

Abstract
Encephalitis induced by reovirus serotype 3 (T3) strains results from the apoptotic death of infected neurons. Extrinsic apoptotic signaling is activated in reovirus-infected neurons in vitro and in vivo, but the role of intrinsic apoptosis signaling during encephalitis is largely unknown. Bax plays a key role in intrinsic apoptotic signaling in neurons by allowing the release of mitochondrial cytochrome c. We found Bax activation and cytochrome c release in neurons following infection of neonatal mice with T3 reoviruses. Bax−/− mice infected with T3 Abney (T3A) have reduced central nervous system (CNS) tissue injury and decreased apoptosis, despite viral replication that is similar to that in wild-type (WT) Bax+/+ mice. In contrast, in the heart, T3A-infected Bax−/− mice have viral growth, caspase activation, and injury comparable to those in WT mice, indicating that the role of Bax in pathogenesis is organ specific. Nonmyocarditic T3 Dearing (T3D)-infected Bax−/− mice had delayed disease and enhanced survival compared to WT mice. T3D-infected Bax−/− mice had significantly lower viral titers and levels of activated caspase 3 in the brain despite unaffected transneuronal spread of virus. Cytochrome c and Smac release occurred in some reovirus-infected neurons in the absence of Bax; however, this was clearly reduced compared to levels seen in Bax+/+ wild-type mice, indicating that Bax is necessary for efficient activation of proapoptotic mitochondrial signaling in infected neurons. Our studies suggest that Bax is important for reovirus growth and pathogenesis in neurons and that the intrinsic pathway of apoptosis, mediated by Bax, is important for full expression of disease, CNS tissue injury, apoptosis, and viral growth in the CNS of reovirus-infected mice.
INTRODUCTION
Acute viral encephalitis is a serious and often fatal disease in humans. Neuronal injury due to encephalitis often leads to permanent alterations in neurologic function, manifested as cognitive impairment, seizures, or focal neurological deficits. Current treatment for most types of viral encephalitis is largely supportive (64). Specific antiviral drugs of proven efficacy are available for only a few agents, exemplified by acyclovir treatment for herpes simplex virus encephalitis, yet even with optimal therapy, morbidity and mortality remain significant. Understanding the cellular mechanisms of virus-induced neuronal injury provides a rational basis for identifying novel targets for potential neuroprotective antiviral therapy.
Apoptosis occurs during encephalitis caused by a diverse group of important human neurotropic viruses, including herpesviruses, flaviviruses, rhabdoviruses, and retroviruses (15, 29, 30). Apoptosis can serve as a host antiviral defense mechanism by triggering death of infected cells before viral replication is complete. Conversely, apoptosis can also enhance pathogenesis and facilitate viral dissemination when it occurs in nonrenewable cell populations, such as neurons, or is triggered after replication is completed (14). Neuronal apoptosis can occur as a direct result of viral infection or via indirect mechanisms, such as target cell lysis by infiltrating inflammatory cells.
Reovirus serotype 3 (T3) infection is a classic experimental system for studying the cellular and molecular basis of viral neuropathogenesis. Infection causes lethal meningoencephalitis in neonatal mice and triggers caspase-dependent apoptosis of neurons, which occurs as a direct consequence of viral infection rather than via immune-mediated processes (65). Caspase-dependent apoptosis can be initiated through extrinsic or intrinsic signaling pathways. The extrinsic pathway becomes activated when a death ligand binds to a cognate death receptor on the cell surface. Ligation of death receptors induces receptor oligomerization and activation of the death receptor-induced signaling complex (DISC), leading to activation of the initiator caspase 8, which subsequently activates the downstream effector caspases 3, 6, and 7 (37). T3 reovirus infection activates the death receptor pathway in neurons in the brain during encephalitis and in both primary neuronal cultures and neuroblastoma cell lines in vitro (13, 62). The FasL/Fas system plays a critical role in T3-induced neuronal apoptosis (13), paralleling that played by the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/DR4/5 system in epithelial cells and cancer cell lines (14).
In the intrinsic pathway, a caspase cascade begins when proapoptotic mitochondrial factors are released into the cytosol. The archetypal proapoptotic mitochondrial factor released is cytochrome c (cyt c), which along with Apaf-1, ATP, and procaspase 9 forms a complex known as the apoptosome (83). Caspase 9, the intrinsic pathway initiator caspase, becomes activated in the apoptosome and then activates downstream effector caspases. Another key proapoptotic mitochondrial protein released is second mitochondrion-derived activator of caspases (Smac), which binds and inactivates inhibitor of apoptosis proteins (IAPs) that normally function to inhibit the activities of caspases (23, 76, 77).
Bcl-2 family proteins are key regulators of the intrinsic pathway and are important for neuronal apoptosis (7). In the “rheostat” model of apoptosis, cell death or survival is determined by the ratio of antiapoptotic to proapoptotic Bcl-2 family proteins at the mitochondria (42). Overexpression of antiapoptotic proteins (including Bcl-2, Bcl-xL, and Mcl-1) protects the cell against apoptosis. In contrast, when the proapoptotic Bcl-2 family proteins are in excess, they release proapoptotic proteins from the mitochondria (12).
Bcl-2-associated X-protein (Bax) and Bcl-associated killer (Bak) protein are proapoptotic executioner proteins that induce apoptosis by forming mitochondrial permeability transition pores and allowing release of cyt c (79). Once cyt c is released, cells, including neurons, are committed to death (57). Though Bax and Bak have the same ultimate function at the mitochondria, one protein may take the dominant role as the executioner within a specific cell type. Bax is thought to be the main executioner in neurons and plays an essential role in neuronal cell death in processes as diverse as developmental apoptosis, hypoxia, ischemia, oxidative stress, nerve growth factor withdrawal, potassium deprivation, p53 overexpression, and nerve injury (10, 18, 26, 31, 50, 54, 57, 68, 69, 80).
Reovirus activates the intrinsic pathway of apoptosis and triggers the release of mitochondrial proapoptotic factors, including cyt c and Smac, in epithelial cells (41). The reovirus protein μ1 associates with lipid membranes when overexpressed and may contribute to mitochondrial permeability during infection (17, 20). Bcl-2 overexpression in reovirus-infected epithelial cells inhibits apoptosis, indicating a role for proapoptotic Bcl-2 family proteins in cell death (40, 63). Caspase 9 activation can be detected in the brain following reovirus infection (4, 13), but this may occur as a consequence of effector caspase activity and is not a reliable indicator of intrinsic pathway activation. To determine if the intrinsic pathway is required for neuronal apoptosis in vivo, we investigated the activation and importance of the intrinsic regulator Bax during reovirus encephalitis. We report that Bax is activated and cyt c is released in neurons in the brains of T3-infected mice. In mice deficient for Bax, virus-induced apoptosis and neurologic disease were inhibited or delayed, indicating that Bax and the intrinsic pathway play a critical role in reovirus-induced neuronal apoptosis and central nervous system (CNS) tissue injury. These effects were organ specific, as the hearts from animals infected intracranially with the neurotropic and myocarditic T3 Abney (T3A) strain showed abundant viral replication, apoptosis, and tissue injury.
MATERIALS AND METHODS
Infection of neonatal mice.
Laboratory stocks of plaque-purified passage 2 (P2) mammalian reovirus type 3 Abney (T3A) or type 3 Dearing (T3D) were generated by infection of L929 cells (ATCC CL1) as previously described (72). Aliquots of the same viral stocks were stored at −80°C and were diluted fresh in phosphate-buffered saline (PBS) for each infection. Two-day-old neonatal mice were injected intracranially (i.c.) with either 10 μl of PBS for mock-infected controls or virus doses of 100 or 1,000 PFU in 10 μl of PBS as previously described (62). NIH Swiss Webster (SW) pregnant dams were obtained from Harlan (Indianapolis, IN). Bax-deficient mice (strain B6.129X1-Baxtm1Sjk/J; Jackson Laboratory, Bar Harbor, ME) were bred as heterozygotes to generate Bax+/+ (wild-type [WT]), Bax+/−, or Bax−/− offspring. Bax pups were placed with a foster SW dam for infections. For survival studies, mice were monitored for 21 days postinfection (dpi) and were sacrificed only if showing signs of severe lethargy during T3A infection as a manifestation of myocarditis or seizures as signs of neurologic disease: the day of sacrifice was considered the day of death. Upon sacrifice, tail snips were taken for genotyping to confirm the Bax genotype. Brains, eyes, and hearts were frozen at −80°C in 1 ml of sterile PBS for titer, immunoblotting, and caspase assays or were fixed in 10% formalin for immunohistochemistry and then stored in PBS at 4°C. All animal experiments were approved by University of Colorado School of Medicine Institutional Animal Care and Use Committee.
Genotyping of Bax mice.
DNA was extracted from tail snips by using a Qiagen blood and tissue kit (Valencia, CA). Genotyping was performed by PCR using primer sequences previously described for Bax exon 5 (forward), Bax intron 5 (reverse), and neomycin (reverse) (80) at 0.3 μM each with native Taq polymerase (catalog no. 18138-018; Invitrogen, Carlsbad, CA). An additional reaction was run to confirm the presence of the neomycin cassette using 0.2 μM forward primer 5′-AGACAATCGGCTGCTCTGAT-3′, 0.2 μM reverse primer 5′-ATACTTTCTCGGCAGGAGCA-3′, 2 mm MgCl2, and 0.2 mM deoxynucleoside triphosphates (dNTPs). The expected product size for the neomycin amplification is 261 bp.
Protein concentrations.
DC protein assay by the microtiter plate method (Bio-Rad, Hercules, CA) and bovine serum albumin (BSA) standards diluted in the appropriate buffer were used to determine protein concentrations. Absorbances were read at 650 nm on an Emax microplate reader (Molecular Devices, Sunnyvale, CA).
Immunoblotting.
Whole-brain lysates were prepared as described previously (28), protein concentrations were determined as described above, and 400 μg of sample was loaded per lane on 15% polyacrylamide–Tricine gels (Hoefer Pharmacia Biotech, San Francisco, CA). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Hybond C nitrocellulose (Amersham Bioscience). Blots were incubated overnight at 4°C with primary antibodies diluted in either 5% bovine serum albumin (BSA) or nonfat dry milk (NFDM) in Tris-buffered saline with 1% Tween (TBST). The antibodies used were anti-Bax polyclonal (2772; Cell Signaling Technology, Beverly, MA) at 1:1,000 in 5% BSA, anti-cleaved caspase 3 (9661; Cell Signaling, Beverly, MA) at 1:1,000 in 5% NFDM, antiactin (CP01; Calbiochem) monoclonal at 1:10,000 in 5% BSA, and mitochondrial marker COX IV (4844; Cell Signaling, Beverly, MA) at 1:1,000 in 5% NFDM. Peroxidase-conjugated secondary antibodies used were goat anti-rabbit at 1:4,000 (111-035-003; Jackson ImmunoResearch, West Grove, PA) and anti-mouse IgM at 1:5,000 (401225; Calbiochem) in 1% NFDM in TBST. Secondary antibodies were incubated for 1 h at room temperature. Immunoblots were developed with Pierce ECL (enhanced chemiluminescence) Western blotting substrate (32106; Thermo Scientific, Rockford, IL) or SuperSignal West Pico chemiluminescent substrate (34080; Thermo Scientific, Rockford, IL).
Brain, heart, and eye titers.
Brains frozen in 1 ml PBS and hearts frozen in 300 μl PBS were thawed and homogenized on ice with 30 strokes of a Dounce homogenizer using a tight pestle. Aliquots of brains or heart homogenates were subject to 2 additional freeze-thaw cycles to −80°C. For eye titers, globes were removed from mice and were stored at −80°C in PBS. Globes were thawed and placed in gel saline, and volume was determined by volume subtraction. Globes were then homogenized with 30 strokes of a Dounce homogenizer using a tight pestle and then subject to three freeze-thaw cycles. All homogenates were sonicated for 30 s at 35% intensity on ice with an ultrasonic processor with a microtip probe (Sonics & Material, Inc., Newton, CT) and diluted in gel saline, and plaque assays were performed with L929 fibroblasts as previously described (72).
Immunohistochemistry.
Tissues were fixed in 10% formalin for 20 h at room temperature and then stored at 4°C in PBS until processing. Tissues were paraffin embedded, cut into 4-μm coronal sections, and stained with hematoxylin and eosin. For immunostaining, sections were deparaffinized in xylene and rehydrated in consecutive washes of 100 to 80% ethanol. Antigen unmasking was performed with 10 mM sodium citrate (pH 6.0) or antigen unmasking solution (Vector, Burlingame, CA). Tissues were permeabilized for 5 min in acetone at −20°C, washed in PBS, outlined with SuperPap Pen (Zymed, Invitrogen Carlsbad, CA), and blocked with either 8% normal goat serum or 8% normal horse serum in PBS for 1 h. Sections were incubated in primary antibodies diluted in 3% BSA–PBS overnight at 4°C and in secondary antibodies diluted 1:100 in the appropriate blocking solution for 1 h at room temperature. Nuclei were stained with Hoechst stain at 1:1,000 (GE Healthcare) for 10 min at room temperature. Slides were washed in PBS and mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA). Imaging was performed using a Zeiss Axioplan 2 digital deconvolution microscope with a Cooke Sensicam 12-bit camera. All exposure times, intensity outputs, and background levels were adjusted the same for mock-infected and infected brains for each imaging experiment with Slidebook Software. Imaging for cytochrome c and Smac release was performed with a Zeiss LSM 510 META on Axiovert 200 M. The antibodies used were Bax NT at 1:100 (06-499; Upstate, Lake Placid NY), reovirus monoclonal 4F2 at 1:200, reovirus rabbit polyclonal antibody at 1:200, rabbit polyclonal μNS at 1:500 (a kind gift from Max Nibert, Harvard University, Boston, MA) (8), NeuN at 1:100 (Millipore, Billerica, MA), cleaved caspase 3 (9661L; Cell Signaling, Beverly, MA) at 1:100, cytochrome c (136F3) (4280; Cell Signaling, Beverly, MA) at 1:50, Smac/DIABLO (612244; BD Biosciences, Franklin Lakes, NJ), and Tom40 (C-15) (sc-11025; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The secondary antibodies from Jackson ImmunoResearch (West Grove, PA) used were Cy3 goat anti-rabbit (111-165-003), Cy3 goat anti-mouse (115-165-146), DyLight 488 donkey anti-mouse (715-485-150), DyLight 649 donkey anti-rabbit, and DyLight 488 donkey anti-mouse (711-495-152). Alexa Fluor 488-conjugated goat anti-mouse (A11029), Alexa Fluor 488 goat anti-rabbit (A11034), and Alexa Fluor 633 goat anti-mouse were from Molecular Probes (Invitrogen, Carlsbad, CA).
Caspase activity assays.
Fluorogenic substrate assays for caspase activity were performed for caspase 3 (BF1100; R&D Systems, Minneapolis, MN) according to the manufacturer's instructions (13), with some modification. Brains frozen in 1 ml PBS or hearts frozen in 300 μl PBS were thawed and then homogenized 30 times with a Dounce homogenizer with a tight pestle on ice, and aliquots were frozen at −80°C. Homogenates were thawed and suspended in the kit lysis buffer, incubated on ice for 10 min, sonicated for 10 s on ice at 35% intensity, and then centrifuged at 720 × g for 10 min at 4°C to remove debris. Protein concentrations were determined as described above. Brain lysates were diluted in lysis buffer to contain 200 μg protein in a 50-μl volume. Samples were run in triplicate in 96-well plates. Negative controls that omitted lysate or substrate were included in all runs. Plates were incubated at 36.5°C for 1 h for brains and 3 h for hearts, and absorbances were read on a CytoFluor fluorescent plate reader (Applied Biosystems, Framingham, MA). Background was defined in no-lysate controls and subtracted from all other fluorescent absorbance readings. Background subtracted fluorescence absorbance values of infected WT or Bax−/− brains were divided by an average value of their mock controls to determine a fold increase in activity compared to that of the mock control. Background subtracted fluorescence values for hearts were normalized by protein concentration, and then values for infected hearts were divided by average mock control values.
Analysis of cytochrome c and Smac intensities.
Pixel intensities of cytochrome c (cyt c) and Smac staining were obtained with Image J software (National Institutes of Health, Bethesda, MD) (1) from immunofluorescent images acquired from coronal sections. Sections for cyt c immunofluorescence were obtained from mock-infected WT (n = 3), T3D-infected WT (n = 3), mock-infected Bax−/− (n = 2), and T3D-infected Bax−/− (n = 3) brains acquired over two separate imaging experiments. For Smac staining, staining was performed for mock-infected WT and Bax−/− sections (n = 3 each) and T3D-infected WT and Bax−/− sections (n = 4 each) over three imaging experiments. Cells were analyzed from one representative imaging experiment with mock-infected WT, mock-infected Bax−/−, and T3D-infected WT mice (n = 1 each) and T3D-infected Bax−/− mice (n = 2). Mock-infected and reovirus antigen-positive cells were outlined around the edges of cell bodies by using the freehand tool as determined by the edge of cyt c or Smac staining for mock or reovirus antigen for infected cells. Color histograms were obtained for individual cells (n = 54 for cyt c, and n = 51 for Smac) for each group, and pixel intensities were given numerical values from 0 to 255, with 0 being black and 255 being bright green. The percentage of pixels at each intensity value from 0 to 255 was calculated based upon the total number of pixels measured per cell. The sum of percentages of pixels at each value from 100 to 255 for cyt c and 10 to 255 for Smac was calculated per cell, and the sums for all cells per group were compared among the different groups for statistical analysis. Standard deviations for Smac staining were obtained directly from the histograms generated in Image J.
Statistical analysis.
Statistical analysis was performed with Prism (Graph Pad Software, Inc., San Diego, CA). For survival of mice, the log-rank (Mantel-Cox) test was used. Student's t test unpaired with two tails was used for all other statistical analyses, unless otherwise noted, and the paired t test was used only when all values within a group were identical. Mean day of death values are listed as ± standard deviations, and all other values are ± standard deviations unless noted. Error bars on graphs represent the standard deviation.
RESULTS
Bax is activated in reovirus-infected neurons in vivo.
When Bax is activated, it undergoes a conformational change that exposes its N terminus, and this alteration can be detected by conformation-specific antibodies (34). To determine if Bax was activated in reovirus-infected cells in the CNS during reovirus encephalitis, we performed immunofluorescence with an antibody specific to the N terminus of Bax on coronal brain sections from neonatal NIH Swiss Webster mice infected intracranially (i.c.) with 1,000 PFU of reovirus T3A and sacrificed at 8 days postinfection (dpi). Bax was found to be activated in specific regions of the brain known to be targeted and damaged by T3 strains, including the cingulate gyrus, hippocampus, and thalamus (52, 62) (Fig. 1). Activated Bax was not found in the brains of mock-infected animals or in regions of the brains of infected animals not known to be infected by reovirus (data not shown). Activated Bax staining was exclusively found in cells staining positive for the neuron-specific nuclear marker NeuN (Fig. 1B), indicating that the cell population containing activated Bax is neuronal. The percentage of cells in specific brain regions containing reovirus antigen or activated Bax is shown in Fig. 1C. Regardless of brain region, the majority (66 to 86%) of reovirus-infected cells contained activated Bax (Fig. 1D). Conversely, the overwhelming majority (91 to 98%) of cells with activated Bax were reovirus infected (data not shown). Total Bax expression was not increased in the brains of infected mice compared to controls by immunoblotting (data not shown), consistent with the absence of changes in Bax gene expression in infected mice (73).
Fig. 1.

Bax is activated in reovirus-infected neurons in the CNS. Two-day-old SW mice were injected with 1,000 PFU T3A. At 8 days postinfection (dpi), coronal sections were analyzed by immunofluorescence for activated Bax (red; Bax NT antibody), reovirus antigen σ3 (green), and Hoechst nuclear stain (blue). (A) Representative high-power (1,000×) images show that Bax is activated in the cingulate of reovirus-infected but not in mock-infected animals, and Bax is activated mainly in reovirus-infected cells. (B) Immunofluorescence staining for neuronal marker NeuN (green) and active Bax (red) in the cingulate of mock- and T3A-infected animals (630×; oil). Active Bax surrounds NeuN-staining nuclei. (C and D) The numbers of reovirus- and active Bax-positive cells were counted per high-power field (400×) per brain region per mouse (n = 4). (C) The graph shows the average percentage of cells expressing reovirus σ3 antigen (black bars) and activated Bax (white bars) per cingulate cortex (Cing), hippocampus (Hippo), and thalamus (Thal) in infected brains. (D) Average percentage of reovirus-infected cells containing active Bax within each brain region. The ranges of cells counted were 81 to 224 per cingulate, 101 to 270 per thalamus, and 87 to 134 per hippocampus per animal.
Bax is important for viral growth and virus-induced tissue injury and pathogenesis during reovirus encephalitis.
Having shown that Bax was activated, we next wished to determine the potential role of Bax in neuropathogenesis. Neonatal Bax−/− mice and their Bax+/− and Bax+/+ (WT) littermates were infected i.c. with 100 PFU of T3A. There were no significant differences in survival rates of Bax−/− mice in comparison to WT littermates: the mean days of death (MDD) were 7.7 dpi for WT (n = 6), 7.8 dpi for Bax+/− (n = 4), and 8.3 dpi for Bax−/− (n = 3) (Fig. 2A). Despite the lack of effect on survival, pathological examination of infected brains from Bax−/− mice showed dramatic reduction in injury compared to WT animals (Fig. 2B). Coronal sections were stained for reovirus protein σ3 by immunofluorescence, and a substantial reduction in the number of antigen-positive cells was seen in all regions of the brains of Bax−/− mice compared to WT controls (Fig. 3A). The thalamus was the brain region with the heaviest antigen burden in both WT and Bax−/− mice, though the amount of viral antigen remained substantially reduced in Bax−/− thalami compared to that in WT thalami. In the Bax−/− brains, infection of ependymal cells was also seen, which was absent in WT mice (Fig. 3A; note the antigen-positive cells lining the ventricle marked *). Cell counts of reovirus antigen-positive cells per high-power field (HPF) revealed that the percentage of reovirus-infected cells was significantly decreased in the cingulate, hippocampus, and thalamus (Fig. 3B). To determine whether the decrease in the number of infected cells was associated with a reduction in whole-brain viral titer, plaque assays were performed to measure viral titer from the brains of WT and Bax−/− mice at 8 dpi with T3A. A slight decrease in viral titer was seen in the brains of the Bax−/− mice compared to WT mice. The average viral titer for WT brains was 109.9 ± 0.5 PFU/ml, and that for Bax−/− brains was 109.3 ± 0.6 PFU/ml (difference not significant) (Fig. 3C). The viral titer was higher than expected in the brains of Bax−/− mice given the marked reduction in the number of virally infected neurons seen in the coronal sections. An alteration in tropism and infection of ependymal cells in Bax−/− mice, which was not seen in WT mice, likely contributes to the higher-than-expected viral titers in whole-brain lysates.
Fig. 2.

Bax is important for reovirus-induced tissue injury in the brain. Neonatal Bax−/− mice and their littermates were injected with 100 PFU T3A i.c. (A) WT (n = 6), Bax+/− (n = 4), and Bax−/− (n = 3) mice from three litters were monitored for survival. (B) T3A-infected WT and Bax−/− mice were sacrificed at 8 dpi. Hematoxylin and eosin stain of cingulate cortex, hippocampus, and thalamus of mock-infected WT mice and T3A-infected WT and Bax−/− mice (100×) shows that tissue injury is decreased in Bax−/− mice.
Fig. 3.

Bax is important for reovirus growth in the brain. WT and Bax−/− mice were injected with 100 PFU T3A i.c. and sacrificed at 8 dpi. (A) Coronal sections were stained by immunofluorescence for reovirus antigen σ3 (green) and Hoechst (blue) in the cingulate cortex (Cing), hippocampus (Hippo), and thalamus (Thal) of mock- or T3A-infected WT and Bax−/− mice (100×). An asterisk indicates the antigen-positive ependymal cells lining the lateral ventricle in the Bax−/− brain. (B) The percentage of reovirus-infected neurons was determined per high-power field (HPF) (400×) in the cingulate (n = 14 for WT [Bax+], and n = 10 for Bax−/− [Bax−]), hippocampus (n = 12 for WT, and n = 10 for Bax−/−), and thalamus (n = 14 for WT, and n = 9 for Bax−/−). Lines between bars indicate comparisons. ***, P < 0.0001. (C) Viral titers from WT (closed circles) and Bax−/− (open circles) brains of mice infected with 100 PFU T3A at 8 dpi. Each circle represents an average viral titer from one mouse brain.
To determine if apoptosis was impaired in the brain in Bax-deficient mice, immunofluorescence was performed to detect the activated form of the executioner caspase, caspase 3, in addition to reovirus antigen. A substantial decrease in the number of apoptotic cells was seen in a focus of infection in the Bax−/− thalamus compared to WT thalamus at 8 dpi with 100 PFU T3A (Fig. 4A). Quantification of cells containing active caspase 3 revealed that there was a significant increase in caspase 3-positive cells in all three regions of T3A-infected WT brains compared to mock-infected controls (Fig. 4B). In contrast, Bax-deficient mice did not have a significant increase in the percentage of caspase 3-positive cells in any brain region following infection, and the percentages in the cingulate and hippocampus were significantly decreased in comparison to those in T3A-infected WT mice. These results indicate that virus-induced activation of caspase 3 is impaired in the CNS in the absence of Bax.
Fig. 4.

Caspase activation is decreased in Bax−/− brains. Immunofluorescence for active caspase 3 (red), reovirus σ3 (green), and Hoechst (blue) was performed on coronal sections from WT and Bax−/− mice infected with 100 PFU T3A at 8 dpi (400×). (A) Panels of cleaved caspase 3 alone and with reovirus antigen show caspase 3 activation is greatly decreased in a focus of infection in the Bax−/− thalamus. (B) The number of caspase 3-positive cells was counted from high-power fields (400×) per brain region in the cingulate, hippocampus, and thalamus. The percentage of cells with cleaved caspase 3 per HPF in mock-infected WT (white bars), T3A-infected WT (black bars), mock-infected Bax−/− (light gray bars), and T3A-infected Bax−/− (dark gray bars) mice is shown per brain region. Lines between bars are comparisons. *, P < 0.05; **, P < 0.01; ns, not significant. The numbers of HPF counted per region were 6 to 8 for mock-infected WT (n = 3) and Bax−/− (n = 2) sections, 12 to 14 HPF for T3A-infected WT (n = 4) sections, and 10 HPF in T3A-infected Bax−/− (n = 4) coronal sections. A range of 60 to 187 cells were counted per HPF.
Bax is dispensable for viral growth, tissue injury, and apoptosis in the heart following reovirus infection.
Having established that Bax plays a key role in the neuropathogenesis of T3A infection, we next wished to determine if this was organ specific. Following i.c. inoculation, in addition to causing encephalitis, T3A is able to spread hematogenously to the heart to also cause severe myocarditis in infected mice. First we confirmed that Bax was activated in the heart by immunofluorescence for the N terminus of Bax in heart sections of WT mice infected with 100 PFU T3A at 8 dpi (Fig. 5A). In contrast to results seen in the brain, in WT and Bax−/− mice infected with T3A, there was no difference in the hearts in the extent or severity of virus-induced injury (Fig. 5B to D). Immunofluorescence in the hearts for reovirus antigen σ3 revealed that the viral burdens in the heart appear similar in WT and Bax−/− hearts (data not shown), and plaque assays were performed to confirm similar viral growth levels in the hearts of WT and Bax−/− mice. The average viral titer in WT hearts at 8 dpi was 108.8 ± 0.9 PFU/heart, and the viral titer in Bax−/− hearts was 109.1 ± 0.9 PFU/heart (differences not significant) (Fig. 5E).
Fig. 5.

Bax is dispensable for viral growth, tissue injury, and apoptosis in the heart following reovirus infection. (A) Bax is activated in reovirus-infected cells in the heart. Hearts from mock-infected and T3A (100 PFU)-infected WT mice at 8 dpi were sectioned and stained for active Bax (red), reovirus antigen (green), and Hoechst nuclear stain (blue) by immunofluorescence. (B to I) WT and Bax−/− mice were injected with 100 PFU T3A i.c. and were sacrificed at 8 dpi. (B to D) Hematoxylin and eosin stain (200×) of hearts from mock-infected WT (B) or T3A-infected WT (C) and Bax−/− (D) mice shows similar amounts of histopathologic injury in WT and Bax−/− hearts. (E) Plaque assays were performed to determine the viral titers in the hearts of WT and Bax−/− mice, and there was no difference between genotypes. (F) The fluorogenic substrate assay for caspase 3 activity was performed with hearts from mock-infected WT (n = 4) and Bax−/− (n = 4) mice and T3A-infected WT (n = 5) and Bax−/− (n = 5) mice. Activity is expressed as a fold increase compared to the average mock-infected value for respective genotype controls. (G) Correlation plot between caspase 3 activity and viral titer shows a correlation between caspase activity and viral titer in the heart (r represents the Pearson correlation for all values). Best-fit lines are shown per genotype, with the lower line representing the WT and the upper line representing Bax−/− values. *, P < 0.05; **, P < 0.01; ns, not significant.
To determine if apoptosis still occurred in the heart in the absence of Bax, activation of the executioner caspase 3 was investigated. Immunofluorescence staining for active caspase 3 showed similar amounts of caspase-positive cells in foci of infection in WT and Bax−/− hearts (data not shown). Fluorogenic substrate assays were performed to analyze caspase 3 activity in the heart. There was no difference in caspase 3 activities in WT and Bax−/− hearts (Fig. 5F). To determine if apoptosis was correlated with viral titer in the heart, we analyzed caspase 3 activity and viral titer by Pearson test for correlation. A significant correlation exists between caspase 3 activity and viral titer in the heart (Pearson r = 0.8666, P = 0.0012) (Fig. 5G), indicating that the amount of viral replication contributes to caspase activation in the heart regardless of the presence or absence of Bax. These results indicate that the role of Bax in reovirus pathogenesis is organ specific and that Bax is not important for viral growth or apoptosis in the heart during infection with T3A.
Bax contributes to mortality during encephalitis with serotype T3D.
WT mice infected with T3A have both severe CNS injury and extensive myocarditis, both of which likely contribute to lethality. We wished to look at a model in which cardiac injury did not occur to investigate the effect of Bax deletion on pathogenesis in CNS disease. Given that the effect of Bax deletion was more pronounced in the brain than in the heart in mice infected with T3A, we infected Bax−/− mice with T3 Dearing (T3D), a virus that is neuropathogenic but not myocarditic (66). Bax−/− mice and their littermates were injected with 100 PFU of T3D i.c. and monitored for survival (Fig. 6A). All of the WT mice infected with 100 PFU T3D died by 11 dpi, with an MDD of 9.7 dpi, which was significantly later than that for WT mice infected with the same dose of T3A (MDD of 7.7 dpi) (Fig. 2A) (P < 0.0001). At 21 dpi, 16% of the T3D-infected Bax−/− mice survived compared to 0% of Bax+/− and WT mice (P < 0.0001). One Bax−/− mouse succumbed at 19 dpi without ever showing signs of neurologic disease. Additionally, there was a significant delay in death in T3D-infected Bax−/− mice compared to WT mice (Table 1). To determine if the effect on survival could be replicated with a higher viral dose, Bax−/− mice and their littermates were challenged with 1,000 PFU of T3D i.c. A significant proportion of Bax−/− mice survived the higher challenge dose, with 20% survival in Bax−/− mice versus 0% in WT mice (P = 0.02) (Fig. 6B). The MDD was also significantly later in Bax−/− mice (P = 0.05) (Table 1). Bax+/− mice infected with 1,000 PFU T3D showed no difference in survival compared to WT mice (Fig. 6B and Table 1). The survival studies indicate that Bax plays an important role in reovirus pathogenesis and contributes to accelerated disease and enhanced mortality during encephalitis with serotype T3D.
Fig. 6.

Bax−/− mice have increased survival following infection with T3D. Bax−/− mice and their Bax+/− and WT littermates were injected i.c. with serotype T3D and monitored for signs of disease and survival until 21 dpi. Mice were sacrificed only if seizing and moribund and were counted as dead on the day of sacrifice. The graphs show the percentage of surviving animals at each dpi. (A) WT (n = 17), Bax+/− (n = 25), and Bax−/− (n = 19) mice from eight litters were infected with 100 PFU T3D. (B) WT (n = 6), Bax+/− (n = 7), and Bax−/− (n = 5) littermates from three litters were infected with 1,000 PFU T3D. *, P < 0.05, and ***, P < 0.0001, for Bax−/− compared to WT by log-rank test.
Table 1.
Bax−/− mice have delayed death following infection with T3D
| T3D dose (PFU) | Mouse genotype | No. of mice surviving/no. infected (%) | MDDa | P valueb |
|---|---|---|---|---|
| 100 | WT | 0/17 (0) | 9.7 ± 0.8 | |
| Bax+/− | 0/25 (0) | 9.7 ± 1.1 | 0.96 | |
| Bax−/− | 3/19 (15.8) | 11.8 ± 1.3c | <0.0001 | |
| 1,000 | WT | 0/6 (0) | 9.3 ± 1.0 | |
| Bax+/− | 0/7 (0) | 8.7 ± 1.0 | 0.28 | |
| Bax−/− | 1/5 (20) | 11.8 ± 2.2 | 0.05 |
Excludes survivors. Mean values ± standard deviations are shown.
P values are in comparison to values for WT mice.
Excludes a mouse that succumbed at 19 dpi.
Bax is important for neurologic injury and viral growth at late time points during encephalitis with serotype T3D.
In order to determine whether the enhanced survival in Bax−/− mice infected with T3D was associated with reduced CNS tissue injury, the extent of injury was evaluated in a blinded fashion with a previously validated histopathological injury grading system (3). Representative images are shown in Fig. 7A. The severity of brain tissue injury was significantly lower in all three evaluated brain regions in Bax−/− mice infected with 100 PFU T3D (P = 0.005 for cingulate and hippocampus and P = 0.04 for thalamus) compared to T3D-infected WT mice (Fig. 7B).
Fig. 7.

Bax is important for reovirus-induced tissue injury in the CNS. Coronal sections of brains of mock- and T3D (100 PFU)-infected WT and Bax−/− mice that were seizing and moribund at 10 to 13 dpi were stained by hematoxylin and eosin for histopathology. Brain regions were scored for injury (n = 3 per group) by two blinded observers as follows: 0, no injury; 1, <10% of region with lesions; 2, 10 to 40% lesions; 3, 40 to 75% lesions; and 4, >75% lesions. (A) Representative images (200×) from the cingulate cortex (Cing), hippocampus (CA3), and thalamus (Thal) are shown from mock-infected Bax−/− mice and T3D-infected WT and Bax−/− animals. The tissue injury score is in the lower right of each picture. (B) The graph shows histopathology scores per cingulate cortex, hippocampus, and thalamus in mock-infected and T3D-infected WT and Bax−/− brains. Tissue injury is greatly decreased in T3D-infected Bax−/− mice exhibiting seizures. Asterisks above bars indicate significance compared to mock infected, and lines between bars indicate a significant difference between groups. *, P < 0.05; **, P < 0.01; and ***, P < 0.005.
To determine if reduced viral injury was associated with diminished replication of virus in the brain, plaque assays were performed to compare reovirus growth rates in the brains of T3D (100 PFU)-infected WT and Bax−/− mice. At 9 dpi, the viral titer in Bax−/− brains was significantly decreased (Fig. 8A). The average viral titer for WT brains at 9 dpi was 108.9 ± 0.2 PFU/ml, while Bax−/− brains had average viral titers of 107.3 ± 0.3 PFU/ml (P = 0.002) (Fig. 8A). Unexpectedly, Bax+/− mice had a significant increase in brain viral titer compared to WT mice, with an average viral titer of 109.4 ± 0.1 PFU/ml (P = 0.02) (Fig. 8A).
Fig. 8.

Reovirus growth is significantly decreased in Bax−/− brains, but transneuronal transport is not impaired. Bax−/− mice and their littermates were infected with 100 PFU T3D. Brains and eyes were harvested at 9 dpi or later and analyzed for viral titer by plaque assay. Each point represents the average viral titer per animal. The graphs show viral growth in brains of mice (n = 5 each) at 9 dpi from four separate litters (A), viral growth in brains of mice showing seizures in WT mice at 10 to 11 dpi (n = 5; 2 litters) and Bax−/− mice at dpi 11 to 13 (n = 9; 5 litters) (B), and viral titers in both eyes of WT (n = 5; 5 litters) and Bax−/− (n = 5; 2 litters) mice at 9 dpi (C). *, P < 0.05; and **, P < 0.005 compared to WT.
In order to determine whether brain titers in later-surviving Bax−/− mice reached levels similar to those seen in moribund WT mice, we examined brain titers in a group of mice surviving 11 to 13 days postinfection (Fig. 8B). The average viral titer in the brain of Bax−/− mice (107.7 ± 0.2 PFU/ml) remained significantly below that seen in WT mice at either 9 dpi (see above) or in the small group of T3D-infected WT mice surviving to 10 to 11 dpi (109.0 ± 0.2 PFU/ml) (P = 0.002).
Axonal transport of virus is not impaired in Bax-deficient mice.
The reduced viral titer in the brain of Bax−/− mice infected with T3D and the decreased number of infected neurons with T3A may reflect decreased viral replication in neurons, diminished viral spread between neurons, or a combination of both factors. Following i.c. inoculation, T3 reoviruses spread to the neuroretina of the eye via transneuronal transport (4, 72). To determine if transneuronal transport was impaired, we measured viral titer in eyes from T3D-infected Bax−/− mice after i.c. inoculation. Although the average viral titers from both eyes of Bax−/− mice were significantly decreased compared to those in WT mice (105.1 ± 0.6 PFU in Bax−/− eyes versus 107.0 ± 0.2 PFU in WT eyes; P = 0.015), the presence of substantial amounts of virus in the eyes of Bax−/− mice indicated that transneuronal spread had occurred (Fig. 8C). The ratio of viral titer in the eye to that in the brain is a measure of the efficiency of transneuronal spread (4), and this was not significantly different in Bax−/− compared to WT mice (brain titer/eye titer, WT, 93 ± 40, and Bax−/−, 405 ± 522; differences not significant) reflecting the fact that lower eye titers in the Bax−/− mice were proportional to the reduction seen in brain titers. This suggests that transneuronal transport occurs in Bax−/− mice and is likely as efficient in disseminating virus as in WT mice.
Bax is important for induction of apoptosis in reovirus-induced encephalitis in vivo.
To determine if reovirus is able to cause apoptosis in the absence of Bax, fluorogenic substrate assays of caspase 3 activity were performed on whole-brain lysates from Bax−/− and WT mice at 9 dpi following infection with 100 PFU T3D i.c. The average caspase 3 activity was 4.9 ± 3.5-fold over mock infected (P = 0.54) in Bax−/− brains compared to 34.4 ± 11.0-fold over mock infected (P = 0.022) in WT brains (P = 0.016 for WT versus T3D-infected Bax−/−) (Fig. 9). A similar reduction was seen in caspase 3 cleavage detected by immunoblotting of whole-brain lysates (data not shown).
Fig. 9.
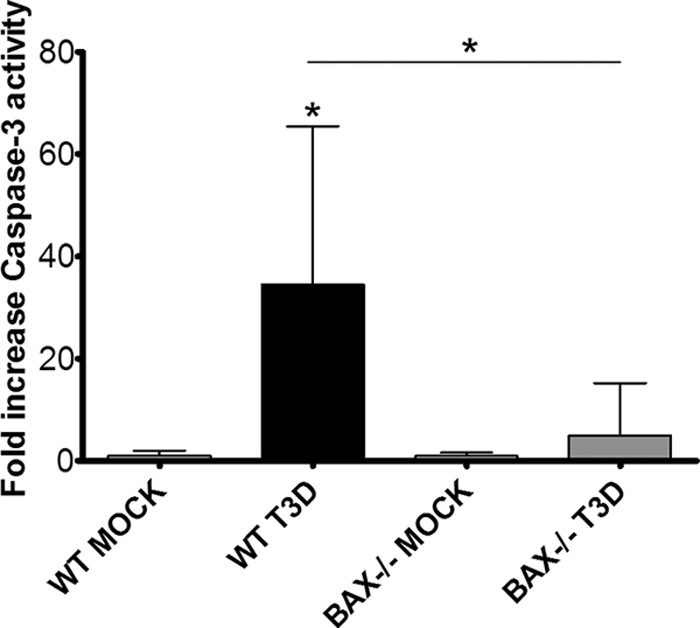
Apoptosis is decreased at 9 dpi in Bax−/− mice infected with T3D. Two-day-old Bax−/− mice and their WT littermates were injected with either PBS or 100 PFU T3D i.c. and sacrificed at 9 dpi. Fluorogenic substrate assays were performed with lysates from mock-infected (n = 6) and T3D-infected WT (n = 8) animals and mock-infected (n = 3) and T3D-infected (n = 9) Bax−/− animals in triplicate. Activity is expressed as fold change over mock infected for that genotype. *, P < 0.05. The asterisk above the bars is compared to mock infected, and that between bars is compared to WT.
Bax is important, but not essential, for cytochrome c and Smac release in reovirus-infected cells in vivo.
Bax activation leads to pore formation in the outer mitochondrial membrane and release of cyt c into the cytoplasm. The release of cyt c has not been investigated in reovirus-infected neurons in vivo; therefore, we performed immunofluorescence imaging to determine the subcellular location of cyt c in infected brains (Fig. 10A). When cyt c is released from the mitochondria, the pattern of staining changes from punctate to diffuse, and in neurons, the release of cyt c can also result in a loss of cyt c staining (82). Additional immunofluorescence staining for translocase of the outer mitochondrial membrane 40 (Tom40) shows that cyt c and Tom40 colocalize in cells in mock-infected brains, but in reovirus-infected neurons, colocalization between cyt c and Tom40 is decreased (Fig. 10A). Interestingly, Tom40 staining is more intense in reovirus-infected cells in the brain and surrounds inclusion bodies. Rater-blinded cell counts at 9 dpi in WT mice infected with 100 PFU of T3D i.c. show that cyt c was released in both antigen-negative and antigen-positive neurons in the cingulate, hippocampus, and thalamus of infected mice. The percentage of mock-infected, antigen-negative cells in T3D-infected brains, and antigen-positive cells with released cyt c was calculated per high-power field (HPF) (Fig. 10B). Virtually every reovirus-infected cell showed either diffuse cyt c or a loss of cyt c staining, which was significant compared to mock infected in all three brain regions examined. Cyt c was also released in a small but significant percentage of antigen-negative neurons, consistent with a small “bystander” effect of apoptosis. Release of cyt c was also investigated in the heart by immunofluorescence for cyt c, Tom40, and reovirus (data not shown), and cyt c release occurs in reovirus-infected cardiac myocytes in both WT and Bax−/− hearts (Fig. 10D). In reovirus-infected WT and Bax−/− cells in the heart, the percentage of cells with released cyt c was significantly increased compared to that in mock controls. Interestingly, there was also significantly more release in reovirus-infected Bax−/− cells compared to WT cells in the heart (P = 0.002). Therefore, Bax is not required for cyt c release in the heart. Myocytes also express Bak, which is likely contributing to release of cyt c in the absence of Bax.
Fig. 10.

Cytochrome c is released in reovirus-infected neurons and cardiac myocytes in vivo. (A to C) Coronal sections taken from brains at 9 dpi from mock-infected WT (n = 3) and Bax−/− (n = 3) mice and T3D (100 PFU)-infected WT (n = 4) and Bax−/− (n = 3) mice were stained for cytochrome c (cyt c; green), Tom40 (red), and reovirus σ3 (gray) by immunofluorescence confocal microscopy (1,000×; oil). (A) Representative images of the cingulate cortex in WT mice and the thalamus in Bax−/− mice show that cyt c is released from the mitochondria in reovirus-infected cells. Released cyt c is exemplified by a diffuse staining pattern (*) or a loss of cyt c staining, while mitochondrial cyt c is punctate. Note that a T3D-infected Bax−/− neuron has released cyt c. (B) cyt c staining patterns were grouped as punctate or diffuse, and the diffuse group includes cells with a loss of cyt c staining. The distributions of cells by average percentage per high-power field (HPF) in the cingulate cortex, hippocampus (CA3), and thalamus of mock-infected (white bars) and antigen-negative (Ag Neg; gray bars) or antigen-positive (Reo; black bars) cells in T3D-infected WT brains are shown. For mock-infected mice, 7, 6, and 11 HPFs were counted in the cingulate, hippocampus, and thalamus, respectively. For T3D-infected WT sections, 11, 6, and 21 HPFs were counted in the cingulate, hippocampus, and thalamus, respectively. (C) Fluorescence intensities of cyt c staining were determined by histogram analysis in 54 cells each of mock- and T3D-infected WT and Bax−/− neurons. The average percentages of pixels with intensities in the range of 100 to 255 are shown per group, which shows that infected WT cells have a significant decrease in intensity of cyt c, while infected Bax−/− neurons do not. (D) Bax−/− mice and their WT littermates were injected with 100 PFU T3A, and hearts were taken at 8 dpi, sectioned, and stained for cyt c, Tom40, and reovirus by immunofluorescence. Rater-blinded cell counts were performed for cyt c release in mock-infected, antigen-negative cells in T3A-infected hearts and reovirus-infected cells per HPF in mock-infected WT (n = 5), T3A-infected WT (n = 15), mock-infected Bax−/− (n = 6), and T3A-infected Bax−/− (n = 11) mice. Hearts were taken from at least two animals per group. The average percentage of cells with released cyt c per HPF is shown. Lines between bars indicate comparisons. *, P < 0.05; **, P < 0.01; ***, P < 0.0001. ns, not significant.
As neurons do not express a full-length form of Bak (70, 71, 75), it is likely that Bax alone is the executioner responsible for cyt c release in neurons, and accordingly, Bax-deficient neurons do not release cyt c during apoptosis induced by intrinsic pathway stimuli (50, 58, 59). Recently it was reported that reovirus protein μ1, an outer capsid viral protein crucial for pathogenesis, is able to release both cyt c and Smac when overexpressed in fibroblasts, and T3D infection of Bax−/− Bak−/− mouse embryonic fibroblasts (MEFs) also results in the release of cyt c (81). To determine if cyt c is released in reovirus-infected neurons in vivo in the absence of Bax, immunofluorescence staining was also performed for cyt c in mock- and T3D-infected Bax−/− mice at 9 dpi (Fig. 10A). In the Bax−/− sections, reovirus-infected neurons were difficult to detect and were mostly found as a few isolated infected cells in the cingulate and in small foci of infection within the thalamus. The inability to detect many infected neurons correlates with the greatly decreased brain titers at this time point (Fig. 8) and was similar to infection with T3A (Fig. 3). Release of cyt c was seen in reovirus-infected Bax−/− neurons (Fig. 10A), indicating that a Bax-independent mechanism of cyt c release exists during reovirus infection of neurons. Many of the Bax−/−-infected neurons did have their inclusion bodies surrounded by Tom40 staining but retained most of their cyt c staining within the cell. A decrease of cyt c signal is commonly used as an indication of release (9, 61, 81); therefore, we measured the intensity of cyt c staining in mock- and reovirus-infected WT and Bax−/− neurons (Fig. 10C). Intensity of staining was determined by color histogram analysis of pixels from single cells as described in Materials and Methods. A significant decrease in cyt c staining intensity was seen between mock- and T3D-infected WT cells, while the difference between mock-infected and infected Bax−/− cells was not significant. Infected Bax−/− neurons retained significantly more intense cyt c staining than infected WT neurons. Due to the limited number of infected Bax-deficient neurons detectable, it is difficult to make a statement regarding the efficiency or timing of cyt c release in the absence of Bax. Thus, while reovirus is capable of releasing cyt c in a Bax-independent manner in infected neurons, cyt c release is impaired in the absence of Bax.
Release of Smac was also investigated by immunofluorescence in infected WT and Bax−/− neurons (Fig. 11A ). Infected WT neurons lost much of their Smac staining, indicating Smac is released. In infected Bax−/− neurons, Smac staining was also decreased compared to the level in mock-infected neurons (Fig. 11A). To quantify a change in fluorescence intensity of Smac, analysis of color histograms was performed. The intensity of Smac staining was decreased in both T3D-infected WT and Bax−/− neurons compared to mock genotype controls (Fig. 11B), confirming that Smac intensity was decreased in infected cells. However, infected Bax−/− cells retained significantly more intense Smac staining than WT cells (Fig. 11B). Smac release can also be assessed by a decrease in standard deviation of pixel intensities as the distribution of Smac becomes diffuse in the cell (61), and the average standard deviations per group are shown in Fig. 11C. Again there was a significant reduction in the standard deviations in infected WT and Bax−/− neurons compared to mock genotype controls, with Bax−/− standard deviations being significantly higher than those of the WT. Thus, it appears that reovirus is able to release both cyt c and Smac independently of Bax, but Bax is an important contributor to release of these proteins from the mitochondria in neurons.
Fig. 11.

Bax contributes to, but is not required for, Smac release in reovirus-infected neurons in vivo. Coronal sections from WT (n = 4) and Bax−/− (n = 4) mice infected with 100 PFU T3D at 9 dpi were analyzed by immunofluorescence for Smac release and were compared to those from mock-infected mice (n = 3 each). (A) Representative image of Smac (green), reovirus σ3 (red), and Hoechst (blue) staining in the thalamus of mock-infected Bax−/− and T3D-infected WT and Bax−/− mice. (B and C) Color histograms from one representative imaging experiment were obtained for individual cells (n = 51 per group) for mock-infected and T3D-infected WT and Bax−/− mice to analyze the intensities of Smac staining. (B) The percentage of pixels between 10 and 255 was determined per cell, and the average is shown per group. (C) The average standard deviation of Smac intensities per cell is shown per group. ***, P < 0.0001.
DISCUSSION
Apoptosis is an important mechanism of neuronal death during reovirus encephalitis. Although there is clear evidence indicating that reovirus-induced neuronal apoptosis is associated with activation of the Fas/FasL extrinsic death receptor pathway (4, 13), the role of the intrinsic pathway in reovirus-induced apoptosis in vivo has not been well characterized. The BH3-only protein Bid is cleaved by the extrinsic pathway initiator caspase 8 and links activation of the extrinsic pathway to the intrinsic pathway via activation of Bax or Bak (25, 78). Bid is cleaved during reovirus infection in vitro and in vivo by the extrinsic pathway and plays an important role in reovirus pathogenesis during encephalitis (13, 21, 40), which suggests that Bid is important for engaging intrinsic pathway activation during reovirus infection. In this report, we show that the intrinsic pathway regulator Bax is activated in the brain following reovirus infection. Bax activation is seen in the same brain regions that are involved in reovirus infection and occurs almost exclusively in infected neurons. We also show that cytochrome c, a critical proapoptotic mitochondrial protein and effector of intrinsic apoptotic signaling, is released from the mitochondria in the brains of reovirus-infected animals. These results establish that the mitochondrial intrinsic apoptotic pathway is activated in the brains of mice during reovirus infection. Our results also demonstrate that reovirus infection of neurons, neuronal apoptosis, and injury within the brain are inhibited in Bax−/− mice, suggesting that Bax is required for the full expression of reovirus-induced pathogenesis during encephalitis. The importance of Bax is organ specific, and in contrast to results found in the brain, we found no difference in rates of viral growth, cyt c release, apoptosis, and injury within the hearts of mice infected with the myocarditic reovirus strain T3A. Our results in the Bax−/− hearts are supported by evidence that intrinsic pathway activation in cardiac myocytes is not required for reovirus-induced apoptosis (22).
In neurons, Bax is required for cyt c release in apoptotic neurons as Bax−/− neurons do not release cyt c in response to intrinsic pathway stimuli (50, 58, 59) and neurons do not express a full-length form of Bak (70, 71, 75). Consistent with results that T3D infection causes release of cyt c in Bax−/− Bak−/− MEFs (81), we found that reovirus-infected neurons were able to release cyt c and Smac in the absence of Bax but that Bax is still important for optimal release of these protein during reovirus infection. The μ1 protein of reovirus can release cyt c and Smac when ectopically expressed (81), but a proapoptotic peptide from the φ fragment that is sufficient and critical for μl to induce apoptosis is not able to release cyt c from isolated mitochondria or when microinjected into cells (39). However, the proapoptotic peptide is able to cause increases in intracellular calcium by an unknown mechanism (39). Both cyt c release from the mitochondria (reviewed in reference 19) and increased intracellular Ca2+ (reviewed in reference 60) potentiate opening of the permeability transition pore complex spanning both the outer and inner mitochondrial membranes, which thus can cause swelling of the matrix and rupture of the outer mitochondrial membrane to release proteins such as cyt c and Smac. Despite the ability of reovirus to release cyt c in the absence of Bax, release was impaired in Bax-deficient neurons compared to the presence of Bax in WT neurons. This suggests that the rate of release of cyt c is delayed in Bax−/− mice, which would delay caspase 3 activation in Bax−/− infected neurons. Recently it was reported that the interferon-stimulated gene (ISG) ISG12b2 localizes to the mitochondria and can release cyt c when expressed in Bax−/− Bak−/− MEFs (46), and reovirus is known to induce ISGs during encephalitis (73), thus offering an additional mechanisms for cyt c release in infected neurons in the absence of Bax. Lysosomal membrane permeabilization and release of lysosomal proteases cathepsins has been shown to be an amplifying signal of apoptosis that is downstream of but requires Bax or Bak (53) and is yet another factor that could cause decreased caspase activity in Bax−/− reovirus-infected neurons.
Bax has other roles within cells aside from that as the mitochondrial executioner of apoptosis. At the endoplasmic reticulum (ER), Bax plays a major role in regulation of intracellular Ca2+ release from the ER and subsequent uptake by mitochondria during apoptosis induced by oxidative stress (reviewed in reference 27). Both Bax and Bak have been implicated in the regulation of mitochondrial dynamic processes of fission and fusion. During apoptosis, mitochondrial fission is increased, which Bax may promote as it is found at fission sites and is complexed with fission machinery (reviewed in reference 27). Conversely, Bax and Bak together are important for mitochondrial fusion, and Bax-deficient cortical neurons have significantly shorted mitochondrial lengths that suggests a defect in mitochondrial fusion in neurons in the absence of Bax (16). We have found that reovirus-infected neurons display alterations in mitochondrial networks, as evidenced by immunofluorescence for the outer mitochondrial membrane protein Tom40. Tom40 staining is enhanced around viral inclusions regardless of the presence of Bax, while cyt c is absent (Fig. 10). Several possibilities exist to explain the enhanced Tom40 staining. The first is that mitochondrial fission is occurring, and there is an increased signal due to an increase in the number of mitochondria, which appears to be unaffected by the absence of Bax. The second is that damaged mitochondria that have already released cyt c are being transported to the perinuclear region for degradation (56).
Many viruses have recently been found to activate Bax, including influenza A virus (49), respiratory syncytial virus (24), hepatitis B virus (44), hepatitis C virus (5), Theiler's murine encephalomyelitis virus (67), West Nile virus (55), rabies virus (36, 74), poliovirus (2), rotavirus (48), and Newcastle disease virus (51). A common link for the ability of many different viruses to activate Bax may be induction of apoptosis via antiviral innate immunity pathways. The double-stranded RNA (dsRNA) sensors RIG-I and MDA-5 were found to initiate intrinsic pathway-mediated apoptosis in melanoma cells when activated by poly(I-C) (6), and reovirus antiviral signaling is dependent on signaling via RIG-I or MDA-5 through their mitochondrial adaptor IPS-1 (45). Recently it was found that IRF-3 is able to activate Bax (11), and activation of IRF-3 downstream of IPS-1 and RIG-I activation in reovirus infection was found to be important for induction of apoptosis and attenuation of reovirus in vivo (32, 33). Innate antiviral signaling pathways may also be a mechanism for Bax activation during reovirus infection.
Our results demonstrate that Bax contributes to reovirus-induced neuronal apoptosis and tissue injury and that reovirus-infected Bax−/− mice show reduced CNS tissue injury, delayed disease, and enhanced survival compared to WT controls. In contrast, although tissue injury and apoptosis were decreased in neonatal Bax−/− mice infected with rabies virus strain RV194-2, there was no difference in disease or survival of Bax−/− mice, and Bax-independent apoptosis in the brainstem was considered to be the lethal lesion in the mice (35). Bax has been shown to be protective against apoptosis during CNS infection by Sindbis virus (38), and 1-week-old Bax−/− mice infected with Sindbis virus had significantly decreased survival compared to their WT littermates (43).
Viral growth and infection of neurons are decreased in the Bax−/− brain following infection with T3 reoviruses. Similar viral titers in the hearts of WT and Bax−/− mice infected with T3A and a lack of difference in viral growth in Bax−/− Bak−/− MEFs (81) indicate that Bax is not directly required for reovirus replication within a cell. We hypothesize that deletion of Bax delays apoptosis of infected cells and slows lateral spread of reovirus to neighboring neurons. Release of reovirus from Ras-transformed cancer cells is caspase dependent (47). Similarly to what we see in T3D-infected Bax−/− mice, viral growth is also decreased in the brains of Bid−/− and caspase 3−/− mice (4, 21), and both Bid and caspase 3 are shown to be important or required for apoptosis signaling during reovirus infection. We also found an unexpected increase in viral titer in the brains of Bax+/− mice infected with 100 PFU T3D at 9 dpi. Considering the rheostat model of Bcl-2 family proteins in apoptosis, it is possible that with only half of the normal amount of cellular Bax available, apoptosis would be delayed in an infected cell. The delay would allow for further viral replication before apoptotic fragmentation of the cell occurs, thereby increasing viral titers. However, by deleting Bax, apoptosis is impaired and this decreases viral spread. Caspase 3 is the main executioner caspase in neurons and is required for pathogenesis of reovirus encephalitis and CNS tissue injury (4). We found that caspase 3 activation is dramatically reduced in the brains of T3D-infected Bax−/− mice at 9 dpi by fluorescent substrate activity and activation is decreased in T3A-infected Bax−/− brains by immunofluorescence for activated caspase 3. Our data indicate that the intrinsic mitochondrial apoptotic signaling pathway plays an important role in augmenting death receptor-initiated apoptosis in the brains of reovirus-infected mice and that reovirus itself augments this pathway as part of its pathogenesis to induce full expression of CNS tissue injury and associated CNS disease.
ACKNOWLEDGMENTS
This work was supported by a VA Merit Grant (K.L.T.) and NIH grants 2T32 AI 052066 (H.M.B.), 5R01NS050138 (K.L.T), and 1R01NS051403 (K.L.T.).
We thank Penny Clarke and J. David Beckham for careful reviews of the manuscript and J. Smith Leser for technical support. Blinded scoring was performed by Stephanie S. Schittone and J. David Beckham. We give thanks to Ron Bouchard at the VA Microscopy Core for expertise in microscopy and the University of Colorado Cancer Center Histology Core for tissue processing.
Footnotes
Published ahead of print on 9 February 2011.
REFERENCES
- 1. Abramoff M. D., Magelhaes P. J., Ram S. J. 2004. Image processing with ImageJ. Biophotonics Intl. 11:36–42 [Google Scholar]
- 2. Autret A., et al. 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 81:7504–7516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beckham J. D., Goody R. J., Clarke P., Bonny C., Tyler K. L. 2007. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J. Virol. 81:6984–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beckham J. D., Tuttle K. D., Tyler K. L. 2010. Caspase-3 activation is required for reovirus-induced encephalitis in vivo. J. Neurovirol. 16:306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benali-Furet N. L., et al. 2005. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24:4921–4933 [DOI] [PubMed] [Google Scholar]
- 6. Besch R., et al. 2009. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Invest. 119:2399–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blomgren K., Leist M., Groc L. 2007. Pathological apoptosis in the developing brain. Apoptosis 12:993–1010 [DOI] [PubMed] [Google Scholar]
- 8. Broering T. J., McCutcheon A. M., Centonze V. E., Nibert M. L. 2000. Reovirus nonstructural protein muNS binds to core particles but does not inhibit their transcription and capping activities. J. Virol. 74:5516–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campos C. B., et al. 2006. Method for monitoring of mitochondrial cytochrome c release during cell death: immunodetection of cytochrome c by flow cytometry after selective permeabilization of the plasma membrane. Cytometry A 69:515–523 [DOI] [PubMed] [Google Scholar]
- 10. Cao G., et al. 2001. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J. Cereb. Blood Flow Metab. 21:321–333 [DOI] [PubMed] [Google Scholar]
- 11. Chattopadhyay S., et al. 2010. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 29:1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng E. H., et al. 2001. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8:705–711 [DOI] [PubMed] [Google Scholar]
- 13. Clarke P., Beckham J. D., Leser J. S., Hoyt C. C., Tyler K. L. 2009. Fas-mediated apoptotic signaling in the mouse brain following reovirus infection. J. Virol. 83:6161–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clarke P., Richardson-Burns S. M., DeBiasi R. L., Tyler K. L. 2005. Mechanisms of apoptosis during reovirus infection. Curr. Top. Microbiol. Immunol. 289:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clarke P., Tyler K. L. 2009. Apoptosis in animal models of virus-induced disease. Nat. Rev. Microbiol. 7:144–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cleland M. M., et al. 2011. Bcl-2 family interaction with the mitochondrial morphogenesis machinery. Cell Death Differ. 18:235–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coffey C. M., et al. 2006. Reovirus outer capsid protein micro1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 80:8422–8438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cregan S. P., et al. 1999. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 19:7860–7869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crompton M. 2003. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr. Med. Chem. 10:1473–1484 [DOI] [PubMed] [Google Scholar]
- 20. Danthi P., Coffey C. M., Parker J. S., Abel T. W., Dermody T. S. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 4:e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danthi P., et al. 2010. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog. 6:e1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DeBiasi R. L., et al. 2010. Critical role for death-receptor mediated apoptotic signaling in viral myocarditis. J. Cardiac Failure 16:901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du C., Fang M., Li Y., Li L., Wang X. 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102:33–42 [DOI] [PubMed] [Google Scholar]
- 24. Eckardt-Michel J., et al. 2008. The fusion protein of respiratory syncytial virus triggers p53-dependent apoptosis. J. Virol. 82:3236–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eskes R., Desagher S., Antonsson B., Martinou J. C. 2000. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 20:929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghatan S., et al. 2000. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J. Cell Biol. 150:335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghibelli L., Diederich M. 2010. Multistep and multitask Bax activation. Mitochondrion 10:604–613 [DOI] [PubMed] [Google Scholar]
- 28. Goody R. J., Hoyt C. C., Tyler K. L. 2005. Reovirus infection of the CNS enhances iNOS expression in areas of virus-induced injury. Exp. Neurol. 195:379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gougeon M. L., Piacentini M. 2009. New insights on the role of apoptosis and autophagy in HIV pathogenesis. Apoptosis 14:501–508 [DOI] [PubMed] [Google Scholar]
- 30. Griffin D. E., Hardwick J. M. 1999. Perspective: virus infections and the death of neurons. Trends Microbiol. 7:155–160 [DOI] [PubMed] [Google Scholar]
- 31. Harris C. A., Johnson E. M., Jr 2001. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J. Biol. Chem. 276:37754–37760 [DOI] [PubMed] [Google Scholar]
- 32. Holm G. H., et al. 2010. Interferon regulatory factor 3 attenuates reovirus myocarditis and contributes to viral clearance. J. Virol. 84:6900–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holm G. H., et al. 2007. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 282:21953–21961 [DOI] [PubMed] [Google Scholar]
- 34. Hsu Y. T., Youle R. J. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272:13829–13834 [DOI] [PubMed] [Google Scholar]
- 35. Jackson A. C. 1999. Apoptosis in experimental rabies in bax-deficient mice. Acta Neuropathol. 98:288–294 [DOI] [PubMed] [Google Scholar]
- 36. Jackson A. C., Rossiter J. P. 1997. Apoptosis plays an important role in experimental rabies virus infection. J. Virol. 71:5603–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin Z., El-Deiry W. S. 2005. Overview of cell death signaling pathways. Cancer Biol. Ther. 4:139–163 [DOI] [PubMed] [Google Scholar]
- 38. Kerr D. A., et al. 2002. BCL-2 and BAX protect adult mice from lethal Sindbis virus infection but do not protect spinal cord motor neurons or prevent paralysis. J. Virol. 76:10393–10400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J. W., Lyi S. M., Parrish C. R., Parker J. S. 2011. A proapoptotic peptide derived from reovirus outer-capsid protein μ1 has membrane-destabilizing activity. J. Virol. 85:1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kominsky D. J., Bickel R. J., Tyler K. L. 2002. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9:926–933 [DOI] [PubMed] [Google Scholar]
- 41. Kominsky D. J., Bickel R. J., Tyler K. L. 2002. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 76:11414–11424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Korsmeyer S. J., Shutter J. R., Veis D. J., Merry D. E., Oltvai Z. N. 1993. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin. Cancer Biol. 4:327–332 [PubMed] [Google Scholar]
- 43. Lewis J., et al. 1999. Inhibition of virus-induced neuronal apoptosis by Bax. Nat. Med. 5:832–835 [DOI] [PubMed] [Google Scholar]
- 44. Liang X., et al. 2007. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through Bax. J. Immunol. 178:503–510 [DOI] [PubMed] [Google Scholar]
- 45. Loo Y. M., et al. 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82:335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu M. Y., Liao F. 10 December 2010, posting date. Interferon-stimulated gene ISG12b2 is localized to the inner mitochondrial membrane and mediates virus-induced cell death. Cell Death Differ. [Epub ahead of print.] doi:10.1038/cdd.2010.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marcato P., Shmulevitz M., Pan D., Stoltz D., Lee P. W. 2007. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 15:1522–1530 [DOI] [PubMed] [Google Scholar]
- 48. Martin-Latil S., Mousson L., Autret A., Colbere-Garapin F., Blondel B. 2007. Bax is activated during rotavirus-induced apoptosis through the mitochondrial pathway. J. Virol. 81:4457–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McLean J. E., Datan E., Matassov D., Zakeri Z. F. 2009. Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 83:8233–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller T. M., et al. 1997. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 139:205–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Molouki A., Hsu Y. T., Jahanshiri F., Rosli R., Yusoff K. 2010. Newcastle disease virus infection promotes Bax redistribution to mitochondria and cell death in HeLa cells. Intervirology 53:87–94 [DOI] [PubMed] [Google Scholar]
- 52. Oberhaus S. M., Smith R. L., Clayton G. H., Dermody T. S., Tyler K. L. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J. Virol. 71:2100–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oberle C., et al. 2010. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 17:1167–1178 [DOI] [PubMed] [Google Scholar]
- 54. Park O. H., et al. 2007. Bax-dependent and -independent death of motoneurons after facial nerve injury in adult mice. Eur. J. Neurosci. 26:1421–1432 [DOI] [PubMed] [Google Scholar]
- 55. Parquet M. C., Kumatori A., Hasebe F., Morita K., Igarashi A. 2001. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 500:17–24 [DOI] [PubMed] [Google Scholar]
- 56. Perlson E., Maday S., Fu M. M., Moughamian A. J., Holzbaur E. L. 2010. Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Putcha G. V., Deshmukh M., Johnson E. M., Jr 1999. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 19:7476–7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Putcha G. V., Deshmukh M., Johnson E. M., Jr 2000. Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J. Cell Biol. 149:1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Putcha G. V., et al. 2002. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J. Cell Biol. 157:441–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rasola A., Bernardi P. 2007. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 12:815–833 [DOI] [PubMed] [Google Scholar]
- 61. Rehm M., Dussmann H., Prehn J. H. 2003. Real-time single cell analysis of Smac/DIABLO release during apoptosis. J. Cell Biol. 162:1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Richardson-Burns S. M., Kominsky D. J., Tyler K. L. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 8:365–380 [DOI] [PubMed] [Google Scholar]
- 63. Rodgers S. E., et al. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 71:2540–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roos K., Tyler K. L. 2008. Meningitis, encephalitis, brain abscess, and empyema, p. 2621–2641 In Fauci A. S., et al. (ed.), Harrison's principles of internal medicine, 17th ed. McGraw Hill, New York, NY [Google Scholar]
- 65. Schiff L. A., Nibert M., Tyler K. L. 2007. Orthoreoviruses and their replication, p. 1853–1915 In Knipe D. M., Howley P. M. (ed.), Fields virology, 5th ed. Lippincott William & Wilkins, Philadelphia, PA [Google Scholar]
- 66. Sherry B., Schoen F. J., Wenske E., Fields B. N. 1989. Derivation and characterization of an efficiently myocarditic reovirus variant. J. Virol. 63:4840–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Son K. N., Becker R. P., Kallio P., Lipton H. L. 2008. Theiler's virus-induced intrinsic apoptosis in M1-D macrophages is Bax mediated and restricts virus infectivity: a mechanism for persistence of a cytolytic virus. J. Virol. 82:4502–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Steckley D., et al. 2007. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 27:12989–12999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun W., et al. 2004. Programmed cell death of adult-generated hippocampal neurons is mediated by the proapoptotic gene Bax. J. Neurosci. 24:11205–11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sun Y. F., Yu L. Y., Saarma M., Arumae U. 2003. Mutational analysis of N-Bak reveals different structural requirements for antiapoptotic activity in neurons and proapoptotic activity in nonneuronal cells. Mol. Cell Neurosci. 23:134–143 [DOI] [PubMed] [Google Scholar]
- 71. Sun Y. F., Yu L. Y., Saarma M., Timmusk T., Arumae U. 2001. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J. Biol. Chem. 276:16240–16247 [DOI] [PubMed] [Google Scholar]
- 72. Tyler K. L., Bronson R. T., Byers K. B., Fields B. 1985. Molecular basis of viral neurotropism: experimental reovirus infection. Neurology 35:88–92 [DOI] [PubMed] [Google Scholar]
- 73. Tyler K. L., Leser J. S., Phang T. L., Clarke P. 2010. Gene expression in the brain during reovirus encephalitis. J. Neurovirol 16:56–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ubol S., Sukwattanapan C., Utaisincharoen P. 1998. Rabies virus replication induces Bax-related, caspase dependent apoptosis in mouse neuroblastoma cells. Virus Res. 56:207–215 [DOI] [PubMed] [Google Scholar]
- 75. Uo T., Kinoshita Y., Morrison R. S. 2005. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J. Biol. Chem. 280:9065–9073 [DOI] [PubMed] [Google Scholar]
- 76. Verhagen A. M., et al. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102:43–53 [DOI] [PubMed] [Google Scholar]
- 77. Verhagen A. M., et al. 2007. Identification of mammalian mitochondrial proteins that interact with IAPs via N-terminal IAP binding motifs. Cell Death Differ. 14:348–357 [DOI] [PubMed] [Google Scholar]
- 78. Wei M. C., et al. 2000. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14:2060–2071 [PMC free article] [PubMed] [Google Scholar]
- 79. Wei M. C., et al. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. White F. A., Keller-Peck C. R., Knudson C. M., Korsmeyer S. J., Snider W. D. 1998. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J. Neurosci. 18:1428–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wisniewski M. L., et al. 2011. Reovirus infection or ectopic expression of outer capsid protein micro1 induces apoptosis independently of the cellular proapoptotic proteins Bax and Bak. J. Virol. 85:296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wyttenbach A., Tolkovsky A. M. 2006. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J. Neurochem. 96:1213–1226 [DOI] [PubMed] [Google Scholar]
- 83. Zou H., Li Y., Liu X., Wang X. 1999. An APAF-1·cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 274:11549–11556 [DOI] [PubMed] [Google Scholar]