in transfected cells is due to the protein expression level, which is higher than that in infected cells.
To test our hypothesis, the expression levels of the P gene products were examined by Western blot analysis using lysates from 293T cells infected with NiV-M or transfected with individual NiV-M genes expressing P, V, and W proteins (Fig. 3). The numbers of cells and percentages of transfected/infected cells remained comparable across the experiment, as determined by immunofluorescence (data not shown). The amounts of P, V, and W proteins present in lysates from transfected cells exceeded those from cells infected with NiV-M (Fig. 3). When levels of protein were semiquantified using ImageJ software (1), the P gene products were shown to be expressed 8-fold (P protein), 15-fold, (V protein), and 40-fold (W protein) more following transfection than following infection. These results support our hypothesis that greater amounts of protein are expressed during transfection, and this is likely to account for the complete block in interferon signaling.
Fig. 3.

Expression of henipavirus P gene products in transfected and infected human cells. 293T cells were infected at an MOI of 2 for 24 h and then lysed with 2% SDS to inactivate virus prior to removal from the biosafety level 4 (BSL4) laboratory. Concurrently, 293T cells were also transfected with pCAGGS expression plasmids (NiV P, V, and W; 1 μg in 2 × 105 cells) and incubated for 24 h. The cell lysates were resolved by 12% SDS-PAGE and detected by Western blotting with rabbit antiserum specific for the N-terminal common region of the P gene proteins (Pn) and rabbit antiserum specific for the unique C-terminal portions of the P (Pc), V, and W proteins. The C-terminal P, V, and W protein antisera were generated against synthesized peptides (GenScript Pty. Ltd.). A monoclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Sigma-Aldrich) antibody was incorporated as a control to determine protein loading and normalization.
As interferon is not produced following henipavirus infection of human cells and interferon signaling is only partially blocked, it could be hypothesized that by stimulating an interferon signaling cascade by other means, the host may be more likely to fight off the infection and survive. To test the hypothesis, HEp-2 cells were infected with HeV at a low MOI and then 6 h later were treated with interferon (Fig. 4). Following interferon treatment, there is an approximately 85% reduction in the number of infected cells and an unambiguous reduction in the size of syncytia. This experiment demonstrates the potential of interferon as a postexposure therapeutic. Further work is necessary to demonstrate in vivo efficacy.
Fig. 4.

Interferon posttreatment as a potential therapeutic following Hendra virus infection. HEp-2 cells were infected with HeV at an MOI of 0.05 for 6 h and then treated with IFN-β (1,000 U) for 24 h. Immunofluorescence was undertaken to determine the level and spread of virus infection using HeV P-specific antiserum and DAPI (4′,6-diamidino-2-phenylindole) staining.
Previous studies have demonstrated a complete block in interferon signaling following single-gene transfection studies of Nipah virus P gene expression plasmids (5, 13, 25). We hypothesize that the differences seen with transfection studies and our results are due to the differing amounts of P gene products available to bind and block the function of STAT proteins. We suggest that a significant amount of STAT1 protein remains unbound by henipavirus proteins during infection and can be activated, which initiates interferon signaling. It is also expected that different cell types will vary in their immune profiles, and this will be reflected in the response to henipavirus infection. Indeed, previous studies suggest that interferon production is not blocked in some epithelial cells after Nipah virus infection (12). This highlights the need for further development of henipavirus reverse genetics systems in order to carry out more biologically significant experiments in vivo.
In conclusion, this study has demonstrated that during infection of human cells with henipaviruses, there are differences in the antagonism of the innate immune response compared to that observed in transfected cells. This demonstrates the importance of confirming results seen in transfected cells in an infection model. While transfection studies may help to elucidate protein function, there are limitations to understanding the true biological story of infection. As the interferon signaling pathway is only partially antagonized following henipavirus infection, this indicates a potential for interferon treatment as a possible postexposure therapeutic preventing systemic infection.
Acknowledgments
This work was supported by a Ph.D. scholarship (E.R.V.) from the Australian Biosecurity Cooperative Research Centre for Emerging Infectious Diseases, an OCE Postdoctoral Fellowship (G.A.M.), and a CEO Science Leader Award (L.-F.W.) from the CSIRO Office of the Chief Executive.
We thank Chris Cowled and Albert Joubert for critical reading of the manuscript.
Footnotes
Published ahead of print on 2 February 2011.
REFERENCES
- 1. Abramoff M. D., Magelhaes P. J., Ram S. J. 2004. Image processing with ImageJ. Biophotonics Int. 11:36–42 [Google Scholar]
- 2. Andrejeva J., et al. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. U. S. A. 101:17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chua K. B., et al. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435 [DOI] [PubMed] [Google Scholar]
- 4. Chua K. B., et al. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–1259 [DOI] [PubMed] [Google Scholar]
- 5. Ciancanelli M. J., Volchkova V. A., Shaw M. L., Volchkov V. E., Basler C. F. 2009. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J. Virol. 83:7828–7841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fontana J. M., Bankamp B., Rota P. A. 2008. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol. Rev. 225:46–67 [DOI] [PubMed] [Google Scholar]
- 7. Goodbourn S., Randall R. E. 2009. The regulation of type I interferon production by paramyxoviruses. J. Interferon Cytokine Res. 29:539–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harcourt B. H., et al. 2005. Genetic characterization of Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis. 11:1594–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Homaira N., et al. 2010. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol. Infect. 138:1630–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horvath C. M. 2004. Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev. 15:117–127 [DOI] [PubMed] [Google Scholar]
- 11. Hsu V. P., et al. 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis. 10:2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lo M. K., et al. 2010. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 404:78–88 [DOI] [PubMed] [Google Scholar]
- 13. Lo M. K., Rota P. A. 2008. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J. Clin. Virol. 43:396–400 [DOI] [PubMed] [Google Scholar]
- 14. Luby S. P., Gurley E. S., Hossain M. J. 2009. Transmission of human infection with Nipah virus. Clin. Infect. Dis. 49:1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luby S. P., et al. 2009. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 15:1229–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marsh G., et al. 2010. Genome sequence conservation of Hendra virus isolates during spillover to horses, Australia. Emerg. Infect. Dis. 16:1767–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murray K., et al. 1995. A novel morbillivirus pneumonia of horses and its transmission to humans. Emerg. Infect. Dis. 1:31–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murray K., et al. 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268:94–97 [DOI] [PubMed] [Google Scholar]
- 19. Nicot N., Hausman J., Hoffmann L., Evers D. 2005. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56:2907–2914 [DOI] [PubMed] [Google Scholar]
- 20. Park M.-S., Garcia-Sastre A., Cros J. F., Basler C. F., Palese P. 2003. Newcastle disease virus V protein is a determinant of host range restriction. J. Virol. 77:9522–9532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez J. J., Horvath C. M. 2004. Host evasion by emerging paramyxoviruses: Hendra virus and Nipah virus V proteins inhibit interferon signaling. Viral Immunol. 17:210–219 [DOI] [PubMed] [Google Scholar]
- 22. Rodriguez J. J., Parisien J. P., Horvath C. M. 2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 76:11476–11483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Selvey L., Sheridan J. 1995. Outbreak of severe respiratory disease in humans and horses due to a previously unrecognized paramyxovirus. J. Travel Med. 2:275. [DOI] [PubMed] [Google Scholar]
- 24. Shaw M. L., Cardenas W. B., Zamarin D., Palese P., Basler C. F. 2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and Toll-like receptor 3-triggered signaling pathways. J. Virol. 79:6078–6088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shaw M. L., Garcia-Sastre A., Palese P., Basler C. F. 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol. 78:5633–5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsutsumi H., Takeuchi R., Ohsaki M., Seki K., Chiba S. 1999. Respiratory syncytial virus infection of human respiratory epithelial cells enhances inducible nitric oxide synthase gene expression. J. Leukoc. Biol. 66:99–104 [PubMed] [Google Scholar]
- 27. Yoneda M., et al. 2010. The nonstructural proteins of Nipah virus play a key role in pathogenicity in experimentally infected animals. PLoS One 5:e12709. [DOI] [PMC free article] [PubMed] [Google Scholar]