

Abstract
The leader proteinase (Lpro) of foot-and-mouth disease virus (FMDV) is a papain-like proteinase that plays an important role in FMDV pathogenesis. Previously, it has been shown that Lpro is involved in the inhibition of the type I interferon (IFN) response by FMDV. However, the underlying mechanisms remain unclear. Here we demonstrate that FMDV Lbpro, a shorter form of Lpro, has deubiquitinating activity. Sequence alignment and structural bioinformatics analyses revealed that the catalytic residues (Cys51 and His148) are highly conserved in FMDV Lbpro of all seven serotypes and that the topology of FMDV Lbpro is remarkably similar to that of ubiquitin-specific protease 14 (USP14), a cellular deubiquitylation enzyme (DUB), and to that of severe acute respiratory syndrome coronavirus (SARS-CoV) papain-like protease (PLpro), a coronaviral DUB. Both purified Lbpro protein and in vivo ectopically expressed Lbpro removed ubiquitin (Ub) moieties from cellular substrates, acting on both lysine-48- and lysine-63-linked polyubiquitin chains. Furthermore, Lbpro significantly inhibited ubiquitination of retinoic acid-inducible gene I (RIG-I), TANK-binding kinase 1 (TBK1), TNF receptor-associated factor 6 (TRAF6), and TRAF3, key signaling molecules in activation of type I IFN response. Mutations in Lbpro that ablate the catalytic activity (C51A or D163N/D164N) or disrupt the SAP (for SAF-A/B, Acinus, and PIAS) domain (I83A/L86A) abrogated the DUB activity of Lbpro as well as its ability to block signaling to the IFN-β promoter. Collectively, these results demonstrate that FMDV Lbpro possesses DUB activity in addition to serving as a viral proteinase and describe a novel mechanism evolved by FMDV to counteract host innate antiviral responses.
INTRODUCTION
Foot-and-mouth disease (FMD) is a highly contagious viral disease of wild and domestic cloven-hoofed animals (22). The etiologic agent, FMD virus (FMDV), is a positive-stranded RNA virus that belongs to the Aphthovirus genus of the Picornaviridae family. The genome of FMDV encodes a polyprotein which is processed into structural and nonstructural proteins by three virus-encoded proteinases, i.e., leader (Lpro), 2A, and 3Cpro (31). Lpro, the first protein to be translated from the FMDV genome, is initiated at two different AUGs that are separated by 84 nucleotides, and this results in two alternative forms of Lpro, termed Labpro and Lbpro. Both forms have been detected in vitro and in infected cells (7, 32). Lpro is a well-characterized papain-like protease that cleaves itself off the nascent polyprotein precursor (36, 39). Using a genetically engineered FMDV lacking the Lpro-coding region (A12-LLV2), de Los Santos et al. demonstrated that Lpro inhibits the induction of beta interferon (IFN-β) and blocks the host innate immune response (9). However, the exact molecular mechanisms underlying the ability of Lpro to inhibit IFN-β induction remain to be elucidated.
It is well known that IFN-β transcription requires the coordinate activation of the latent transcription factors NF-κB, interferon regulatory factors (IRFs), and ATF2-c-Jun (AP-1) and their subsequent binding to the IFN-β enhancer elements (47). Many of the signaling events that link the sensors to the transcription factors are mediated by the activities of kinases and ubiquitinating enzymes that modify and activate critical intermediates in the signaling cascade (3, 4, 25). Type I IFNs trigger signals that culminate in expression of IFN-stimulated genes (ISGs) and inflammatory cytokines, which suppress the replication of invading pathogens and also facilitate the development of adaptive immune responses (37, 48). However, the ubiquitin (Ub) chains conjugated to signaling molecules during activation of each pathway can be inactivated by cellular deubiquitylation enzymes (DUBs) such as A20, CYLD, and DUBA (17, 26, 52), suggesting that ubiquitin modification enzymes and DUBs play critical roles in modulating the immune responses.
Recent work has further revealed that many viruses have evolved elaborate strategies to counteract innate antiviral immune signaling pathways by redirecting or inhibiting the ubiquitination machinery of the host for their survival (49). For example, HIV-1 prevents the antiviral interferon response via Vpr- and Vif-directed ubiquitin-mediated degradation of IRF-3 (35), the N-terminal protease (Npro) of bovine viral diarrhea virus interacts with IRF-3 and promotes its polyubiquitination and degradation through the proteasome (5, 23), and the murid herpesvirus 4 (MuHV-4) latency-associated protein ORF73 associates with the host ubiquitin-ligase complex to promote polyubiquitination and subsequent proteasomal degradation of p65/RelA, which inhibits the activity of NF-κB that facilitates MuHV-4 latency (40). Recent studies showed that the papain-like protease (PLpro) domains of many coronaviruses, such as severe acute respiratory syndrome coronavirus (SARS-CoV), human coronavirus (HCoV) NL63, and mouse hepatitis virus (MHV) A59, act as both papain-like proteases and deubiquitinating enzymes that block type I IFN induction (2, 6, 8, 13, 18, 28, 54). The FMDV Lpro is a papain-like protease and has been shown to inhibit the induction of transcription of IFN-β (9, 10, 39, 51). However, the precise mechanism(s) by which FMDV Lpro exerts this effect remains unclear. In this work, sequence alignment and structural bioinformatics analyses suggested that the topology of FMDV Lbpro is remarkably similar to that of ubiquitin-specific protease 14 (USP14) (24), a cellular DUB, and to that of SARS-CoV PLpro (38), a coronaviral DUB. We then conducted experiments to demonstrate that the papain-like protease of FMDV, Lbpro, is also a novel viral DUB. Furthermore, we found that Lbpro cleaves ubiquitin moieties from critical signaling proteins of the type I IFN signaling pathway, such as retinoic acid-inducible gene I (RIG-I), TANK-binding kinase 1 (TBK1), tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3), and TRAF6. In addition, mutations that ablate the catalytic activity or disrupt the SAP (for SAF-A/B, Acinus, and PIAS) domain of Lbpro abrogate the DUB activity and also the ability of Lbpro to block IFN-β induction.
MATERIALS AND METHODS
Cells and virus.
HEK293T cells (human embryonic kidney epithelial cells) were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% heated-inactivated fetal calf serum (FCS), 100 U/ml penicillin, and 10 μg/ml streptomycin sulfate at 37°C in a humidified 5% CO2 incubator. Porcine kidney (IBRS-2) cells were grown in Eagle minimal essential medium (MEM) supplemented with 10% heated-inactivated FCS, 100 U/ml penicillin, and 10 μg/ml streptomycin sulfate. FMDV strain O/ES/2001 was propagated in IBRS-2 cells, and the supernatants of infected cells were clarified and stored at −80°C. Sendai virus (SEV) was obtained from the Centre of Virus Resource and Information, Wuhan Institute of Virology, Chinese Academy of Sciences.
Plasmids.
Full-length hemagglutinin (HA)-tagged ubiquitin (Ub) plasmid (HA-Ub) and HA-Ub mutants in which all but one Lys residue (HA-K48-Ub or HA-K63-Ub) was replaced with Arg were gifts of Tomohiko Ohta (St. Marianna University School of Medicine, Japan) (34). pcDNA3.1-Flag-Ub was previously described (8). The expression plasmids for wild-type (WT) RIG-I (pEF-Flag-RIG-I), its constitutively active mutant (pEF-Flag-RIG-IN), and p125-Luc (IFN-β-Luc) were kindly provided by T. Fujita (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). The TBK1 expression vector was kindly provided by Himanshu Kuma and Shizuo Akira (Immunology Frontier Research Center, Osaka University, Japan), The TRAF3 and TRAF6 expression vectors were gifts from Edward W. Harhaj (University of Miami School of Medicine, Miami, FL).
For construction of pcDNA3.1-V5/His/Lbpro, the cDNA fragment encoding the full-length Lbpro of FMDV was amplified by PCR from the cDNA of FMDV O/ES/2001 (GenBank accession no. AY686687) and subcloned into the pcDNA3.1-V5/His B vector (Invitrogen). Mutagenesis of individual amino acid residues (C51A, I83A, L86A, D163N, and D164N) in Lbpro was conducted using overlap extension PCR. Detailed sequences of the specific primers used are available upon request. All constructs were validated by DNA sequencing.
In vitro deubiquitination assay.
The FMDV Lbpro protein was purified from cells transfected with pcDNA3.1-V5/His/Lbpro using Ni Sepharose 6 Fast Flow (GE Healthcare) according to the manufacturer's protocol. As a negative control, Ni Sepharose 6 Fast Flow was also used to isolate the proteins from empty-vector-transfected cells. The polyubiquitin chains were purchased from Boston Biochem (K48-Ub2-7 [catalog no. UC-230] and K63-Ub2-7 [catalog no. UC-330]). The purified products were incubated with 3 μg of K48-Ub2-7 chains or K48-Ub2-7 chains at 37°C in a 30-μl reaction mixture containing 25 mM NaCl, 100 mg/ml bovine serum albumin (BSA), and 2 mM dithiothreitol (DTT). A control reaction mixture was incubated under identical conditions with the exclusion of enzyme. Reactions were terminated by addition of 5× SDS-PAGE sample loading buffer (Beyotime, China) followed by heat treatment at 100°C for 5 min. The samples were analyzed by electrophoresis on a 15% SDS-polyacrylamide gel and stained with Coomassie blue dye.
Assay of deubiquitination activity in vivo.
The effect of FMDV Lbpro on ubiquitinated cellular proteins in vivo was assessed as described previously (14). HEK293T cells cultured in 60-mm dishes were cotransfected with 1 μg of HA-Ub, HA-K48-Ub, or HA-K63-Ub plus appropriate amounts of constructs containing FMDV Lbpro or the corresponding mutants using Lipofectamine 2000. Where applicable, the empty pcDNA3.1/V5-HisB vector was supplemented to keep the total amount of DNA transfected constant. After 30 h, cells were harvested by adding 250 μl 2× lysis buffer A (LBA) (65 mM Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate, 3% dl-dithiothreitol, and 40% glycerol) containing 20 mM N-ethylmaleimide (NEM) (Sigma) and 20 mM iodoacetamine (Sigma). Cell lysates were then analyzed for ubiquitin-conjugated proteins by Western blotting with anti-HA antibody (1:1,000) (MBL, Japan). To confirm the expression levels of FMDV Lbpro and the mutants, anti-V5 antibody (MBL, Japan) was used to detect the V5-tagged proteins. Beta-actin was detected with anti-beta-actin monoclonal antibody (MAb) (Beyotime, China) to demonstrate equal protein sample loading.
Luciferase reporter gene assay.
HEK293T cells grown in 24-well plates were cotransfected with 0.1 μg/well of IFN-β-Luc along with 0.05 μg/well of pRL-TK plasmid (Promega) (for normalization of transfection efficiency) and various other expression plasmids or an empty control plasmid. In some experiments, cells were further infected or mock infected with SEV at 24 h after the initial cotransfection. Cells were harvested 12 h later, and firefly luciferase and Renilla luciferase activities were determined using the dual-luciferase reporter assay system (Promega) according to the manufacturer's protocol. Data represent relative firefly luciferase activity normalized to Renilla luciferase activity and are representative of three independently conducted experiments. Data are presented as means ± standard deviations (SD). A P value of less than 0.01 was considered highly statistically significant.
Coimmunoprecipitation and immunoblot analysis.
Transient transfection of HEK293T cells with the indicated plasmids was performed routinely using Lipofectamine 2000 as per the manufacturer's instructions (Invitrogen). Transfected HEK293T cells from each 100-mm dish were lysed in l ml lysis buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 20 nM phenylmethylsulfonyl fluoride [PMSF]), and the protein concentration was measured and adjusted. For each immunoprecipitation, 500 μg of cell lysate protein was incubated with 0.5 μg of the indicated antibody and 25 μl of protein A+G-agarose (Beyotime, China) overnight at 4°C. The Sepharose beads were then washed three times with 1 ml lysis buffer. The precipitates were subjected to 10% SDS-PAGE and subsequent immunoblot analysis using the indicated antibodies.
RESULTS
Bioinformatics analysis predicts FMDV Lbpro to be a viral DUB.
Based on the structures of their catalytic domains, the human DUBs have been classified into five subfamilies, most of which exhibit a high degree of homology mainly in two regions known as Cys and His boxes (C and H boxes, respectively) that surround the catalytic Cys and His residues (33, 53). FMDV Lpro is a well-characterized papain-like proteinase that also possesses the catalytic Cys and His residues (21, 43). Sequence alignment showed that Cys51 and His148 (numbering based on FMDV type O Labpro) are highly conserved among all seven serotypes of FMDV (Fig. 1A). To predict whether FMDV Lbpro has the structural characteristics of a deubiquitinating enzyme, the structure comparison service Secondary-Structure Matching (SSM) (http://www.ebi.ac.uk/msd-srv/ssm/cgi-bin/ssmserver) was used by clustering FMDV Lbpro with USP14 (24), a known cellular DUB, and SARS-CoV PLpro (38), a known viral DUB. As shown in Fig. 1B, the topology of FMDV Lbpro is remarkably similar to those of SARS-CoV PLpro and USP14, with corresponding root mean square (RMS) deviations of alignment of 2.845 Å and 2.776 Å, respectively, despite the fact that FMDV Lbpro has low amino acid sequence homology with SARS-CoV PLpro and USP14 (9.2% and 12.3%, respectively). These data suggested that FMDV Lbpro is likely a viral DUB.
Fig. 1.

Structure analysis of FMDV Lbpro. (A) Amino acid alignment of the conserved region surrounding Cys51 and His148 (numbering based on Labpro) in the Lbpro proteins of the seven types of sequenced FMDV genomes. Black boxes indicate conserved enzymatic proteolysis residues. The sequences were derived from GenBank entries with the following accession numbers: FMDV type O, NC_004004; FMDV type A, NC_011450; FMDV type C, NC_002554; FMDV type Asia 1, NC_004915; FMDV type SAT1, NC_011451; FMDV type SAT2, NC_003992; and FMDV type SAT3, NC_011452. (B) Comparison of FMDV Lbpro (Protein Data Bank [PDB] code 1QOL) with SARS-CoV PLpro (PDB code 3E9S) and the cellular DUB USP14 (PDB code 2AYN). The topologies of the proteins were structurally aligned and superimposed using the Web-based server SSM (http://www.ebi.ac.uk/msd-srv/ssm/cgi-bin/ssmserver). A ribbon diagram of conserved topologies shows FMDV Lbpro, SARS-CoV PLpro, and USP14 in color; the rest is represented by gray.
FMDV Lbpro processes K48-linked and K63-linked polyubiquitin in vitro.
As mentioned in the introduction, initiation of FMDV protein synthesis can occur at the first or the second AUG codon (7, 32). To avoid initiation of translation at both sites, we subcloned the FMDV Lbpro fragment into pcDNA3.1-V5/His B in frame with C-terminal V5-6× His tags and transfected the construct into HEK293T cells. To further determine if FMDV Lbpro has DUB activity, a DNA construct expressing FMDV Lbpro was transiently transfected into HEK293T cells and the recombinant Lbpro was purified from cell lysates using Ni Sepharose 6 Fast Flow (Fig. 2A). When incubated with K48- and K63-linked polyubiquitin chains in vitro, the purified Lbpro was able to cleave both substrates (Fig. 2B and C). Upon prolonged incubation, Lbpro completely processed both substrates to monoubiquitin (data not shown). These data show that both of the two major forms of polyubiquitin chains, K48 and K63, serve as in vitro substrates for recombinant FMDV Lbpro.
Fig. 2.
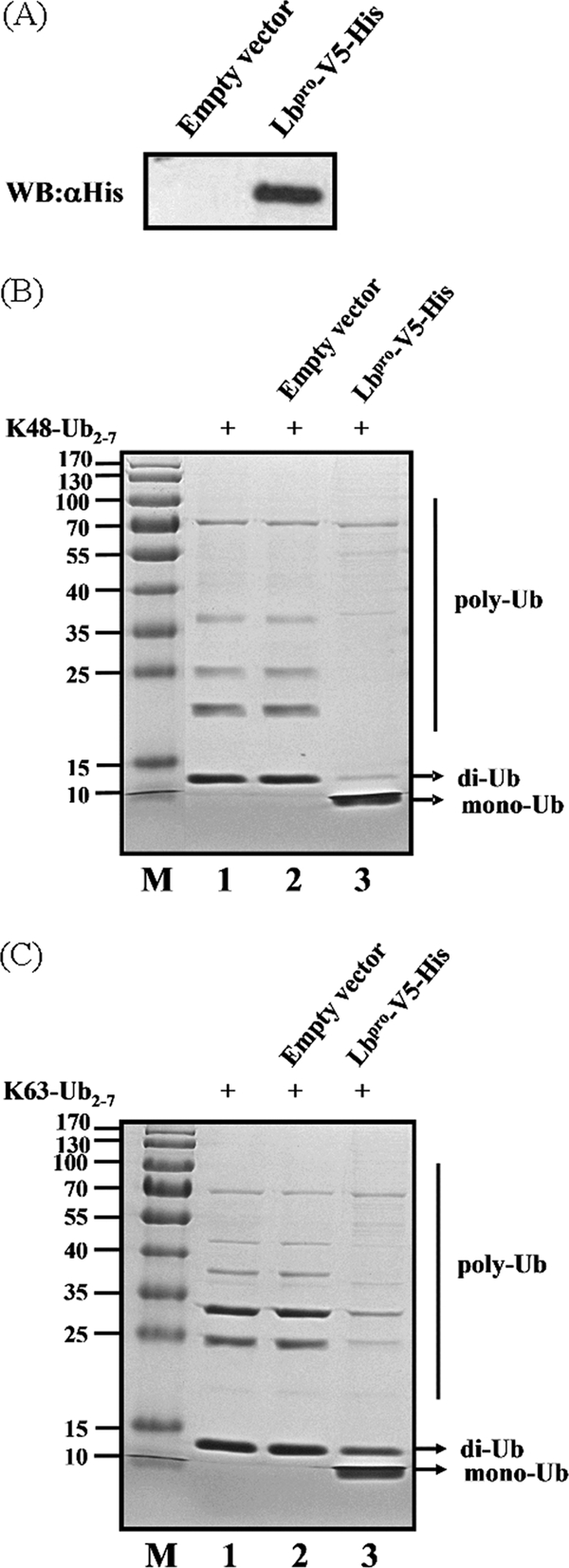
Processing of K48- and K63-linked polyubiquitin chains by FMDV Lbpro in vitro. (A) The protein was obtained from FMDV Lbpro-transfected or mock-transfected 293T cells using Ni Sepharose 6 Fast Flow and analyzed for His-tagged FMDV Lbpro-conjugated proteins by Western blotting (WB) with an anti-His antibody. (B) In vitro K48-linked polyubiquitin deconjugation assay. K48-linked polyubiquitin was incubated with the protein obtained from mock-transfected (lane 2) or FMDV Lbpro-transfected (lane 3) HEK293T cells at 37°C for 1 h before being analyzed by SDS-PAGE. Lane 1, uncleaved K48-linked polyubiquitin chain (K48-Ub2-7). M, molecular mass markers, including 170-, 130-, 100-, 70-, 55-, 40-, 35-, 25-, 15-, and 10-kDa bands. (C) In vitro K63-linked polyubiquitin deconjugation assay. The experiment was performed similarly to that described for panel B except that the K63-linked polyubiquitin chain (K63-Ub2-7) was used.
FMDV Lbpro has DUB activity in vivo.
To determine whether Lbpro functioned as a DUB in a cell-based assay, HEK293T cells were transfected with the empty vector or increasing amounts of plasmid DNA encoding Lbpro along with HA-tagged ubiquitin vector (HA-Ub), and the effect of Lbpro on all ubiquitinated cellular proteins was assessed via Western blotting with an anti-HA antibody. As shown in Fig. 3A, expression of Lbpro resulted in a dose-dependent reduction in the level of ubiquitinated cellular proteins compared to that in the control vector-transfected cells. To further identify which Ub linkage type is targeted by Lbpro in vivo, HEK293T cells were transfected with HA-K48-Ub or HA-K63-Ub in lieu of HA-Ub. These constructs allow solely the formation of K48- and K63-linked polyubiquitin chains, respectively (34). Notably, both K48- and K63-linked Ub chains were processed by Lbpro in a dose-dependent manner, with no apparent preference between them (Fig. 3B and C). These results confirm our earlier results that FMDV Lbpro is a potent DUB that removes ubiquitin conjugates formed through either K48 or K63 linkage from many cellular substrates.
Fig. 3.

FMDV Lbpro has a dose-dependent deubiquitinating activity in vivo. (A) HEK293T cells grown in 60-mm dishes were transfected with HA-tagged Ub expression plasmids (1.2 μg), along with increasing quantities (0, 0.008, 0.04, 0.2, or 1 μg) of plasmid encoding Lbpro, using Lipofectamine 2000. Cell lysates were prepared at 30 h posttransfection and analyzed for Ub-conjugated proteins by Western blotting with an anti-HA antibody. Western blotting with anti-V5 antibody shows expression of Lbpro, and Western blotting for beta-actin serves as a protein loading control. (B and C) FMDV Lbpro effectively cleaves both K48 and K63 Ub linkages in vivo. The experiment was performed similarly to that described for panel A except that HA-K48-Ub or HA-K63-Ub was used in lieu of HA-Ub. The asterisk indicates a nonspecific band.
To exclude the possibility that the DUB activity of Lbpro observed was an artificial effect of plasmid overexpression in cell culture, we analyzed the DUB activity in the context of FMDV infection. IBRS-2 cells were transfected with the HA-Ub vector for 24 h and then infected with FMDV at increasing multiplicities of infection (MOIs). As shown in Fig. 4, compared to those in uninfected cells, the levels of ubiquitinated cellular proteins were reduced in a dose-dependent manner in FMDV-infected cells. Similar results were obtained when K48-Ub or K63-Ub was transfected in place of HA-Ub prior to FMDV infection (data not shown). In aggregate, these data suggest that Lbpro, expressed from replicating FMDV, possesses DUB activities toward both K48- and K63-linked ubiquitin chains.
Fig. 4.
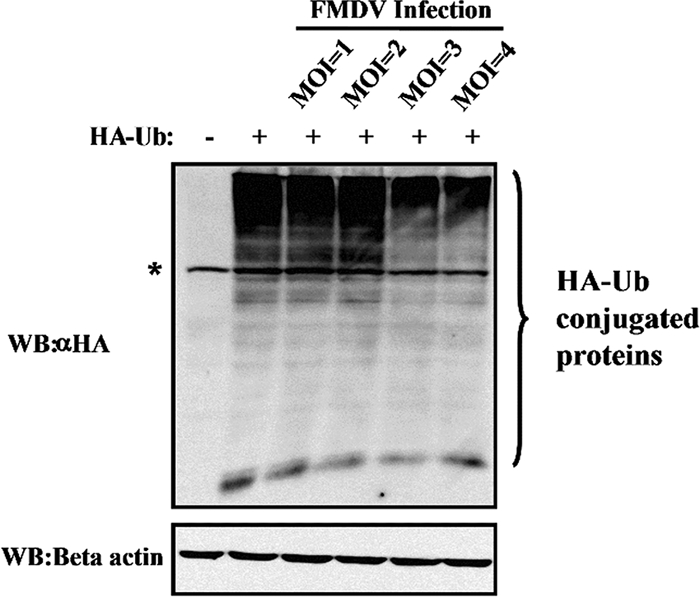
DUB activity assay during FMDV infection. IBRS-2 cells cultured in 60-mm dishes were transfected with HA-tagged ubiquitin and infected with FMDV at different MOIs at 24 h posttransfection. Cells were lysed at 12 h postinfection and assayed for anti-HA and anti-beta-actin staining by Western blotting. The asterisk indicates a nonspecific band.
The DUB activity of FMDV Lbpro is uncoupled from its ability to process eIF-4G.
It is established that Lpro possesses the ability to cleave the translation initiation factor eIF-4G and shuts off host cell translation (15). We considered the possibility that the DUB activity of Lbpro is coupled with or dependent upon its ability to cleave eIF-4G. Previous studies revealed that mutations in amino acid residues C51, D163, and D164 in Lpro partially reduced (D163N and D164N) or completely eliminated (C51A and D163N/D164N) the abilities of Lpro to process itself from viral polyprotein and to cleave eIF-4G (15, 41). Such activities of Lpro, however, were not affected upon disruption of the SAP domain by double amino acid substitutions at residues 83 and 86 (I83A/L86A) (11). Based on these previous findings, we constructed these Lbpro mutants and compared them with WT Lbpro for DUB activity and the ability to cleave eIF-4G. In agreement with the previous studies, the I83A/L86A double mutant retained the ability to cleave eIF-4G (Fig. 5A, lane 7), while the D163N/D164N double mutant (lane 10) and the C51A mutant (lane 4) completely lost such activity. These Lbpro mutants were then examined for DUB activity in HEK293T cells. To this end, HEK293T cells were cotransfected with the Flag-Ub vector and the indicated Lbpro expression plasmids. As shown in Fig. 5B, overexpression of WT Lbpro almost completely eliminated Flag-Ub-reactive protein bands (lane 3). The catalytically inactive mutants (C51A and D163N/D164N) lost the DUB activity (lanes 4 and 10, respectively), as did the SAP domain I83A/L86A double mutant (lane 7), which is capable of eIF-4G cleavage. These data suggest that although the catalytic activity of Lbpro is important for its DUB activity, the latter does not depend on the ability of Lbpro to process eIF-4G (i.e., proteolytic activity). Further confirming this notion, the D164N mutant was less active in cleaving eIF-4G than was the I83A/L86A double mutant (Fig. 5A, compare lanes 9 and 7) but had substantially better DUB activity than the latter (Fig. 5B, compare lanes 9 and 7). Similar results were obtained when K48-Ub or K63-Ub was used in lieu of HA-Ub to compare the DUB activities of WT and mutant Lbpro (data not shown).
Fig. 5.

The ability of Lbpro to process eIF-4G is not necessary for its DUB activity. (A) HEK293T cells cultured in 60-mm dishes were transfected with the indicated Lbpro expression plasmids (2 μg). Cell lysates were prepared at 30 h posttransfection and analyzed for cellular eIF-4G proteins with anti-eIF-4G antibody (Cellsignal, catalog no. 2498) by Western blotting. (B) HEK293T cells cultured in 60-mm dishes were transfected with Flag-tagged Ub expression plasmid (pcDNA3.1-Flag-Ub) (1.2 μg), along with indicated Lbpro expression plasmids (1 μg). Cell lysates were prepared at 30 h posttransfection and analyzed for Flag-Ub and Lbpro-V5 conjugated proteins by Western blotting. Anti-beta-actin antibody was used to detect beta-actin, which serves as a protein loading control.
DUB activity is essential for FMDV Lbpro to block type I IFN induction.
Ubiquitination is essential for the activation of many components of the type I IFN signaling pathway, such as RIG-I, TBK1, TRAF3, and TRAF6 (12, 19, 20, 30, 50). In addition, the negative feedback regulation of these molecules depends in part on DUBs (25, 44, 49). Previous studies have shown that several viral proteins that contain a conserved DUB motif can block the IFN-β response, including SARS-CoV PLpro, HCoV NL63 PLP2, and MHV A59 PLP2 (8, 13, 18, 54). To determine whether Lbpro also can block the type I IFN signaling pathway, HEK293T cells were transfected with an Lbpro expression construct together with a luciferase reporter plasmid with the IFN-β promoter and pRL-TK, followed by SEV infection. As shown in Fig. 6A, Lbpro downregulated SEV-induced IFN-β promoter in a dose-dependent manner; these data were consistent with the report that Lpro inhibits IFN-β transcription (9, 10, 51). We further investigated whether overexpression of Lbpro inhibits RIG-I-, TBK1-, and TRAF6-mediated activation of the IFN-β promoter. To this end, HEK293T cells were transfected with DNA constructs encoding RIG-I, TBK1, or TRAF6, together with IFN-β-Luc. As shown in Fig. 6B, overexpression of RIG-I, TBK1, or TRAF6 significantly activated the IFN-β promoter compared with cells transfected with the empty vector control. However, such effects were all substantially reduced in the presence of Lbpro.
Fig. 6.

Lbpro inhibits type I interferon induction. (A) HEK293T cells grown in 24-well plates were transfected with 0.1 μg/well of IFN-β-Luc reporter plasmid, along with 0.05 μg/well of pRL-TK plasmid and increasing quantities (0, 0.004, 0.02, 0.1, or 0.5 μg) of plasmid encoding Lbpro, using Lipofectamine 2000. Twenty-four hours after the initial transfection, the cells were further infected with SEV or mock infected. Luciferase assays were performed at 18 h after infection. Results represent the means and standard deviations from three independent experiments. The relative firefly luciferase activity was normalized to the Renilla reniformis luciferase, and the untreated empty-vector control value was set to 1. (B) HEK293T cells were cotransfected with the IFN-β-Luc reporter plasmid (0.1 μg), pRL-TK plasmid (0.05 μg), and 0.5 μg of plasmid encoding Lbpro together with the RIG-I, RIG-I N, TBK1, or TRAF6 expression vector (0.5 μg). Luciferase assays were performed at 36 h after transfection. (C) HEK293T cells were cotransfected with the IFN-β-Luc reporter plasmid (0.1 μg), 0.05 μg of pRL-TK, and the designated Lbpro expression plasmids (0.5 μg). An empty vector (pcDNA3.1-V5/His B) was used as a control. Twenty-four hours after the initial transfection, the cells were further infected with SEV or mock infected. Cell extracts were collected at 18 h after infection and analyzed for firefly and Renilla luciferase expression. *, P < 0.01 compared with vector plus SEV.
To determine whether the DUB activity of Lbpro is involved in Lbpro inhibition of type I IFN induction, various Lbpro mutants with differing DUB activities (Fig. 5B) were analyzed for their ability to impair signaling to the SEV-induced IFN-β promoter in HEK293T cells. As shown in Fig. 6C, the catalytically inactive mutants (C51A and D163N/D164N) devoid of DUB activity completely lost the ability to block viral activation of the IFN-β promoter, as did the proteolytically active, SAP domain I83A/L86A double mutant, which was also defective for DUB activity. Thus, the DUB activity, but not the proteolytic activities toward viral polyprotein and eIF-4G, is critical for the function of Lbpro as an IFN antagonist. Our data also revealed that the D163N and D164N mutants were still able to significantly inhibit IFN induction (to similar extents), although both were less effective than WT Lbpro (Fig. 6C). This provides further support for the notion that the ability of FMDV Lbpro to block viral induction of IFN-β transcription is proportional to its DUB activity instead of the proteolytic activity, because these two mutants differed in their proteolytic activity (Fig. 5A, lanes 8 and 9) yet had similar reduced DUB activity compared with WT Lbpro (Fig. 5B, lanes 8 and 9). We found that these Lbpro mutants acted in a similar fashion when the IFN-β promoter was activated by ectopic expression of RIG-I, TBK1, and TRAF6 (data not shown).
FMDV Lbpro deubiquitinates RIG-I, TBK1, TRAF3, and TRAF6.
We further investigated whether the IFN antagonist function of Lbpro is associated with the deubiquitination of RIG-I, TBK1, TRAF6, and TRAF3, which are essential signaling components in the type I IFN pathway activated by virus infection. We found that overexpression of Lbpro significantly inhibited ubiquitination of RIG-I (Fig. 7A), TBK1 (Fig. 7B), TRAF6 (Fig. 7C), and TRAF3 (Fig. 7D). In contrast, the Lbpro mutants (C51A, D163N/D164N, and I83A/L86A) lacking DUB activity (Fig. 5B) and incapable of inhibiting IFN induction (Fig. 6C) had no such effects. Taken together, our results suggest that the DUB activity of FMDV Lbpro was pivotal for the inhibition of type I IFN induction.
Fig. 7.

FMDV Lbpro inhibits ubiquitination of RIG-I, TBK1, TRAF6, and TRAF3. HEK293T cells cultured in 100-mm dishes were cotransfected with HA-tagged Ub expression plasmid (1.5 μg), 0.5 μg of the indicated plasmids encoding Lbpro, and the RIG-I (A), TBK1 (B), TRAF6 (C), or TRAF3 (D) expression vector (4 μg). MG132 (20 nM) was added at 30 h after transfection. Cell lysates were prepared at 4 h after treatment and immunoprecipitated with anti-Flag antibody (Macgene, China), and ubiquitin conjugation of protein was verified by immunoblotting with anti-HA antibody. The input tagged proteins were verified with the indicated antibodies.
DISCUSSION
FMD is one of the most contagious diseases of cloven-hoofed animals. The responsible agent, FMDV, rapidly replicates and disseminates within the infected animal and is able to spread quickly to susceptible animals that are in close contact (22). Previous studies have revealed that FMDV Lpro, a papain-like proteinase, is involved in antagonizing the innate immune responses by inhibiting type I interferon production, which is thought to play an important role in FMDV pathogenesis and virulence (10, 39, 51). Precisely how Lpro accomplishes this important function, however, remains unclear. Various mechanisms have been proposed, including the blockade of cap-dependent translation of cellular mRNAs (including those of IFNs) through Lpro-mediated cleavage of eIF-4G and the inhibition of induction of type I IFN transcription via an as-yet unclear mechanism(s) (9, 10, 15, 51). In this study, we provide biochemical and molecular evidence that FMDV Lbpro is a novel viral deubiquitinating enzyme. Our study also uncovers a novel mechanism by which FMDV Lbpro antagonizes type I IFN induction, i.e., by deubiquitinating the critical signaling components RIG-I, TBK1, TRAF6, and TRAF3. Our data comparing various Lbpro mutants suggest that the DUB activity of Lbpro, but not its classical proteolytic activity toward eIF-4G and viral polyprotein, governs the ability of Lbpro to block induction of the IFN-β promoter.
DUB activity was recently demonstrated for many viral proteins encoded by distinct viruses, such as human adenovirus, herpesvirus, coronavirus, and bunyavirus, and these enzymes play specific roles in regulation of viral infection (1, 2, 8, 16, 28, 42, 45, 54). Here, three strategies were used to demonstrate FMDV Lbpro to be a novel viral DUB: (i) a bioinformatics approach to predict FMDV Lbpro to be a DUB, (ii) an in vitro deubiquitination assay, and (iii) an assay of deubiquitination activity in vivo. First, we found that FMDV Lbpro proteins of seven serotypes share highly conserved Cys and His residues and flanking regions, which exist in many human DUBs. Indeed, structure bioinformatic studies indicated that the topology of FMDV Lbpro is highly similar to those of USP14 and SARS-CoV PLpro, suggesting that FMDV Lbpro may have deubiquitinating activity. In subsequent experiments, we showed that Lbpro indeed had DUB activity both in vitro and in vivo. Importantly, we demonstrated that Lbpro expressed from viral polyprotein in the context of FMDV infection possessed DUB activity, confirming the biological relevance of this finding. Furthermore, we revealed that FMDV Lbpro could act on both K48- and K63-linked Ub polymers, a feature shared by other known viral DUBs, such as human cytomegalovirus (HCMV) UL48 (27), herpes simplex virus type 1 (HSV-1) UL36 (27), and SARS-CoV PLpro (29).
Many cysteine proteases encoded by RNA viruses, which generate mature viral proteins from viral polyprotein that are necessary for virus replication, have been found to be multifunctional proteins. Like the coronaviral papain-like proteases, including SARS-CoV PLpro, MHV A59 PLP2, and HCoV NL63 PLP2, and the cysteine protease of an arterivirus, i.e., the nonstructural protein 2 (NSP2) of porcine reproductive and respiratory syndrome virus, Lbpro possesses deubiquitination activity and is able to antagonize innate immune induction of type I interferon (2, 6, 8, 13, 18, 28, 45, 54). Using an IFN-β promoter reporter assay, we also demonstrated that Lbpro, as a novel picornavirus DUB, significantly blocked SEV-induced IFN-β expression in a dose-dependent manner. These data are consistent with previous reports that Lpro is an antagonist of IFN-β (9, 10, 51). Recent studies indicated that a SAP domain exists in Lpro (11). Initially, we would like to know which one of the three functions/domains of Lpro, namely, cysteine protease, the SAP domain, and DUB activity, is sufficient for the inhibition of viral induction of IFN-β transcription. Although we cannot exclude the possibility that the protease activity is also involved, our data clearly demonstrated that the ability to block activation of the IFN-β promoter correlated with the DUB activity of Lbpro but not its proteolytic activity (toward eIF-4G). At present, we do not know whether the SAP domain also contributes to inhibition of IFN induction by Lbpro, as the SAP domain mutants also were impaired/defective for DUB activity. Future investigation to identify SAP mutants that disrupt this domain but do not affect the DUB activity will be required.
Ubiquitination and deubiquitination are critically involved in virus-induced type I IFN signaling pathways. Several ubiquitin ligase enzymes have been found to regulate these processes (3, 25, 44). For example, ubiquitination of RIG-I by the E3 ubiquitin ligase TRIM25 is necessary and sufficient to activate VISA/IPS-1/MAVS/Cardif, which triggers the downstream signaling cascade to produce type I IFN (19). Nrdp1, as an E3 ligase for TBK1, interacts with TBK1 and promotes ubiquitination of TBK1, leading to TBK1 and IRF-3 activation (50). In addition, virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of IFN-β and the cellular antiviral response (30). However, a few cellular DUBs are known to negatively regulate type I IFN signaling pathways. A20, DUBA, and CYLD were found to target RIG-I, TRAF3, and TBK1, respectively, for deubiquitination, thereby functioning as negative regulators of innate immune responses (17, 26, 52). In addition, the bacterial virulence factor YopJ is a deubiquitinating protease that acts on TRAF proteins to prevent or remove the ubiquitin conjugates required for signal transduction (46). To our knowledge, thus far there has been no direct demonstration of whether viral DUBs can remove ubiquitin chains from RIG-I, TBK1, and TRAF3/6, which in turn negatively regulates type I IFN induction. We report here that FMDV Lbpro significantly inhibits ubiquitination of RIG-I, TBK1, and TRAF3/6, which is essential for activation of type I IFN signaling. Both catalytically inactive mutants and double SAP domain mutants that are defective for DUB activity lost the capability of reducing the ubiquitinated RIG-I, TBK1, and TRAF3/6, indicating that the DUB activity of FMDV Lbpro is directly involved in the inhibition of type I IFN induction. Thus, FMDV Lpro is a multifunctional protein that blocks the IFN antiviral response through multiple distinct mechanisms: (i) Lpro cleaves the translation initiation factor eIF-4G and shuts off host cell translation, resulting in lower levels of IFN protein expression (15); (ii) Lpro represses the transcription of IFN-β not only by inhibiting activation of NF-κB but also by decreasing IRF-3/7 protein expression (10, 51); and (iii) Lpro acts as a DUB that cleaves ubiquitin chains from RIG-I, TBK1, TRAF6, and TRAF3, thereby inhibiting the activation of type I IFN signaling (this study). The identification of FMDV Lpro as a viral DUB reveals the multilayered counteracting of host defense by a picornaviral leader protein and opens new research avenues to develop effective new strategies that target Lpro for control of FMDV infections.
ACKNOWLEDGMENTS
We thank H. Kuma, S. Akira, E. W. Harhaj, T. Fujita, A. Marchese, and T. Ohta for providing reporter plasmids and expression constructs.
This work was supported by the New Century Excellent Talent Project (NCET-07-0347), the Program for Changjiang Scholars and Innovative Research Team in the University of China (IRT0726), the National Natural Science Foundation of China (30870536 and 30972761), the National S&T Major Project (2008ZX10004-015), and the Beijing Municipal Natural Science Foundation (7092075).
Footnotes
Published ahead of print on 9 February 2011.
REFERENCES
- 1. Balakirev M. Y., Jaquinod M., Haas A. L., Chroboczek J. 2002. Deubiquitinating function of adenovirus proteinase. J. Virol. 76:6323–6331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barretto N., et al. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189–15198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhoj V. G., Chen Z. J. 2009. Ubiquitylation in innate and adaptive immunity. Nature 458:430–437 [DOI] [PubMed] [Google Scholar]
- 4. Bibeau-Poirier A., Servant M. J. 2008. Roles of ubiquitination in pattern-recognition receptors and type I interferon receptor signaling. Cytokine 43:359–367 [DOI] [PubMed] [Google Scholar]
- 5. Chen Z., et al. 2007. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366:277–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Z., et al. 2007. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 81:6007–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clarke B. E., et al. 1985. Two initiation sites for foot-and-mouth disease virus polyprotein in vivo. J. Gen. Virol. 66:2615–2626 [DOI] [PubMed] [Google Scholar]
- 8. Clementz M. A., et al. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84:4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Los Santos T., de Avila Botton S., Weiblen R., Grubman M. J. 2006. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J. Virol. 80:1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Los Santos T., Diaz-San Segundo F., Grubman M. J. 2007. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 81:12803–12815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de los Santos T., et al. 2009. A conserved domain in the leader proteinase of foot-and-mouth disease virus is required for proper subcellular localization and function. J. Virol. 83:1800–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng L., et al. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361 [DOI] [PubMed] [Google Scholar]
- 13. Devaraj S. G., et al. 2007. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282:32208–32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evans P. C., et al. 2004. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem. J. 378:727–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foeger N., Glaser W., Skern T. 2002. Recognition of eukaryotic initiation factor 4G isoforms by picornaviral proteinases. J. Biol. Chem. 277:44300–44309 [DOI] [PubMed] [Google Scholar]
- 16. Frias-Staheli N., et al. 2007. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2:404–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friedman C. S., et al. 2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frieman M., Ratia K., Johnston R. E., Mesecar A. D., Baric R. S. 2009. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83:6689–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gack M. U., et al. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920 [DOI] [PubMed] [Google Scholar]
- 20. Gatot J. S., et al. 2007. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J. Biol. Chem. 282:31131–31146 [DOI] [PubMed] [Google Scholar]
- 21. Gorbalenya A. E., Koonin E. V., Lai M. M. 1991. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 288:201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grubman M. J., Baxt B. 2004. Foot-and-mouth disease. Clin. Microbiol. Rev. 17:465–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hilton L., et al. 2006. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 80:11723–11732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu M., et al. 2005. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 24:3747–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isaacson M. K., Ploegh H. L. 2009. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5:559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kayagaki N., et al. 2007. DUBA: a deubiquitinase that regulates type I interferon production. Science 318:1628–1632 [DOI] [PubMed] [Google Scholar]
- 27. Kim E. T., Oh S. E., Lee Y. O., Gibson W., Ahn J. H. 2009. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 83:12046–12056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindner H. A., et al. 2005. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79:15199–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindner H. A., et al. 2007. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 466:8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mao A. P., et al. 2010. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 285:9470–9476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mason P. W., Grubman M. J., Baxt B. 2003. Molecular basis of pathogenesis of FMDV. Virus Res. 91:9–32 [DOI] [PubMed] [Google Scholar]
- 32. Medina M., Domingo E., Brangwyn J. K., Belsham G. J. 1993. The two species of the foot-and-mouth disease virus leader protein, expressed individually, exhibit the same activities. Virology 194:355–359 [DOI] [PubMed] [Google Scholar]
- 33. Nijman S. M., et al. 2005. A genomic and functional inventory of deubiquitinating enzymes. Cell 123:773–786 [DOI] [PubMed] [Google Scholar]
- 34. Nishikawa H., et al. 2004. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 279:3916–3924 [DOI] [PubMed] [Google Scholar]
- 35. Okumura A., et al. 2008. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373:85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Piccone M. E., Rieder E., Mason P. W., Grubman M. J. 1995. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J. Virol. 69:5376–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Randall R. E., Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 38. Ratia K., et al. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 103:5717–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roberts P. J., Belsham G. J. 1995. Identification of critical amino acids within the foot-and-mouth disease virus leader protein, a cysteine protease. Virology 213:140–146 [DOI] [PubMed] [Google Scholar]
- 40. Rodrigues L., et al. 2009. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 28:1283–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlick P., Kronovetr J., Hampoelz B., Skern T. 2002. Modulation of the electrostatic charge at the active site of foot-and-mouth-disease-virus leader proteinase, an unusual papain-like enzyme. Biochem. J. 363:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schlieker C., Korbel G. A., Kattenhorn L. M., Ploegh H. L. 2005. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 79:15582–15585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skern T., Fita I., Guarne A. 1998. A structural model of picornavirus leader proteinases based on papain and bleomycin hydrolase. J. Gen. Virol. 79:301–307 [DOI] [PubMed] [Google Scholar]
- 44. Sun S. C. 2008. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8:501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun Z., Chen Z., Lawson S. R., Fang Y. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 84:7832–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sweet C. R., Conlon J., Golenbock D. T., Goguen J., Silverman N. 2007. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell. Microbiol. 9:2700–2715 [DOI] [PubMed] [Google Scholar]
- 47. Thanos D., Maniatis T. 1995. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100 [DOI] [PubMed] [Google Scholar]
- 48. Theofilopoulos A. N., Baccala R., Beutler B., Kono D. H. 2005. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23:307–336 [DOI] [PubMed] [Google Scholar]
- 49. Viswanathan K., Fruh K., DeFilippis V. 2010. Viral hijacking of the host ubiquitin system to evade interferon responses. Curr. Opin. Microbiol. 13:517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C., et al. 2009. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 10:744–752 [DOI] [PubMed] [Google Scholar]
- 51. Wang D., et al. 2010. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 399:72–78 [DOI] [PubMed] [Google Scholar]
- 52. Wertz I. E., et al. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430:694–699 [DOI] [PubMed] [Google Scholar]
- 53. Wilkinson K. D., et al. 1995. Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 34:14535–14546 [DOI] [PubMed] [Google Scholar]
- 54. Zheng D., Chen G., Guo B., Cheng G., Tang H. 2008. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 18:1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]