

Abstract
Induction of apoptosis in cells infected by Sendai virus (SeV), which triggers the cytosolic RIG-I pathway, requires the presence of interferon regulatory factor 3 (IRF-3). Independent of IRF-3's transcriptional role, a novel IRF-3 activation pathway causes its interaction with the proapoptotic protein Bax and its mitochondrial translocation to induce apoptosis. Here we report that two other RNA viruses, vesicular stomatitis virus (VSV) and encephalomyocarditis virus (EMCV), may also activate the same pathway. Moreover, cytosolic DNA, produced by adenovirus or introduced by transfection, activated the pathway in an RNA polymerase III-dependent fashion. To evaluate the contribution of this newly discovered apoptotic pathway to the host's overall antiviral response, we measured the efficiencies of replication of various viruses in vitro and viral pathogenesis in vivo, using cells and mice that are selectively deficient in components required for the apoptotic pathway of IRF-3. Our results clearly demonstrate that the IRF-3/Bax-mediated apoptotic signaling branch contributes significantly to the host's protection from viral infection and consequent pathogenesis.
INTRODUCTION
The cellular innate immune response is the first line of host defense against viruses and other pathogens. Innate immune response genes, including interferon and interferon-stimulated genes, play essential roles in the host antiviral response. Double-stranded RNA (dsRNA), generated during infection with both RNA and DNA viruses, is a strong inducer of host antiviral responses. Mammals have several families of pattern recognition receptors (PRRs), e.g., Toll-like receptors (TLRs), RIG-I-like helicases (RLHs), and Nod-like receptors (NLRs), to recognize viral dsRNA (2, 15, 17, 27). The cytosolic RLH receptors RIG-I, MDA-5, and LGP2, upon activation by dsRNA, interact with the mitochondrial adaptor protein IPS-1, which recruits signaling complexes consisting of TRAF3 and TBK1/IKKε, and activate interferon regulatory factor 3 (IRF-3) (16–18, 29, 30). IRF-3 remains in an inactive monomeric state in the cytosol; its phosphorylation by TBK1/IKKε leads to dimerization and translocation to the nucleus, where it binds to the ISRE sites in the promoters of antiviral genes and drives their transcription (21).
To suppress viral replication and spread, host cells often undergo premature cell death by triggering apoptosis. Apoptosis is therefore considered a potent antiviral defense mechanism by which infected cells are eliminated from the host. IRF-3 has been shown to be a key player in the induction of apoptosis by Sendai virus (SeV), a member of the paramyxovirus family (5, 11, 25). SeV infection triggers cytosolic RIG-I signaling, activating IRF-3, which mediates cellular apoptosis in addition to inducing antiviral genes. SeV-induced apoptosis is a relatively slow process, and inhibition of phosphatidylinositol 3-kinase activity accelerates the apoptotic effect (25). Our studies revealed a physiological connection between apoptosis and SeV replication: cells deficient in IRF-3 (or RIG-I activity) fail to trigger apoptosis and establish viral persistence (25). IRF-3-deficient cells continuously produce infectious virus particles, and reintroduction of IRF-3 into these cells leads to apoptosis. IRF-3 is therefore a critical cellular factor that dictates cell fate during SeV infection by causing either apoptotic cell death or viral persistence.
We have recently shown that IRF-3 activated by dsRNA exhibits its proapoptotic activity by a distinct mechanism that is independent of its role as a transcription factor (5, 6). Although RIG-I signaling is critical, the apoptotic pathway requires additional proteins, TRAF2 and TRAF6, which are specific for the IRF-3-mediated apoptotic, but not transcriptional, pathway. Activated IRF-3 executes its direct apoptotic effect by interacting with the proapoptotic protein Bax and translocating to the mitochondrial membrane. The interaction of IRF-3 and Bax, followed by their translocation to the mitochondria, triggers the mitochondrial intrinsic apoptotic pathway by the release of cytochrome c into the cytosol and subsequent activation of caspase-9 (5). Our study demonstrated that IRF-3 interacts with cytosolic Bax by using a BH3-like domain. As expected, Bax deficiency causes inhibition of viral apoptosis.
Infection by DNA viruses can also trigger efficient innate immune responses by the induction of antiviral genes and proinflammatory cytokines. DNA viruses often lead to the accumulation of cytosolic DNA, which can be recognized by the cytoplasmic DNA sensor DAI and activate downstream signaling by interacting with TBK1 and IRF-3, leading to the induction of interferons (IFNs) (31). However, subsequent gene knockout studies demonstrated that DAI deficiency had no effect on DNA-induced IFN production, indicating redundant roles of additional sensors in the recognition of viral DNA. A recent report identified LRRFIP1 as a sensor of cytosolic dsDNA for the induction of IFN by a unique activation mechanism of IRF-3 (34). Moreover, two independent studies have identified RNA polymerase III as a sensor of cytosolic dsDNA whose RNA transcript activates RIG-I-mediated antiviral responses (1, 7). Cytosolic dsDNA can also be recognized by AIM2, which can cause inflammasome-mediated activation of proinflammatory cytokines; however, this pathway is apparently independent of IFN induction (4, 9, 13).
In the present study, we investigated whether cytosolic dsDNA produced by DNA viruses, like cytosolic dsRNA produced by RNA viruses, could also trigger the IRF-3-mediated cellular apoptotic response. We observed that RIG-I-mediated activation of IRF-3 was required for the apoptotic effect of adenovirus infection. Using genetically defective cellular and mouse models, we evaluated the role of the newly discovered IRF-3-mediated apoptotic pathway in inhibiting virus replication and viral pathogenesis. We measured the efficiencies of replication of several viruses in vitro and viral pathogenesis in vivo, using cells and mice that are selectively deficient in components required for the apoptotic but not the gene induction branch of IRF-3 signaling. Our results clearly demonstrate that the IRF-3/Bax axis contributes significantly to host protection from viral infection and consequent pathogenesis.
MATERIALS AND METHODS
Cells and reagents.
HT1080, HT1080/si-IRF-3, HT1080/RIG-Ic, P2.1, and IRF-3−/−, TRAF2−/−, TRAF6−/−, and Bax−/− MEF cells have been described before (5, 25). The cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Primary peritoneal macrophages and bone marrow-derived dendritic cells were isolated from wild-type (wt) and Bax−/− mice by using procedures described elsewhere (5). Adenovirus strain Ad5 was a kind gift from Thomas Shenk (Princeton University, NJ), green fluorescent protein-expressing vesicular stomatitis virus (GFP-VSV) was a kind gift from Curt Horvath (Northwestern University, Chicago, IL), and encephalomyocarditis virus (EMCV) was a kind gift from Robert Silverman (Lerner Research Institute, Cleveland, OH). Antibodies against human IRF-3 were a kind gift from Michael David (University of California, San Diego, CA), Sendai virus C protein was a kind gift from Atsushi Kato (National Institute of Infectious Diseases, Tokyo, Japan), P54, P56, and P60 were raised in our laboratory, poly(ADP-ribose) polymerase (PARP), caspase-3, and caspase-1 were from Cell Signaling, Bax and HDAC1 were from Santa Cruz, porin and tubulin were from Calbiochem, and actin was from Sigma. Poly(I:C) was purchased from GE Healthcare, and poly(dA-dT), cycloheximide (CHX), actinomycin D (ActD), and 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB) were purchased from Sigma-Aldrich. FuGENE 6 and Lipofectamine 2000 were purchased from Roche Diagnostics and Invitrogen, respectively. An inhibitor of RNA polymerase III (ML-60218, a cell-permeating indazolo-sulfonamide compound) and caspases were obtained from Calbiochem. Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining reagent was obtained from Promega.
Poly(I:C) and poly(dA-dT) transfections.
Poly(I:C) transfections were done as described previously (5). Poly(dA-dT) was resuspended in sterile phosphate-buffered saline (PBS) and transfected using Lipofectamine 2000 reagent. Briefly, 2 μg of poly(dA-dT) (or as indicated in the figure legends) was complexed with 5 μl of Lipofectamine 2000 in Opti-MEM and applied to the cells.
Caspase activity.
Caspase-3/7 activity of the cell lysates was determined as described previously (5). Briefly, the cells were treated or infected with viruses as indicated, all treatments were performed on biological triplicates, and the results shown represent the averages and standard deviations for the three independent samples. After the treatment, the cells were lysed in lysis buffer containing protease inhibitors, and protein extracts were used to measure caspase activity, using the Apo-ONE homogeneous caspase-3/7 assay according to protocols provided by the manufacturer (Promega, Madison, WI). The fold induction of caspase activity was obtained by subtracting the caspase activity in treated samples from the value obtained from untreated cells and converted to the amount per μg of protein extract. Induction of caspase activity for wt cells was considered 100, and all other values were normalized to this value.
For measurement of caspase-1 activity, the cells were lysed in lysis buffer containing protease inhibitors, and cell lysates were used to measure caspase-1 activity, using a caspase-1 fluorometric assay reagent (R&D) according to the protocol provided by the manufacturer.
Cell fractionation.
Mitochondrial and cytosolic fractions were isolated using a mitochondrial isolation kit (Pierce Biotechnology) following the manufacturer's instructions. Isolated mitochondrial fractions were washed with PBS and extracted in lysis buffer for analysis.
siRNA experiments.
HT1080/si-IRF-3 cells were described previously (25). A small interfering RNA (siRNA) against Bax was obtained from Dharmacon and was transfected using DharmaFECT 4 reagent for 48 h, after which the cells were used for transfection of poly(dA-dT), as indicated in the figure legends.
Western blotting.
Western blotting was performed as described before (5). Briefly, cells were lysed in 50 mM Tris buffer, pH 7.4, containing 150 mM NaCl, 0.1% Triton X-100, 1 mM sodium orthovanadate, 10 mM NaF, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, and protease inhibitors (Roche). The total protein extracts were analyzed by SDS-PAGE followed by Western blotting.
Immunostaining.
GFP-IRF-3-expressing IRF-3−/− MEF cells were either mock infected or infected with EMCV, and Mitotracker Red (Invitrogen) was used only during the last hour of the infection. Cells were fixed in 4% paraformaldehyde and then permeabilized by 0.1% Triton X-100. Fixed cells were analyzed by confocal microscopy. All images were taken by confocal laser scanning microscopy, and the images were then processed by Adobe Photoshop software.
Virus infections and mice.
The following protocol was used for all virus infections described in this study. The cells were washed two times with virus infection medium (DMEM supplemented with 2% FBS; no serum was used for adenovirus infection) and then placed in a minimal amount of virus infection medium containing the virus (at the multiplicities of infection [MOIs] indicated in the figure legends). Cells were incubated with virus for 1 h, with gentle agitation every 10 min. The virus was removed, and cells were washed twice with complete medium. The cells were placed in complete medium until they were harvested. Sendai virus (Cantell strain) was obtained from Charles Rivers Spafas (Preston, CT), and all other viruses were obtained as mentioned before. For virus titration, previously described procedures were followed (5). Adenovirus titration was carried out in A549 cells by a standard plaque assay. Bax−/− mice were obtained from Jackson Laboratories and maintained by breeding the heterozygous mice. Wild-type and Bax−/− mice of 6 to 8 weeks of age were used for EMCV infection. Briefly, the mice were infected with EMCV (500 PFU) intraperitoneally, monitored every 12 h for any symptoms of paralysis or disease, and subsequently euthanized. For quantification of the infectious virus particles in the organs, tissues were collected at 48 h post-EMCV infection and homogenized in PBS, and viral titers were determined by plaque assay. All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee.
RESULTS
Adenovirus infection and cytosolic DNA trigger RIG-I/IRF-3-mediated apoptotic response.
We recently demonstrated that infection of cells with RNA viruses triggers cellular apoptosis by RLH signaling, which causes mitochondrial translocation of IRF-3 and subsequent activation of the intrinsic apoptotic pathway (5). To establish the generality of the phenomenon, we tested whether DNA viruses could also trigger a similar apoptotic response. Adenovirus, a double-stranded DNA virus, caused cell death in a dose-dependent manner (Fig. 1A); this cell death was apoptotic, as demonstrated by the cleavage of PARP, a prominent marker of apoptosis (Fig. 1B). Adenovirus infection also led to transcriptional activation of IRF-3, as indicated by the induction of P60, an IRF-3-dependent gene product, although with slower kinetics (Fig. 1C). Infection with adenovirus is known to cause accumulation of cytosolic DNA, which can activate cellular innate immune responses. Indeed, transfection of a dsDNA, poly(dA-dT), triggered efficient cell death in human cells (Fig. 1D) and caused rapid cleavage of PARP, as early as 4 h posttransfection (Fig. 1E). dsDNA also caused transcriptional activation of IRF-3; consequently, proteins encoded by IRF-3-dependent genes, such as P56, were strongly induced (Fig. 1E). A dsRNA, poly(I:C), served as the positive control in these experiments.
Fig. 1.

Induction of apoptosis by adenovirus infection and cytosolic DNA. (A) HT1080 cells were infected with adenovirus (Ad5) at the indicated MOIs. Representative culture fields are shown for 96 h postinfection. (B) HT1080 cells were infected with adenovirus (Ad5) (at an MOI of 10) for the indicated times, when the cell lysates were analyzed for cleaved PARP (C-PARP). (C) HT1080 cells were infected with adenovirus (at an MOI of 10) for the indicated times, when the cell lysates were analyzed for induction of P60, an IRF-3-dependent gene product. (D) HT1080 cells were transfected with poly(dA-dT) [p(dA-dT)] at 1 and 5 μg/ml, as indicated, or with poly(I:C) [p(I:C)] at 2 μg/ml. Representative culture fields are shown for 24 h posttransfection. (E) HT1080 cells were transfected with poly(dA-dT) (1 μg/ml) or poly(I:C) (2 μg/ml) by use of Lipofectamine 2000 (Lipo), and at the indicated times, cell lysates were analyzed for PARP cleavage (FL, full length; CL, cleaved) and induction of P56, an IRF-3-dependent gene product, by Western blotting.
The observed induction of apoptosis by cytosolic DNA and adenovirus infection led us to assess a possible need for IRF-3 in the process. Apoptosis triggered by adenovirus required the presence of IRF-3. HT1080 cells were killed by adenovirus infection, but not when IRF-3 expression was ablated by siRNA (Fig. 2A). Similarly, HT1080-derived P2.1 cells, which are low in IRF-3 expression, were resistant to cytosolic DNA-induced cell death (see Fig. S1A in the supplemental material). To investigate the role of the signaling pathway that activates IRF-3 in the observed apoptotic effect, we tested a human cell line that expresses a dominant-negative mutant of RIG-I (RIG-Ic), and these cells were resistant to cell death caused by adenovirus or cytosolic DNA (Fig. 2A; see Fig. S1A). As expected, PARP cleavage did not occur in the absence of IRF-3 and RIG-I action in both adenovirus-infected and dsDNA-transfected cells (Fig. 2B, D, and E; see Fig. S1B). However, staurosporine, an unrelated stimulus, did induce apoptosis in HT1080/si-IRF-3 cells, indicating the involvement of IRF-3 in RIG-I-induced apoptosis only (Fig. 2C and D). These results suggest that RIG-I signaling-mediated activation of IRF-3 is required for dsDNA-induced apoptosis, similar to dsRNA-induced apoptosis. Adenovirus replication, measured by the number of infectious virus particles produced, was significantly higher in cells with either ablated IRF-3 expression or expression of a dominant-negative mutant of RIG-I (RIG-Ic) than in wt control cells (Fig. 2F). It is unclear how DNA activates RIG-I; there is evidence that RIG-I can either directly bind to cytosolic DNA or recognize an intermediate dsRNA, transcribed by DNA-dependent RNA polymerase III, to mediate transcriptional activation of IRF-3 (1, 7, 8). In our system, the second mechanism was operative, as a chemical inhibitor of RNA polymerase III (ML-60218, an indazolo-sulfonamide compound), but not two chemical inhibitors of RNA polymerase II, ActD and DRB, blocked dsDNA-induced apoptosis and gene induction (Fig. 3A and B). To test whether IRF-3-dependent apoptosis by dsDNA might be driven by IRF-3-induced proapoptotic proteins, we used a pharmacological inhibitor of translation, CHX, which blocks the synthesis of any new protein. As expected, in the presence of CHX, P56 was not induced by dsDNA; however, apoptosis triggered by dsDNA (or dsRNA) was unaffected by blocking the induced protein synthesis (Fig. 3C and D). These results clearly show that dsDNA-induced apoptosis is independent of de novo protein synthesis but dependent on the activity of RNA polymerase III.
Fig. 2.

Adenovirus- and cytosolic DNA-induced apoptosis is dependent on RIG-I and IRF-3. (A) HT1080 (wt), HT1080/si-IRF-3 (IRF-3 knockdown), or HT1080/RIG-Ic (expressing a dominant-negative mutant of RIG-I) cells were either mock infected or infected with adenovirus (at an MOI of 10). Pictures of the culture fields were taken at 96 h postinfection. (B) HT1080, HT1080/si-IRF-3, or HT1080/RIG-Ic cells were infected with adenovirus (at an MOI of 10) for 96 h, when the cell lysates were analyzed for cleaved PARP (C-PARP) by Western blotting. (C) HT1080/si-IRF-3 cells or HT1080/si-con cells (cells expressing a nontargeting short hairpin RNA [Sigma] by lentiviral transduction) were either left untreated (Unt), transfected with poly(dA-dT), or treated with staurosporine (1 μM) (Stauro). Representative culture fields are shown for 18 h posttreatment. (D) HT1080/si-IRF-3 or HT1080/si-con cells were either left untreated, transfected with poly(dA-dT), or treated with staurosporine (1 μM). Cell lysates from 12 h posttreatment were analyzed for PARP cleavage by Western blotting. (E) HT1080 (wt) or HT1080/RIG-Ic cells were transfected with poly(dA-dT), and at 8 h posttransfection, the cell lysates were analyzed for PARP cleavage and induction of P56 by Western blotting. (F) HT1080 (wt), HT1080/si-IRF-3, or HT1080-RIG-Ic cells were infected with adenovirus (at an MOI of 10), and at 4 days postinfection, the cells and the culture medium were analyzed for infectious virus particles.
Fig. 3.

dsDNA-induced apoptosis is mediated by RNA polymerase III but independent of de novo protein synthesis. (A) HT1080 cells were pretreated with an inhibitor of RNA polymerase III (ML-60218) at the indicated dose or with a vehicle control (dimethyl sulfoxide [DMSO]) overnight and then transfected with poly(dA-dT). Representative culture fields are shown for 24 h posttransfection. (B) HT1080 cells were pretreated with an inhibitor of RNA polymerase III (Pol III-inh; 20 μM), actinomycin D (ActD; 0.5 μg/ml), or DRB (50 μM) and then transfected with poly(dA-dT) for 8 h, when the cell lysates were analyzed for cleaved PARP and induction of P56 by Western blotting. (C) HT1080 cells were transfected with poly(dA-dT) or poly(I:C) in the absence or presence of CHX (1 and 5 μg/ml, as indicated), and 8 h posttransfection, the cell lysates were analyzed for cleaved PARP and induction of P56 by Western blotting. (D) HT1080 cells were transfected with poly(dA-dT) in the absence or presence of CHX (1 μg/ml, as in panel C). Representative culture fields are shown for 18 h posttransfection.
Cytosolic DNA-induced apoptosis is mediated by IRF-3- and Bax-mediated activation of the intrinsic mitochondrial pathway.
In the next series of experiments, we further characterized the nature of the apoptotic pathway triggered by dsDNA. In some cell types, dsDNA is known to activate inflammatory responses by activating caspase-1. However, in HT1080 cells, upon dsDNA stimulation, we observed no activation of caspase-1 but efficient activation of caspase-3 (Fig. 4A). Consequently, inhibition of caspase-9, an upstream activator of caspase-3, but not that of caspase-1, blocked apoptosis (Fig. 4B). The efficiency of the caspase-1 inhibitor was verified in primary peritoneal macrophages treated with lipopolysaccharide-ATP (LPS-ATP), a strong inducer of caspase-1 activity (see Fig. S2 in the supplemental material). These results indicate that dsDNA-induced apoptosis is mediated by the intrinsic mitochondrial pathway. We previously demonstrated that in RNA virus-infected cells, the intrinsic mitochondrial pathway can be activated by the translocation of activated IRF-3 to mitochondria; similarly, the levels of IRF-3 were increased in the mitochondrial fractions of the cells transfected with dsDNA (Fig. 5A). Moreover, dsDNA caused concomitant translocation of Bax to mitochondria (Fig. 5A). When we knocked down Bax expression in cells, dsDNA could not trigger apoptosis (Fig. 5B). These results demonstrate that both cytosolic RNA and DNA trigger mitochondrial translocation of IRF-3 and Bax, which mediates the apoptotic response.
Fig. 4.
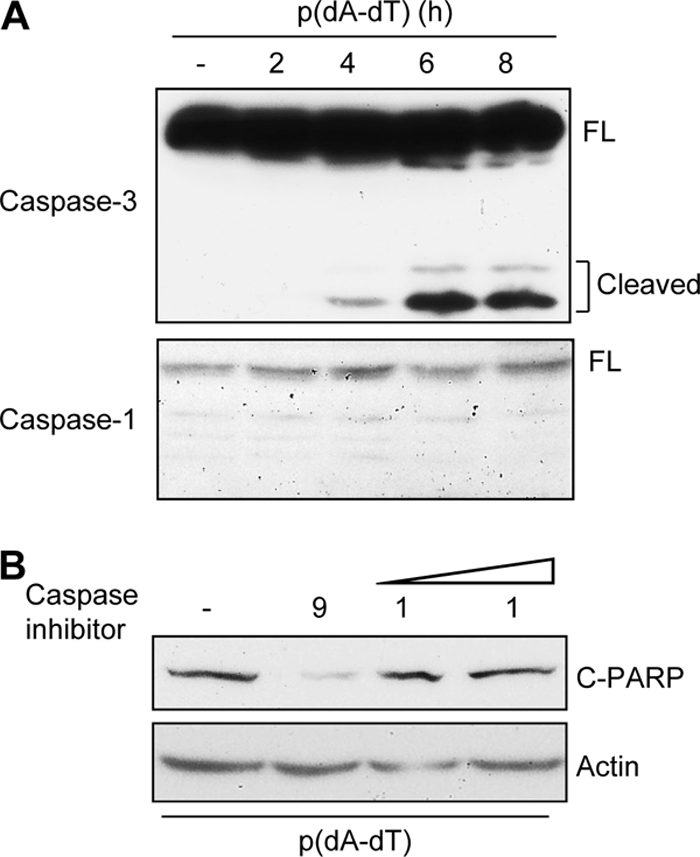
Apoptosis is triggered by activation of caspase-9 and caspase-3 but not caspase-1. (A) HT1080 cells were transfected with poly(dA-dT) (1 μg/ml) for the indicated times, and the cell lysates were analyzed for the cleavage of caspase-3 and caspase-1 by Western blotting. (B) HT1080 cells were transfected with poly(dA-dT) in the absence or presence of an inhibitor of caspase-9 (10 μg/ml) or caspase-1 (10 and 25 μg/ml, as shown by increasing dose), and at 8 h posttransfection, cell lysates were analyzed for cleaved PARP by Western blotting.
Fig. 5.

dsDNA-induced apoptosis requires translocation of IRF-3 and Bax to mitochondria. (A) HT1080/si-IRF-3 cells reconstituted with wt IRF-3 were transfected with poly(dA-dT) for the indicated times, when the mitochondrial fractions were isolated and analyzed for IRF-3, Bax, porin (a mitochondrial marker protein), tubulin (a cytosolic marker protein), and HDAC1 (a nuclear marker protein) by Western blotting. (B) RNA interference-mediated knockdown of Bax was carried out by transfection of HT1080 cells with an siRNA against Bax (Dharmacon) or a nontargeting siRNA (Con) (Dharmacon), using DharmaFECT 4 reagent (Dharmacon). Forty-eight hours later, these cells were transfected with poly(dA-dT) for 8 h, and the cell extracts were analyzed for cleaved PARP by Western blotting.
IRF-3-mediated apoptosis suppresses virus replication.
Once we established that both dsDNA and dsRNA in cytosol could trigger the newly discovered apoptotic pathway, we wanted to investigate whether this pathway was a part of the cellular antiviral response. For experimental convenience, we chose to focus on its role in RNA virus replication and resultant pathogenesis in mice. In the first set of experiments, we examined the status of this pathway in various primary mouse cells. In IRF-3−/− MEFs, dsRNA did not induce apoptosis as measured by TUNEL assay (Fig. 6A). The involvement of Bax was evaluated in primary immune cells derived from Bax−/− mice. Activation of RIG-I signaling by SeV infection or dsRNA transfection effectively triggered apoptosis in primary bone marrow-derived dendritic cells or peritoneal macrophages derived from wt mice; however, the cells derived from Bax−/− mice were resistant to apoptosis (Fig. 6B and C). On the other hand, gene induction by RIG-I signaling was unaffected in the Bax−/− immune cells (Fig. 6D and E), indicating that Bax is specifically involved in the apoptotic, not gene induction, pathway of RIG-I signaling. The role of the apoptotic pathway in impairing viral gene expression and replication was assessed by using MEFs derived from mice deficient in different components of the pathway. In SeV-infected IRF-3−/− cells, more viral C protein was expressed than in wt cells (Fig. 7A). The specific contribution of the IRF-3-driven apoptotic pathway to inhibition of viral gene expression was analyzed in MEFs deficient in TRAF2, TRAF6, and Bax, proteins that are required for IRF-3-mediated apoptosis but not antiviral gene induction (5). All three knockout MEFs had enhanced levels of SeV C protein expression (Fig. 7B and C), indicating that the apoptotic branch contributes significantly to the host's ability to suppress viral gene expression. Consequently, Bax deficiency led to elevated production of infectious viruses: MEFs missing Bax produced about 40 times more SeV than wt cells did (Fig. 7D). Similarly, primary bone marrow-derived dendritic cells produced more infectious SeV than wt cells did (Fig. 7E).
Fig. 6.

Bax is required for apoptosis, but not for gene induction, in mouse primary cells. (A) wt or IRF-3−/− MEF cells were grown on coverslips and either left untreated or transfected with poly(I:C) for 16 h, after which the cells were stained with fluorescein isothiocyanate (FITC)-conjugated TUNEL reagent and analyzed microscopically. Representative culture fields are shown. Average TUNEL positivity rates for poly(I:C)-transfected cells in multiple fields were 36.1% ± 3.6% for wt cells and 1.8% ± 0.6% for IRF-3−/− cells. DAPI, 4′,6-diamidino-2-phenylindole. (B) Primary bone marrow-derived dendritic cells from wt or Bax−/− mice were infected with SeV (at an MOI of 10), and culture fields were photographed after 96 h. (C) Primary peritoneal macrophages from wt or Bax−/− mice were transfected with poly(I:C) for 16 h, and caspase activity was measured. (D) Primary bone marrow-derived dendritic cells from wt or Bax−/− mice were transfected with poly(I:C) for 8 h, and then induction of P54 was analyzed by Western blotting. (E) Primary peritoneal macrophages from wt or Bax−/− mice were transfected with poly(I:C), and induction of P54 was analyzed by Western blotting after 8 h.
Fig. 7.

Deficiency of apoptotic components leads to enhanced SeV replication. (A) wt or IRF-3−/− MEF cells were infected with SeV for 16 h, after which the cell lysates were analyzed for SeV C protein expression by Western blotting. (B) wt, TRAF2−/−, or TRAF6−/− MEF cells were infected with SeV for 16 h, after which the cell lysates were analyzed for SeV C protein expression by Western blotting. (C) wt or Bax−/− MEF cells were infected with SeV for the indicated times, after which the cell lysates were analyzed for SeV C protein expression by Western blotting. (D) wt or Bax−/− MEF cells were infected with SeV (at an MOI of 10) for the indicated times. The cells and the culture medium were analyzed together to determine the titer of infectious virus particles by plaque assay. (E) Primary bone marrow-derived dendritic cells from wt or Bax−/− mice were infected with SeV (at an MOI of 10). The cells and the culture medium were analyzed to determine the titer of infectious virus particles.
The generality of our observation was tested using viruses of other families. A recombinant VSV expressing GFP showed enhanced replication in Bax-deficient MEFs, primary dendritic cells, and macrophages (Fig. 8A, B, and C). Similarly, primary dendritic cells derived from IRF-3−/− mice showed enhanced VSV replication (Fig. 8B). The enhanced expression of GFP was not due only to the accumulation of GFP proteins in the surviving cells, as the Bax-deficient cells produced more infectious virus particles from infected dendritic cells and macrophages, as analyzed by plaque assays (Fig. 8B and C).
Fig. 8.

Deficiency of apoptotic components leads to enhanced VSV replication. (A) wt or Bax−/− MEF cells were infected with GFP-VSV (at an MOI of 10). Representative GFP-positive culture fields are shown for 12 h postinfection. (B) Primary bone marrow-derived dendritic cells (DCs) from wt or Bax−/− mice were infected with GFP-VSV (at an MOI of 10) for the indicated times, after which the cells and culture medium were analyzed to determine the titer of infectious virus particles by plaque assay. Representative GFP-positive culture fields for wt, IRF-3−/−, and Bax−/− DCs infected with GFP-VSV are shown for 12 h postinfection. (C) Primary peritoneal macrophages (Mφ) derived from wt or Bax−/− mice were infected with GFP-VSV (at an MOI of 10), and the cells and culture medium were analyzed at 16 h postinfection to determine the viral titer. Representative GFP-positive culture fields are shown for 16 h postinfection.
Bax deficiency leads to enhanced EMCV replication and pathogenesis in mice.
To study whether a direct correlation exists between the apoptotic effect and viral pathogenesis, we chose the picornavirus EMCV because it causes lethal infection in mice. We observed that EMCV infection caused activation of IRF-3 in mouse cells: IRF-3 was translocated to nuclear and mitochondrial fractions upon EMCV infection, as shown by microscopic data (Fig. 9A). Also, EMCV failed to induce an apoptotic effect in the absence of Bax. Upon EMCV infection, Bax−/− MEF cells showed a marked reduction in caspase activation compared to the wt control cells (Fig. 9B). Consequently, there was enhanced production of infectious virus by Bax−/− cells compared to wt control cells (Fig. 9C). To examine the role of the Bax-mediated apoptotic antiviral branch in preventing pathogenesis, we used Bax−/− mice, which have no defect in the IRF-3-mediated antiviral gene induction branch. These mice were highly sensitive to EMCV-mediated pathogenesis; at the dose of virus tested, all experimental Bax−/− mice succumbed by day 4 of EMCV infection, whereas 60% of wt mice were still alive at this time (Fig. 9D). The difference in lethality was due to enhanced replication of the virus in Bax−/− mice: the viral titer in the brains of Bax−/− mice was 150 times higher than that in wt mice (Fig. 9E). These results strongly indicate that the Bax-mediated apoptotic pathway contributes significantly to the host's ability to block virus replication and resultant pathogenesis.
Fig. 9.

Bax deficiency leads to enhanced EMCV replication and pathogenesis in mice. (A) IRF-3−/− MEF cells expressing GFP-IRF-3 were either mock infected or infected with EMCV (at an MOI of 1), and at 8 h postinfection (Mitotracker Red was used only for the final hour of infection), cells were fixed and analyzed by confocal microscopy. The EMCV-infected cell shows clear nuclear localization of IRF-3 and strong colocalization of GFP with Mitotracker Red (yellow staining), indicating mitochondrial localization. (B) wt or Bax−/− MEF cells were infected with EMCV (at an MOI of 0.01), and the cell lysates were analyzed for caspase activity at 6 h postinfection. (C) wt or Bax−/− MEF cells were infected with EMCV (at an MOI of 0.01), and the cells and culture medium were analyzed to determine the titer of infectious virus particles at 6 h postinfection. (D) Six- to 8-week-old wt (n = 11) or Bax−/− (n = 8) mice were infected with EMCV (500 PFU) intraperitoneally and assessed for survival (P < 0.005). (E) Six- to 8-week-old wt or Bax−/− mice (five mice for each genotype) were infected with EMCV as described above, and brain tissues were collected at 48 h postinfection, homogenized in PBS, and analyzed to determine the titer of infectious virus particles by plaque assay (viral titers are presented as PFU/g of brain tissue).
DISCUSSION
The results presented here demonstrate that infection by DNA viruses, like that by RNA viruses, can trigger an IRF-3-dependent cellular apoptotic pathway (Fig. 10). DNA virus-induced apoptosis is also dependent on mitochondrial activation by IRF-3 and Bax. Moreover, the newly discovered IRF-3-induced apoptotic pathway contributes significantly to the overall antiviral responses of the host (Fig. 10). Multiple innate immune signaling pathways are activated upon infection by DNA viruses and other microbes. Endosomal TLR9, cytosolic DAI, LRRFIP1, RNA polymerase III, AIM2, etc., have been implicated in the recognition of dsDNA, which is often generated by DNA virus infection. Although dsDNA-mediated signaling can activate IRF-3- and NF-κB-dependent gene expression or inflammasome formation, its role in induction of apoptosis has not been studied. Our results clearly indicate that infection by adenovirus or transfection of dsDNA can activate cellular apoptosis. In addition to its role in the induction of genes, IRF-3 plays a critical role in DNA virus-induced apoptosis. Cytosolic RIG-I signaling is essential for the apoptotic effect, indicating that a RIG-I-mediated activation of IRF-3 is required for DNA-dependent apoptosis. Our results further suggest that the activity of RNA polymerase III is critical for the apoptotic pathway induced by IRF-3, identifying RNA polymerase III as a mediator of the cellular response to cytosolic dsDNA. Our observation supports the recently revealed role of RNA polymerase III in the dsDNA-induced transcriptional activity of IRF-3. In addition to the induction of antiviral genes, IRF-3 drives the transcription of a variety of proapoptotic genes (3, 10, 26). However, the apoptotic effect of cytosolic dsDNA (and dsRNA) does not require the synthesis of any new induced protein or mRNA, thus demonstrating that a distinct apoptotic pathway is triggered by IRF-3. This distinction is further supported by the fact that TLR3 signaling, which activates IRF-3 to transcribe similarly induced genes, is incapable of inducing an apoptotic effect, as the apoptotic pathway is not activated by TLR3. Because cellular protein synthesis is often shut off by virus infection (32, 33), the existence of a transcription-independent apoptotic mechanism ensures that the cell death-mediated antiviral response is still operative in the absence of the host protein synthesis machinery.
Fig. 10.

IRF-3- and Bax-mediated apoptosis acts as a host antiviral response. Infection of cells by DNA or RNA viruses triggers the cellular apoptotic pathway by RIG-I signaling-mediated activation of IRF-3 and Bax to cause their translocation to mitochondria, which in turn activates the apoptotic pathway. Cytosolic DNA, generated upon infection by DNA viruses or other microbes, activates RIG-I signaling via an intermediate dsRNA generated by RNA polymerase III. The IRF-3-mediated apoptotic pathway, in addition to the gene induction pathway, significantly contributes to the host antiviral response.
Activation of IRF-3 has been described primarily as its phosphorylation, dimerization, and nuclear translocation upon signaling by dsRNA receptors, e.g., TLR3 and RLHs, and dsDNA sensors, e.g., DAI, followed by its binding to promoters containing ISRE binding sites. Here we have identified a new activation mechanism of IRF-3 by dsDNA signaling, which causes its translocation to the mitochondrial membrane. These results indicate a commonality of IRF-3 activation by both dsRNA and dsDNA in triggering the mitochondrial apoptotic pathway. The DNA-mediated apoptotic pathway also requires the presence of Bax, which cotranslocates to the mitochondrial membrane with IRF-3, in a signal-dependent manner. DNA signaling in a variety of cell types has been associated with activation of caspase-1 via interaction of AIM2 and the inflammasome (9, 13). However, such a mechanism was not operative in our system: no caspase-1 activation was detected, and inhibition of caspase-1 had no effect on the DNA-induced apoptosis. Moreover, caspase-9 activity was essential for the activation of the executioner caspase, caspase-3, supporting the role of the mitochondrial apoptotic pathway.
The IRF-3-mediated antiviral response is essential for inhibition of virus replication and prevention of viral pathogenesis. Our previous studies demonstrated a critical role of IRF-3 in inducing either cellular apoptosis or viral persistence in SeV-infected cells (25). IRF-3-deficient cells remain persistently infected by SeV and produce infectious virus continuously. IRF-3 knockout mice are highly susceptible to infection by different viruses. IRF-3-dependent induction of interferon genes and other antiviral genes plays a critical role in the inhibition of viral pathogenesis. However, the results presented here demonstrate that the IRF3/Bax-mediated apoptotic pathway plays a critical role in mediating antiviral responses as well. Cells defective in the apoptotic but not the gene-inducing pathway showed a significant increase in viral replication. We chose a group of RNA viruses on the basis of their ability to activate the RIG-I signaling pathway. The replication of these viruses was augmented substantially in the absence of the IRF-3-induced apoptotic pathway. SeV replication was significantly enhanced in cells deficient in the components of the IRF-3-mediated apoptotic pathway. Replication of VSV was significantly enhanced in the absence of Bax, a specific component of the IRF-3-induced apoptotic pathway. Our results identified a new role of Bax in EMCV-mediated apoptosis and viral pathogenesis. Bax-deficient MEFs were refractory to EMCV-induced apoptosis and consequently produced higher levels of progeny virus, and Bax-deficient mice, which are defective only in the apoptotic pathway activated by IRF-3, were more susceptible to EMCV-mediated lethal infection. These observations demonstrated a contribution of the IRF-3/Bax-mediated apoptotic pathway to the host's overall protection against virus infection. Although IRF-3−/− mice are susceptible to EMCV infection, these mice produce similar levels of interferons to those in wt mice (12). Hence, it is intriguing to speculate that the enhanced viral pathogenesis observed in the IRF-3−/− mice might be due primarily to the absence of IRF-3's apoptotic effect.
Apoptosis, as a host antiviral strategy to eliminate virus-infected cells, is often counteracted by virus-encoded antiapoptotic proteins that allow the cells to complete the replication cycle before their destruction. For example, cytomegalovirus uses specific viral proteins to block cellular apoptosis by inhibiting the activation of Bax in response to various stimuli (22). It will be interesting to investigate in future studies whether such viral inhibitory mechanisms exist to target the IRF-3/Bax-mediated apoptotic pathway. The host often uses apoptosis to block viral spread. For example, virus-induced apoptosis of peripheral neuronal cells can be a protective host response that blocks transmission of herpes simplex virus 2 (HSV-2) to the central nervous system (CNS) (24). In contrast, late in infection, apoptotic death of the infected cell may facilitate virus egress and spread of the infection; thus, the timing of apoptosis is a critical factor in determining whether it is pro- or antiviral. Therefore, it is not surprising that inhibition of viral apoptosis has mixed effects on viral pathogenesis. Caspase-3−/− mice infected with West Nile virus have less severe disease and apoptosis than wt mice, but the CNS viral titers remain similar (28). Bax−/− mice are highly susceptible to infection by Sindbis and rabies viruses; however, viral replication in the brain is similar to that in wt mice (14, 19, 20). Apoptosis and disease severity are reduced and viral replication is decreased in mice infected with a Bcl-2-expressing recombinant Sindbis virus (20). Influenza virus, on the other hand, requires Bax for efficient virus replication, although apoptosis of the infected cells is mediated by Bax (23).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Thomas Shenk, Curt Horvath, Michael David, Atsushi Kato, and Robert Silverman for important reagents for this study. We also thank Christine White for assistance with dendritic cell isolation.
This work was supported by National Institutes of Health grant AI073303 to G.C.S.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 9 February 2011.
REFERENCES
- 1. Ablasser A., et al. 2009. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10:1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexopoulou L., Holt A. C., Medzhitov R., Flavell R. A. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738 [DOI] [PubMed] [Google Scholar]
- 3. Andersen J., VanScoy S., Cheng T. F., Gomez D., Reich N. C. 2008. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 9:168–175 [DOI] [PubMed] [Google Scholar]
- 4. Burckstummer T., et al. 2009. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10:266–272 [DOI] [PubMed] [Google Scholar]
- 5. Chattopadhyay S., et al. 2010. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29:1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chattopadhyay S., Sen G. C. 2010. IRF-3 and Bax: a deadly affair. Cell Cycle 9:2479–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiu Y. H., Macmillan J. B., Chen Z. J. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi M. K., et al. 2009. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. U. S. A. 106:17870–17875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goubau D., et al. 2009. Transcriptional re-programming of primary macrophages reveals distinct apoptotic and anti-tumoral functions of IRF-3 and IRF-7. Eur. J. Immunol. 39:527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heylbroeck C., et al. 2000. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 74:3781–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Honda K., et al. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777 [DOI] [PubMed] [Google Scholar]
- 13. Hornung V., et al. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson A. C. 1999. Apoptosis in experimental rabies in Bax-deficient mice. Acta Neuropathol. 98:288–294 [DOI] [PubMed] [Google Scholar]
- 15. Kato H., et al. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28 [DOI] [PubMed] [Google Scholar]
- 16. Kato H., et al. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kato H., et al. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105 [DOI] [PubMed] [Google Scholar]
- 18. Kawai T., et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 19. Kerr D. A., et al. 2002. BCL-2 and BAX protect adult mice from lethal Sindbis virus infection but do not protect spinal cord motor neurons or prevent paralysis. J. Virol. 76:10393–10400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis J., et al. 1999. Inhibition of virus-induced neuronal apoptosis by Bax. Nat. Med. 5:832–835 [DOI] [PubMed] [Google Scholar]
- 21. Lin R., Heylbroeck C., Pitha P. M., Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manzur M., Fleming P., Huang D. C., Degli-Esposti M. A., Andoniou C. E. 2009. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 16:312–320 [DOI] [PubMed] [Google Scholar]
- 23. McLean J. E., Datan E., Matassov D., Zakeri Z. F. 2009. Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 83:8233–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mori I., et al. 2006. Herpes simplex virus US3 protein kinase regulates virus-induced apoptosis in olfactory and vomeronasal chemosensory neurons in vivo. Microbes Infect. 8:1806–1812 [DOI] [PubMed] [Google Scholar]
- 25. Peters K., Chattopadhyay S., Sen G. C. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J. Virol. 82:3500–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romieu-Mourez R., et al. 2006. Distinct roles for IFN regulatory factor (IRF)-3 and IRF-7 in the activation of antitumor properties of human macrophages. Cancer Res. 66:10576–10585 [DOI] [PubMed] [Google Scholar]
- 27. Sabbah A., et al. 2009. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 10:1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samuel M. A., Morrey J. D., Diamond M. S. 2007. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 81:2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satoh T., et al. 2010. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U. S. A. 107:1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seth R. B., Sun L., Ea C. K., Chen Z. J. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 [DOI] [PubMed] [Google Scholar]
- 31. Takaoka A., et al. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505 [DOI] [PubMed] [Google Scholar]
- 32. Toribio R., Ventoso I. 2010. Inhibition of host translation by virus infection in vivo. Proc. Natl. Acad. Sci. U. S. A. 107:9837–9842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weber F., Kochs G., Haller O. 2004. Inverse interference: how viruses fight the interferon system. Viral Immunol. 17:498–515 [DOI] [PubMed] [Google Scholar]
- 34. Yang P., et al. 2010. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 11:487–494 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.