

Abstract
The paramyxovirus fusion (F) glycoprotein is anchored in the virion membrane in a metastable, pretriggered form. Once triggered, the F protein undergoes a dramatic conformational extension that inserts its hydrophobic fusion peptide into the target cell membrane, then folds back on itself to bring the membranes together and initiate fusion. Unlike most other paramyxoviruses, the respiratory syncytial virus (RSV) F protein alone is sufficient to mediate membrane fusion and virus infection. To study the triggering mechanism of the RSV F protein, we have generated a soluble F (sF) protein by replacing the transmembrane and cytoplasmic tail domains with a 6His tag. The sF protein is secreted efficiently from 293T cells in a fully cleaved form. It is recognized by neutralizing monoclonal antibodies, appears spherical by electron microscopic analysis, and is not aggregated, all consistent with a native, pretriggered trimer. The sF protein was purified on a Ni+2 column and eluted with 50 mM phosphate buffer containing 500 mM NaCl and 250 mM imidazole. Dialysis against 10 mM buffer caused the sF protein to trigger, forming “hat pin”-shaped molecules that aggregated as rosettes, characteristic of the posttriggered form. Further dialysis experiments indicated that the efficiency of triggering correlated well with the reduction of buffer molarity. Reduction of buffer molarity by dilution also resulted in exposure of the fusion peptide, as detected by liposome association, confirming sF protein triggering. Mutation of the furin cleavage site adjacent to the fusion peptide prevented liposome association, further confirming that association is via the fusion peptide.
INTRODUCTION
Respiratory syncytial virus (RSV) is a major pathogen in infants and young children worldwide, causing bronchiolitis and pneumonia in 1% to 5% of this population (36). It is also a significant cause of mortality in the elderly and in immunocompromised patients (9, 20). A humanized monoclonal antibody (MAb) is used to protect premature infants, who are at highest risk for severe RSV disease (30). Despite extensive efforts, a vaccine is not yet available.
RSV is a member of the Mononegavirales in the Paramyxoviridae family, Pneumovirinae subfamily (9). Other viruses in the Paramyxovirinae subfamily require their homologous attachment glycoprotein in addition to the fusion (F) glycoprotein to initiate fusion between the virus membrane and the target cell membrane. In these viruses, the attachment protein not only binds to the target cell receptor but also triggers the F protein, resulting in fusion (14, 17, 23, 37). Fusion allows the viral nucleocapsid to enter the cytoplasm and initiate infection.
The F protein of RSV is unique in that it is capable of causing membrane fusion and initiating virus infection in immortalized, cultured cells in the absence of its attachment G glycoprotein (25, 26, 38). The RSV F protein has been a major target for vaccine and antiviral drug development because of its importance in the viral replication cycle, its conserved sequence and structure, its exposed position in the virion, and its strong immunogenicity (9, 24, 46).
The RSV F protein expressed in nonpolarized cultured cells causes cell-cell fusion at neutral pH, leading to the characteristic syncytia or multinucleated giant cells. Since low pH is not required for cell-cell fusion by the RSV F protein, unlike the influenza virus HA protein, for instance, and since the G attachment glycoprotein is not required for F protein triggering (2, 38), it is not clear what triggers the F protein.
Like other class I viral fusion proteins, the RSV F protein is a trimer. Each monomer is produced in a precursor, F0, form that is modified by N-linked glycans. During passage through the Golgi apparatus, these glycans mature and the protein is cleaved by a furin-like protease in two positions, releasing a 27-amino-acid peptide (pep27) with attached glycans (47, 48). The resulting F protein is composed of the N-terminal F2 protein linked by one, and probably two, disulfide bonds to the F1 transmembrane protein (13). The highly hydrophobic fusion peptide resides at the cleavage-created N terminus of the F1 protein (9). This cleaved form of the RSV F protein is fully active and able to cause cell-cell fusion once it reaches the plasma membrane and to cause virion-cell fusion.
Initially, the paramyxovirus F protein is in a metastable, pretriggered trimer form (46). Upon triggering, it is thought to undergo a dramatic and irreversible conformational change, extending the α-helical region (HRA) that follows the N-terminal fusion peptide, thereby inserting the fusion peptide into the target cell membrane (7). The three F1 monomers then fold back on themselves, juxtaposing HRA with a second α-helix (HRB) adjacent to the transmembrane domain of F1 and forming a highly stable six-helix bundle (6-HB) (7, 18). This action pulls the two membranes together to initiate membrane fusion (18).
The complete X-ray crystal structures of two paramyxovirus soluble F (sF) proteins, one in a pre- and one in a posttriggered conformation, have been solved. The first, the parainfluenza virus (PIV) type 3 F protein, which lacked the transmembrane and cytoplasmic domains, was in the posttriggered form (43). In an attempt to stabilize the pretriggered form for crystallization, the PIV5 sF protein was modified with a trimerization domain at its C terminus (44). The PIV5 sF protein was in a very different, apparently pretriggered form, suggesting that stabilization of the C terminus of the sF protein is important for maintenance of this form. The PIV5 sF protein could be triggered by the addition of a surrogate trigger, heat (11). The natural mechanism by which the PIV5 F protein causes membrane fusion requires the homologous attachment protein. Following receptor binding, a conformational change within the attachment protein is thought to exert a destabilizing effect on the F protein that initiates its refolding (10).
In the present study, we have produced fully cleaved, pretriggered RSV sF protein by removing the transmembrane and cytoplasmic domains and replacing them with tags for purification and detection. This sF protein had a spherical shape and was not aggregated, consistent with it being in the pretriggered form. Dialysis into a low-molarity buffer converted the sF protein to a “hat pin” shape that aggregated in rosettes, characteristic of the posttriggered form of the sF protein. Likewise, dilution of the sF protein with low-molarity buffer in the presence of liposomes resulted in similar hat pin spikes embedded in the lipid membranes. We conclude that low-molarity buffer functions as a surrogate, or perhaps physiological, trigger for the RSV F protein.
MATERIALS AND METHODS
Cells.
Human kidney epithelial 293T cells were grown in Dulbecco modified Eagle medium (DMEM), 10% fetal calf serum (FCS) at 37°C in a humidified chamber with 5% CO2. During sF protein production, transfected 293T cells were grown at 33°C.
Construction of the sF protein and mutant genes.
A codon-optimized version of the RSV D53 F protein gene, similar to that of strain A2, was used to construct the SC-2 soluble F protein gene. Codons 1 to 524 of the F open reading frame, encoding the ectodomain of the F protein, were fused to the FLAG tag followed by the 6His tag by inverted PCR mutagenesis (5), effectively deleting the transmembrane and cytoplasmic domains.
sF mutant proteins with furin cleavage site 1 (fcs-1) (i.e., at the downstream side of pep27; parental sequence KKRKRR), fcs-2 (at the upstream side of pep27; parental sequence RARR), or both sites ablated were generated by inverted PCR mutagenesis. The fcs-1 site was replaced by the enterokinase cleavage site (DDDDK), and the fcs-2 site was replaced with a trypsin cleavage site (NATR) to create the SC-33 and SC-31 sF genes, respectively. The SC-35 sF gene had both fcs sites replaced. The production of all sF proteins was driven by a cytomegalovirus (CMV) promoter from the pcDNA3.1 expression plasmid (Invitrogen).
Production and purification of the sF proteins.
Plasmids containing the sF gene were transfected into 293T cells. The sF protein was purified from the medium harvested at 48 h posttransfection, by using passage over a Ni2+ column (Probond purification system; Invitrogen) and elution with 250 mM imidazole in either 50 mM phosphate buffer (pH 8.0) with 500 mM NaCl or 10 mM HEPES buffer (pH 8.0) without additional NaCl. Samples were reduced and analyzed by Western blotting with 5His MAb (Invitrogen), anti-mouse antibody–horseradish peroxidase (Kirkegaard & Perry Laboratories), and Lumi-Light Western blotting substrate (Roche).
Velocity gradient centrifugation.
Purified sF protein was layered over an 11-ml 20-to-55% (wt/wt) linear sucrose gradient (in HBSS buffer with Ca2+ and Mg2+) and centrifuged at 41,000 rpm for 20 h in an SW41Ti rotor (Beckman Instruments). When indicated below, purified sF protein was incubated at 50°C for 1 h prior to centrifugation. Gradients were collected from the bottom with a peristaltic pump and a fraction collector. Two micrograms of bovine serum albumin was added as carrier, and the proteins were precipitated with 15% (vol/vol) trichloroacetic acid (TCA) and analyzed by Western blotting with anti-5His MAb.
Radioimmunoprecipitation.
293T cells were transfected with plasmids expressing the SC-2 protein and metabolically labeled with [35S]methionine-cysteine (MP Biomedical, Solon, OH) at 100 μCi/ml for 1 h at 37°C, beginning at 46 h posttransfection. The medium was removed, complete DMEM with 10% FCS was added, and incubated continued for another 1 h. Then the medium from these cells was harvested, clarified, and incubated for 1 h at 4°C or 50°C, followed by immunoprecipitation overnight with protein G Plus-Sepharose beads (Calbiochem) precoated with neutralizing anti-RSV F MAbs. The beads were washed six times with 0.01% Triton X-100 in phosphate-buffered saline and resuspended in Laemmli protein loading buffer before boiling for 5 min. Proteins were separated by SDS-PAGE, and protein bands were visualized using a Typhoon PhosphorImager and quantified using ImageQuant TL software (GE Healthcare).
Protein dialysis.
One milliliter of freshly prepared and purified SC-2 sF protein in 50 mM phosphate buffer containing 500 mM NaCl and 250 mM imidazole was promptly dialyzed against six different buffers (pH 8.0) described in the figure legends for 18 h at 4°C using dialysis cassette units (Slide-A-Lyzer; Thermo Scientific). Dialyzed protein samples were analyzed by sucrose velocity gradient and electron microscopy (EM).
Lipsosome association assay.
Liposomes were prepared from an a 8:2:5 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and cholesterol (Avanti Polar Lipids) dissolved in chloroform-methanol (2:1). Lipid films were deposited on glass tubes by removing most of the solvent with a stream of argon and removing any that remained by evaporation under vacuum (Freezone 2.5; Labconco). Lipid films were stored at −20°C under argon for up to 1 month. Before use, the dried lipid film was resuspended in 1 ml of buffer, then taken through five freeze-thaw cycles with vortexing at maximum speed between cycles. The suspension of multilayer liposomes was extruded 40 times through 100-μm filters with a miniextruder (Avanti Polar Lipids). In each protein-liposome reaction mixture, 2.2 μg of sF protein was mixed with 20 μl of 20 mM liposome (4 mmol of total lipids), and the same buffer used in the liposome preparation was added to bring the volume up to 80 μl. The samples were then incubated at 4°C for 30 min. As a control, protein alone was incubated at 4°C for 30 min. When indicated, 80 μl of 100 mM sodium carbonate (pH 11) was added at 21°C for 10 min before sucrose gradient flotation.
Each reaction mixture was combined with 840 μl of 60% sucrose made in the same buffer used in the liposome preparation step to obtain a final sucrose concentration of 50%, and the mixture was overlaid with 1 ml each of 40% sucrose, 30% sucrose, 20% sucrose, and 10 mM HEPES. Following centrifugation in an SW55Ti rotor at 55,000 rpm for 2 h 20 min at 4°C, the gradients were collected in 1.2-ml fractions as described above. Lipids were solubilized in 0.5% Triton X-100 and TCA precipitated as described above. Proteins were analyzed by Western blotting with anti-5His MAb.
Electron microscopy.
sF protein samples were adsorbed onto 300-mesh carbon-coated copper grids freshly treated by glow discharge and stained with fresh 1% sodium uranyl formate, pH 4.5 to 5. An FEI Technai G2 Spirit transmission electron microscope operating at 80 kV was used to examine the sample. For sF protein-liposome mixtures, the sF protein was mixed with liposome in 10 mM HEPES containing 0.8 mM lipid to a final protein concentration of 12.8 μg/ml and incubated at 4°C for 30 min. Samples were examined by EM as described above.
RESULTS
Production of RSV sF protein.
We constructed a full-length, codon-optimized synthetic gene for the RSV F protein to enable plasmid expression from the nucleus (39). Transient expression in 293T cells resulted in syncytium formation (data not shown), demonstrating that the optimized F gene produced a functional product. To facilitate structural and functional studies of the RSV F protein, we modified the gene sequence by removing the transmembrane and cytoplasmic domains to produce an anchorless sF protein, SC-2 (Fig. 1). FLAG and 6His tags were linked to the C terminus to facilitate purification, concentration, and detection. This sF protein sequence retained the entire F protein ectodomain, with its functional furin cleavage sites.
Fig. 1.

The wild-type RSV F protein and the cDNA-expressed SC-2 sF protein generated from it. The wild-type F0 precursor is cleaved intracellularly by a furin-like protease at two sites to generate the mature F1 and F2 subunits, which remain linked by probably two disulfide bonds. The two heptad repeats, HRA and HRB, interact to complete the conformational changes that result in the final posttriggered F protein 6-HB. To generate the cDNA-expressed sF protein, the cytoplasmic and transmembrane (TM) domains were replaced with the FLAG and 6His tags, and the sF protein was expressed from pcDNA3.1.
A plasmid containing the SC-2 sF gene, driven by a CMV promoter, was transfected into 293T cells, and the sF protein was detected in the cell culture medium at 48 h posttransfection (Fig. 2). Samples from the cell lysate (C) and the medium (M) were reduced before loading onto the SDS-polyacrylamide gel for electrophoresis. Both the precursor, sF0, and the fully cleaved sF1 protein were detected intracellularly. However, only the sF1 protein was detected in the medium, indicating that only completely cleaved sF protein was secreted. Because the cell lysates were loaded at a 10-fold-greater level than the medium and the total signals in these lanes were similar, it appears that at least 90% of the sF protein was released into the medium. Thus, the SC-2 sF protein was efficiently cleaved and released from 293T-transfected cells in its fully cleaved form.
Fig. 2.
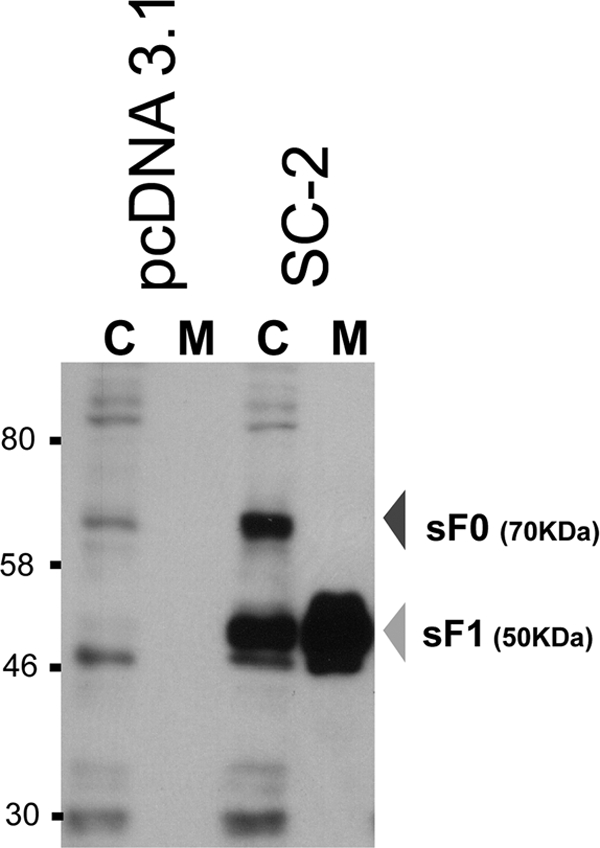
Western blot analysis of the SC-2 sF protein produced from transfected 293T cells at 48 h posttransfection. The sF protein from cell lysates (C) and media (M) were stained with the 5His MAb. The C lanes represent 10-fold more cell equivalents than the M lanes. pcDNA3.1 was included as the empty vector control.
The sF protein is produced in the pretriggered form.
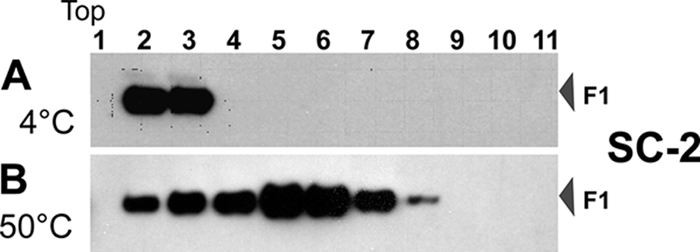
If the SC-2 sF protein represents the pretriggered form of the F protein, MAbs that recognize the RSV F protein and are capable of neutralizing RSV infectivity in cell culture should recognize it. To test this possibility, transfected cells were metabolically labeled with [35S]methionine-cysteine, and the medium was immunoprecipitated individually with 11 neutralizing MAbs representing four antigenic sites with multiple epitopes in each site. All 11 of these MAbs precipitated the sF protein. The findings with one MAb from each antigenic site are presented in Fig. 3. These results suggest that this sF protein is in its native conformation, at least relative to these antigenic sites and their epitopes. The reduced reactivity of these MAbs with heated (50°C) sF protein, presented in Fig. 3, is discussed below.
Fig. 3.

MAb immunoprecipitation of the SC-2 sF protein after incubation at 4°C or 50°C for 1 h. 293T cells were transfected with plasmids expressing SC-2 sF protein and metabolically labeled with [35S]methionine-cysteine. The medium from these cells was incubated for 1 h at 4°C or 50°C, followed by immunoprecipitation with individual MAbs. The antigenic sites for the MAbs that have been mapped by competition (3) are indicated across the top.
Affinity-purified RSV SC-2 sF protein in 50 mM phosphate or 10 mM HEPES buffer was examined by negative staining with EM. It appeared primarily as a sphere with a diameter of 10.9 to 15.2 nm, often with a dark center (Fig. 4A). This shape and the dark center were similar to that previously reported for the pretriggered PIV5 sF protein (10, 11). However, the PIV5 sF protein, whose C terminus was linked to a GCNt self-trimerizing domain, was reported to be somewhat smaller, 7 to 11 nm in diameter, and often included an attached stem. The SC-2 sF protein did not include such a stem, probably because we did not link a trimerization domain to it. The dark center of these sF proteins probably represents a collection of the negative stain in the hollow sF protein head.
Fig. 4.

Electron micrographs of the SC-2 sF protein. Ni2+ affinity-purified SC-2 sF protein was analyzed by negative staining EM using 1% uranyl formate. (A) SC-2 sF protein eluted from the Ni2+ beads in either 50 mM phosphate buffer, 500 mM NaCl, 250 mM imidazole (images in sections 1 and 3), or 10 mM HEPES buffer (section 2) with imidazole. (B) The SC-2 protein in 50 mM phosphate buffer from panel A, heated to 50°C for 30 min. (C) SC-2 protein released from Ni2+ beads in 50 mM phosphate buffer, 500 mM NaCl, and 250 mM imidazole and dialyzed against 10 mM HEPES buffer for 18 h at 4°C.
A small number of rosette aggregates were detected in the RSV sF protein preparations (Fig. 4A, section 3, final panel with asterisk next to it). The individual molecules in these rosettes have a “hat pin” shape with a much smaller head and a thin, long body compared to the majority of the sF molecules, which appear as spheres. Similar hat pin-shaped or cone-shaped structures were previously observed for the posttriggered RSV sF protein (6), the PIV3 sF protein, and for the heat-triggered PIV5 sF protein (11). These structures likely represent the posttriggered form of the sF protein, aggregated via their highly hydrophobic fusion peptides. This low level of triggering presumably occurred nonspecifically during preparation. The effects of heat and dialysis on the sF protein presented in this figure are discussed below.
Test of mild heat as a surrogate trigger for the RSV sF protein.
To determine whether mild heat acts as a surrogate trigger for the SC-2 sF protein as it had for the PIV5 sF protein (11), we compared SC-2 sF protein maintained at 4°C or at 50°C for 1 h by analysis on sucrose gradients. If the unheated SC-2 sF protein were in the pretriggered form, trimers would remain unaggregated and near the top of a velocity sedimentation sucrose gradient (4, 11). Aggregates, including posttriggered trimers, would move further into the gradient. The SC-2 sF protein was found in fractions 2 and 3 (Fig. 5A), near the top of the gradient, indicating that it was not in an aggregated form. This conclusion is consistent with the EM identification of individual pretriggered sF protein spheres.
Fig. 5.
Analysis of the SC-2 sF protein aggregation state by velocity sucrose gradient centrifugation. Freshly prepared and purified SC-2 was incubated at 4°C or 50°C for 1 h before loading on top of a linear 25%-to-55% sucrose gradient for ultracentrifugation in an SW41 rotor at 41,000 rpm for 20 h. The protein in each fraction was TCA precipitated, separated by SDS-PAGE, and detected by Western blotting with 5His MAb. The top fraction of the gradient of the gradient is indicated.
In contrast, the heated SC-2 sF protein migrated further into the gradient, with a peak in fractions 5 and 6 (Fig. 5B). By EM, most of the heated sF protein appeared to be in larger aggregates with no obvious definition (Fig. 4B, section 1), probably due to heat-induced denaturation and aggregation. A smaller portion of the heat-treated sF protein appeared as rosettes of hat pins with their thin ends pointing toward the center (Fig. 4B, section 2), indicative of posttriggered sF proteins aggregated by their hydrophobic fusion peptides. These rosettes ranged from 36 to 43 nm in diameter (Fig. 4B, section 2).
Consistent with this interpretation, 50°C treatment resulted in the loss of nearly all reactivity with the F-specific MAbs (Fig. 3, + lanes). Together, these results confirmed that mild heating induces a dramatic change in the RSV sF protein conformation, resulting in aggregation that destroys or hides all of the antigenic sites in most of the protein molecules rather than causing an orderly triggering process.
Effect of dialysis on protein triggering.
With the exception of the immunoprecipitation experiment (Fig. 3), the sF protein was partially purified and concentrated by Ni2+ column chromatography. It was released from the column by 250 mM imidazole in 50 mM phosphate, 500 mM NaCl buffer. To reduce the salt concentration and remove the imidazole from the purified sF protein solution, we dialyzed the solution against 10 mM HEPES buffer. Surprisingly, dialysis converted most of the sF protein spheres to rosettes (Fig. 4C).
We reasoned that the sF protein must have been triggered by one of three changes: exposure to the low-molarity buffer (10 mM HEPES); the loss of imidazole; or contact with the dialysis membrane. To distinguish among these possibilities, we dialyzed the purified sF protein (50 mM phosphate, 500 mM NaCl, 250 mM imidazole) with three buffers, each with or without 250 mM imidazole, for a total of six conditions. The three buffers were the same buffer the sF protein was in, 10 mM phosphate with 100 mM NaCl, and 10 mM HEPES. Dialyzed samples were examined by EM and by sucrose velocity sedimentation to determine which variable caused the change in protein morphology and aggregation.
When 50 mM phosphate, 500 mM NaCl buffer, with or without imidazole, was used for dialysis, the sF protein retained its spherical shape and was not aggregated (Fig. 6A and B), corresponding to the predialysis unaggregated pretriggered protein (Fig. 5A). These results indicate that contact with the dialysis membrane did not cause the sF protein to trigger. Also, removal of the imidazole failed to cause the sF protein to trigger, indicating that imidazole is not essential for maintaining the pretriggered form.
Fig. 6.

Sucrose velocity gradients and electron micrographs of the SC-2 sF protein following dialysis with six different buffers. A 1-ml aliquot of freshly prepared and purified SC-2 in 50 mM phosphate (PO4) buffer containing 500 mM NaCl and 250 mM imidazole was dialyzed against six different buffers for 18 h at 4°C before sucrose velocity gradient and EM analyses. The buffers and the net salt molarity are listed above each sucrose gradient panel. Proteins were detected by Western blotting as described in the legend to Fig. 5.
Dialysis against either 10 mM phosphate buffer with 100 mM NaCl or 10 mM HEPES buffer, both in the presence of 250 mM imidazole, converted a small amount of the sF protein spheres to rosettes, consistent with the migration of a small amount of the protein further into the gradient (Fig. 6C and D). In the absence of imidazole, more of the sF protein was converted to the posttriggered form when dialyzed against 10 mM phosphate buffer, 100 mM NaCl (Fig. 6E), and nearly all of it was converted to the posttriggered form when dialyzed against 10 mM HEPES buffer (Fig. 6F), as shown above in Fig. 4C. Since the removal of imidazole was ruled out (see above) as the single cause of triggering, these results indicated that low buffer molarity is responsible for triggering the sF protein. Imidazole contributes to this molarity effect in conjunction with both the 10 mM phosphate, 100 mM NaCl, and the 10 mM HEPES buffers.
This posttriggered sF protein retained reactivity with all five of the anti-F MAbs (data not shown) that were demonstrated in Fig. 3 to react with the pretriggered sF protein. Apparently none of these antigenic sites was altered by triggering. None of these MAbs bound to the sF protein in a Western blot assay (data not shown), indicating that their corresponding antigenic sites are conformational.
Association of the sF protein with liposomes.
The classical method for detecting triggering of a viral fusion protein is by liposome association and coflotation. A soluble fusion protein does not insert its fusion peptide into the liposomes until an agent that triggers the fusion protein is added (12, 16, 22). Liposome association is detected by coflotation of the fusion protein with the liposomes. For instance, a mixture of trypsin-treated PIV5 sF protein and liposomes treated with a surrogate stimulus, mild heat (60°C for 30 min), causes triggering, liposome association, and coflotation (11).
To confirm that triggering is induced by low molarity, we prepared large unilamellar liposomes in each of the six buffers used above. We mixed 30 μl of sF protein with 50 μl of liposomes in each buffer and incubated the mixtures at 4°C for 30 min. To separate the free sF protein from the liposomes, we added sucrose to a final concentration of 50%, overlaid with solutions of 40%, 30%, 20%, and 0% sucrose, and subjected the gradient to ultracentrifugation. Protein that associates with liposomes will float with them into the two less-dense sucrose fractions.
Without liposomes, the sF protein remained in the bottom two sucrose gradient fractions (Fig. 7G). With liposomes in the high-molarity 50 mM phosphate, 500 mM NaCl buffer, most of the sF protein did not associate with the liposomes, remaining in the bottom two fractions regardless of whether imidazole was included (Fig. 7A and B), indicating that the high-molarity buffer did not cause triggering and fusion peptide exposure. With liposomes in the intermediate-molarity buffers, 10 mM phosphate, 100 mM NaCl or 10 mM HEPES in the presence of 250 mM imidazole, approximately half of the sF protein associated with the liposomes (Fig. 7C and D).
Fig. 7.

Association of the RSV sF protein with POPC-POPE-cholesterol (8:2:5) large unilamellar liposomes. The SC-2 sF protein (30 μl; 2.2 μg) was mixed with liposomes (4.4 mmol) in the presence of the six different buffers and incubated for 30 min at 4°C (lane groups A to F) in two aliquots. One aliquot was treated with carbonate buffer at pH 11 for 10 min at 20°C to release any superficially bound sF protein. Both aliquots were adjusted to a final concentration of 50% sucrose, overlaid with steps of 40%, 30%, and 20% sucrose, and ultracentrifuged in an SW55Ti rotor at 55,000 rpm for 2.5 h. (G) The sF protein was also incubated for 1 h at 4°C without liposomes and analyzed on a sucrose flotation gradient. The protein in each fraction was precipitated and analyzed as described in the legend to Fig. 5. The top fraction of the gradient is indicated.
With liposomes in lower-molarity, 10 mM phosphate, 100 mM NaCl buffer without imidazole, most of the sF protein associated with the liposomes (Fig. 7E). In the lowest-molarity buffer, 10 mM HEPES, nearly all of the sF protein associated with liposomes (Fig. 7F). The interactions between the sF protein and liposomes were not disrupted by pH 11 treatment (Fig. 7, bottom row), indicating that, in all cases, the sF protein was stably inserted into the liposome membrane rather than peripherally associated. As in the EM and velocity gradient assays discussed above, the association with liposomes correlated closely with the molarity of the buffer, confirming that low buffer molarity triggers the sF protein.
If the RSV sF protein were indeed triggered by low-molarity buffer conditions and associated with the liposomes via its fusion peptide, as supposed, we should have been able to visualize the posttriggered sF protein projecting from the liposome-like spikes. Such spikes have been demonstrated in class II membrane fusion proteins from Semliki Forest virus and Dengue virus, in which triggering has been reported to be dependent on low pH and cholesterol (28, 35). Consistent with this expectation, when the sF protein was incubated at 4°C for 30 min with liposomes in 10 mM HEPES buffer, protein spikes were found inserted into the liposome membranes (Fig. 8). These molecules had the same hat pin shape and size (14 to 16 nm by 5.4 nm at the top) of the posttriggered sF protein found in the rosettes (Fig. 4), with the narrow end of the hat pin inserted in the membrane. This is the end that should contain the fusion peptide. These results again indicate that low-molarity buffer causes the sF protein to trigger, with exposure of its fusion peptide and insertion into a lipid membrane.
Fig. 8.

EM of SC-2 sF protein inserted into liposomes. The purified SC-2 protein was incubated with liposomes in 10 mM HEPES at 4°C for 30 min before negative staining and EM examination. A diagram resembling SC-2 spikes inserted into a liposome membrane is shown at the bottom right.
Involvement of the fusion peptide in liposome association.
To confirm that the association between the SC-2 sF protein and the liposome membrane occurred specifically through insertion of the exposed hydrophobic fusion peptide, rather than by a hydrophobic patch on the sF molecule, we generated three RSV sF proteins with mutations in one or the other, or both, furin cleavage sites. Specifically, in SC-31, the upstream cleavage sequence was changed from RARR to NATR; in SC-33, the downstream cleavage site (at the F2-F1 junction, and thus proximal to the fusion peptide) was changed from KKRKRR to DDDDK, and in SC-35, both changes were made.
All three sF cleavage mutant proteins were cloned and produced in 293T cells by transient transfection, as described above for the parental SC-2 sF protein (Fig. 2). Western blot analysis showed that all of these sF protein mutants were efficiently synthesized and released from 293T-transfected cells (Fig. 9A). These results indicated that cleavage is not required for transit through the cell and release into the medium. Under reducing conditions, the SC-33 and SC-35 mutants migrated with a higher molecular mass than the parental SC-2 sF protein, because their F1 proteins retain the pep27 fragment or the pep27+F2 fragment, respectively. All five of the anti-F MAbs shown in Fig. 3 recognized the three cleavage site mutants (data not shown), indicating that, at least as far as these antigenic sites are concerned, these sF mutant proteins are in the native conformation.
Fig. 9.

Efficient secretion of sF protein cleavage mutants and the requirement of cleavage at the furin site adjacent to the fusion peptide for liposome association. (A) Western blot analysis of the SC-2-derived cleavage site mutant produced from transfected 293T cells. The sF protein from cell lysates (C) and media (M) were stained with the 5His MAb. The C lanes represent 10-fold more cell equivalents than the M lanes. pcDNA3.1 is the empty vector control, and SC-2 is the positive-control parental sF protein. (B) Association of furin cleavage site sF mutants with liposomes. Each sF protein was incubated with liposomes in 10 mM HEPES buffer without imidazole at the indicated temperatures for 30 min. The mixtures were then treated with carbonate buffer, pH 11, for 10 min at 20°C to release any superficially bound sF protein before the flotation analysis. The mixtures were analyzed by using a flotation gradient as described in the legend to Fig. 7. The top fraction of the gradient is indicated. As indicated in the cartoons to the left of each panel, SC-2 is the parental protein, with both cleavage sites intact. In SC-31 the upstream cleavage site has been ablated, in SC-33 the downstream, fusion peptide-proximal site has been ablated, and in SC-35 both sites have been ablated.
These cleavage site mutants were tested for liposome association under the most effective triggering condition identified above, 10 mM HEPES buffer. Only SC-31, the sF protein mutant that retained the cleavage site proximal to the fusion peptide, associated with liposomes (Fig. 9B, panel ii), like the parental SC-2 (Fig. 9B, panel i). The two mutants lacking the fusion peptide-proximal cleavage site, SC-33 and SC-35, did not associate with liposomes (Fig. 9B, panels iii and iv). These results confirm that the RSV sF protein associates with liposomes via its fusion peptide, but only after the fusion peptide is released by cleavage, becoming the N terminus of the F1 protein.
DISCUSSION
The RSV F protein appears to be the simplest paramyxovirus fusion protein for study of the triggering process, because it does not need its attachment protein to cause membrane fusion. It is also the major target for vaccine development and for small-molecule antiviral drug development regarding this medically important virus. To initiate studies on the RSV F protein triggering process, we have generated an sF protein that lacks the transmembrane and cytoplasmic domains. This sF protein was secreted in the fully cleaved, pretriggered conformation, as confirmed by its MAb reactivity, spherical morphology, and unaggregated state. The addition of an artificial trimerization domain was not required, as had been found for the only other pretriggered paramyxovirus sF protein that had previously been produced, the PIV5 sF protein (44). We have identified a treatment that triggers the metastable RSV sF protein: placement in a buffer of low molarity. It is not yet clear if this treatment initiates triggering in a manner similar to the physiological triggering of the F protein on the cell or virion surface.
In previous work by other investigators, the sF protein of a different strain of RSV, the Long strain, was generated by removing the transmembrane and cytoplasmic domains (21) in a manner similar to the methods in the present study. However, most of that sF protein aggregated in posttriggered rosettes. That portion of the sF protein population was efficiently cleaved, whereas the sF population that remained unaggregated retained some uncleaved molecules. The addition of trypsin to the unaggregated, uncleaved sF molecules caused them to form rosettes. These results led the authors to speculate that the final proteolytic cleavage triggers the F protein trimers and that in the virion this final cleavage might take place on the surface of the target cell during entry, initiating fusion and infection (4). However, both their supposed pretriggered and posttriggered forms had a very similar appearance by EM, a small round head with a relatively long stem, and both were very stable to heating, showing no evidence for conformational change until the temperature reached approximately 90°C (33). Extreme heat stability is not a characteristic of metastable proteins like the F protein, including the pretriggered form of the PIV5 sF protein (11). However, the posttriggered, 6-HB form of the F protein is very heat stable (1). In hindsight, it is likely that both the uncleaved and cleaved forms of their RSV sF protein were in fact already triggered, as discussed below for the PIV3 sF protein.
The use in the previous study of the F protein from the Long strain, with 6 amino acid differences from the D53 strain that we used, could have contributed to the differences in our results. It is also possible that the triggered sF protein observed in the previous study was due to a difference in the handling of the sF protein. The low pH of the glycine-HCl buffer used to elute their purified sF protein from the MAb column, or its molarity (100 mM), may have caused triggering. We have shown here that a 10 mM phosphate, 100 mM NaCl buffer converts a large proportion of the sF protein to posttriggered rosettes. In addition, it is possible that other differences in the treatment of the sF protein during isolation and storage were responsible. Their sF protein was frozen, thawed, and precipitated with 65% ammonium sulfate prior to analysis (6). We have found that freezing and thawing causes the sF protein to trigger (S. Chaiwatpongsakorn and M. E. Peeples, unpublished data). In contrast, the pretriggered sF protein that we produced and rapidly purified was surprisingly stable when stored in 50 mM phosphate (pH 8.0), 500 mM NaCl, 250 mM imidazole buffer at 4°C, converting to the posttriggered form with a half-life of approximately 3 weeks (Chaiwatpongsakorn and Peeples, unpublished data).
Our ability to produce the RSV sF protein in the pretriggered form, despite complete proteolytic cleavage, indicates that cleavage does not cause triggering of the RSV F protein. This conclusion is consistent with the results of the three successful X-ray crystallographic studies of other paramyxovirus sF proteins, all of which started with a mutant sF protein that could not be cleaved by furin during transport through the cell, to avoid triggering. The structures of both the Newcastle disease virus (8) and the PIV3 (43) sF proteins were, surprisingly, in the posttriggered form. Trypsin treatment of this already-triggered PIV3 sF protein resulted in cleavage at the former furin site and rosette formation (11). Clearly, rosette formation was the result of fusion peptide exposure, not of triggering, because this PIV3 sF protein was already in the posttriggered form before trypsin treatment. Proteolytic cleavage clearly does not serve as a triggering mechanism, as was previously proposed (21).
Removal of the membrane anchor of the PIV3 F protein led to its triggering (43). To avoid this, Yin et al. (44) fused a trimerization domain from a foreign protein, GCNt, to the C terminus of the PIV5 sF protein. This PIV5 sF protein appeared by EM as a sphere with a short stem composed of its HRB and the GCNt α-helical extension (10, 11), similar to the shape found in its pretriggered crystal structure (44). Proteolytic cleavage of this sF protein at the site between F1 and F2 did not trigger a conformational change, reinforcing the idea that cleavage is not the trigger. However, once it is cleaved, the PIV5 sF protein can be triggered by heat, adopting a different structure with a smaller head and a longer stem, similar to the posttriggered 6-HB form of the PIV3 sF protein (11), which forms rosettes. Therefore, the PIV5 sF protein was originally in the pretriggered form, and heat served as a surrogate trigger. In membrane fusion caused by PIV3 and PIV5, engagement of the attachment protein with its receptor is thought to lead to a conformational change within the attachment protein that destabilizes and triggers the F protein, resulting in refolding and leading to membrane fusion (10). As reported here, neither the membrane anchor nor a substitute trimerization domain is necessary to maintain the RSV sF protein in its pretriggered form, as found for the PIV3 and PIV5 sF proteins.
Here we have identified low molarity as a novel method for triggering the RSV sF protein. It is not clear why reduction in buffer molarity causes sF protein triggering, but buffers are known to play a major role in protein stability and conformational change (40). Modifying the ionic strength or molarity of a solution can affect the stability of a protein (32, 41), depending on its charge distribution (27). Similarly charged residues in a protein, which are screened in solutions containing salt, repel each other in low-salt environments (15, 19, 31), thereby destabilizing it.
We found that the RSV sF protein association with liposomes was not temperature dependent, occurring equally well at 37°C and 4°C (Fig. 9). In contrast, the avian leukemia and sarcoma virus (ALSV) attachment glycoprotein associates with liposomes in a temperature-dependent manner, with very poor association at 4°C (12, 22). The influenza virus HA also associates with liposomes in a temperature-dependent manner if the liposomes are composed of dimyristoylphosphatidylcholine (DMPC). However, with liposomes composed of egg phosphatidylcholine (PC), there is no temperature effect, probably because egg PC melts below 0°C and therefore remains fluid at 4°C while DMPC melts at 24°C and therefore would not remain fluid at 4°C (16). From these earlier studies it appears that the melting temperature of the lipids is critical in liposome-fusion protein binding experiments. In other words, the lack of ALSV glycoprotein insertion into the liposomes at low temperature may have been caused by the state of the lipids rather than the lack of glycoprotein triggering. The major lipid in the liposomes that we used in the present studies was POPC, a PC derivative with a low melting temperature similar to PC. It would also be fluid at 4°C.
The addition of heat has been reported to be a surrogate trigger for many full-length paramyxovirus fusion proteins (11, 29, 34, 42, 45). The mechanism probably involves thermal destabilization of the metastable F protein, initiating its conformational change. Yunis et al. (45) found that heating RSV-infected cells to 45 to 55°C enhanced antibody recognition of the membrane-anchored F protein by antiserum specific for the posttriggered 6-HB structure, indicating that elevated temperature can act as a surrogate trigger for the transmembrane RSV F protein. Raising the temperature to 60°C has also been shown to act as a surrogate trigger for the PIV5 sF protein, resulting in liposome association (11). However, in our present study heating the sF protein to 50°C destroyed its ability to react with neutralizing MAbs and caused it to form aggregates, both apparently due to denaturation rather than triggering. The difference between the RSV transmembrane F protein and the released sF protein is the membrane anchor, and so it is possible that when mild heat is used as a surrogate stimulus, the membrane anchor regulates or stabilizes the refolding of the F protein, enabling it to refold properly, resulting in the posttriggered 6-HB form, while without the anchor, the sF protein denatures.
Here, we have produced a fully cleaved paramyxoviral sF protein in its pretriggered form for the first time without the addition of a foreign trimerization domain. We also identified molarity of the solution containing the sF protein as an unexpected but critical component in the production of the pretriggered RSV sF protein and used it as a novel, surrogate method for triggering a viral fusion protein. Although it is not clear whether this triggering method mimics the physiological triggering of the intact F protein, it confirms that the SC-2 sF protein is produced in the pretriggered form. The availability of pretriggered sF protein should enable studies of the RSV sF protein attachment and triggering mechanisms.
ACKNOWLEDGMENTS
We thank Judy Beeler of the WHO Programme for Vaccine Development Reagent Bank for Respiratory Syncytial and Parainfluenza Type 3 Viruses and Edward Walsh for monoclonal antibodies, William Ray for guidance on the structure of the F protein, Gregory Melikian for helpful discussions, and Barbara Newton for excellent technical support.
This work was supported by grant AI47213 from the National Institutes of Health and funds from The Research Institute at Nationwide Children's Hospital. R.M.E. was supported by grant MOP 86608 from the Canadian Institutes of Health Research. P.L.C. was supported by the Intramural Program of NIAID, NIH.
Footnotes
Published ahead of print on 9 February 2011.
REFERENCES
- 1. Baker K. A., Dutch R. E., Lamb R. A., Jardetzky T. S. 1999. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 3:309–319 [DOI] [PubMed] [Google Scholar]
- 2. Barretto N., Hallak L. K., Peeples M. E. 2003. Neuraminidase treatment of respiratory syncytial virus-infected cells or virions, but not target cells, enhances cell-cell fusion and infection. Virology 313:33–43 [DOI] [PubMed] [Google Scholar]
- 3. Beeler J. A., van Wyke Coelingh K. 1989. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: effect of mutation upon fusion function. J. Virol. 63:2941–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Begona Ruiz-Arguello M., et al. 2002. Effect of proteolytic processing at two distinct sites on shape and aggregation of an anchorless fusion protein of human respiratory syncytial virus and fate of the intervening segment. Virology 298:317–326 [DOI] [PubMed] [Google Scholar]
- 5. Byrappa S., Gavin D. K., Gupta K. C. 1995. A highly efficient procedure for site-specific mutagenesis of full-length plasmids using Vent DNA polymerase. Genome Res. 5:404–407 [DOI] [PubMed] [Google Scholar]
- 6. Calder L. J., et al. 2000. Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology 271:122–131 [DOI] [PubMed] [Google Scholar]
- 7. Carr C. M., Chaudhry C., Kim P. S. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U. S. A. 94:14306–14313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L., et al. 2001. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure (Camb.) 9:255–266 [DOI] [PubMed] [Google Scholar]
- 9. Collins P. L., Crowe J. E. J. 2007. Respiratory syncytial virus and metapneumovirus, p. 1601–1646 In Knipe D. M., Howley P. M. (ed.), Fields virology, 5th ed., vol. 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 10. Connolly S. A., Leser G. P., Jardetzky T. S., Lamb R. A. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J. Virol. 83:10857–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly S. A., Leser G. P., Yin H. S., Jardetzky T. S., Lamb R. A. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Damico R. L., Crane J., Bates P. 1998. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 95:2580–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Day N. D., et al. 2006. Contribution of cysteine residues in the extracellular domain of the F protein of human respiratory syncytial virus to its function. Virol. J. 3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng R., Wang Z., Mirza A. M., Iorio R. M. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469 [DOI] [PubMed] [Google Scholar]
- 15. Dominy B. N., Perl D., Schmid F. X., Brooks C. L., III 2002. The effects of ionic strength on protein stability: the cold shock protein family. J. Mol. Biol. 319:541–554 [DOI] [PubMed] [Google Scholar]
- 16. Doms R. W., Helenius A., White J. 1985. Membrane fusion activity of the influenza virus hemagglutinin. The low pH-induced conformational change. J. Biol. Chem. 260:2973–2981 [PubMed] [Google Scholar]
- 17. Ebata S. N., Cote M. J., Kang C. Y., Dimock K. 1991. The fusion and hemagglutinin-neuraminidase glycoproteins of human parainfluenza virus 3 are both required for fusion. Virology 183:437–441 [DOI] [PubMed] [Google Scholar]
- 18. Eckert D. M., Kim P. S. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810 [DOI] [PubMed] [Google Scholar]
- 19. Elcock A. H., McCammon J. A. 1998. Electrostatic contributions to the stability of halophilic proteins. J. Mol. Biol. 280:731–748 [DOI] [PubMed] [Google Scholar]
- 20. Falsey A. R., Hennessey P. A., Formica M. A., Cox C., Walsh E. E. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759 [DOI] [PubMed] [Google Scholar]
- 21. Gonzalez-Reyes L., et al. 2001. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 98:9859–9864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hernandez L. D., et al. 1997. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 139:1455–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu X. L., Ray R., Compans R. W. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J. Virol. 66:1528–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson P. R., Jr., et al. 1987. Antigenic relatedness between glycoproteins of human respiratory syncytial virus subgroups A and B: evaluation of the contributions of F and G glycoproteins to immunity. J. Virol. 61:3163–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kahn J. S., Schnell M. J., Buonocore L., Rose J. K. 1999. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology 254:81–91 [DOI] [PubMed] [Google Scholar]
- 26. Karron R. A., et al. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U. S. A. 94:13961–13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kohn W. D., Kay C. M., Hodges R. S. 1997. Salt effects on protein stability: two-stranded alpha-helical coiled-coils containing interor intrahelical ion pairs. J. Mol. Biol. 267:1039–1052 [DOI] [PubMed] [Google Scholar]
- 28. Liao M., Sanchez-San Martin C., Zheng A., Kielian M. 2010. In vitro reconstitution reveals key intermediate states of trimer formation by the dengue virus membrane fusion protein. J. Virol. 84:5730–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paterson R. G., Russell C. J., Lamb R. A. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30 [DOI] [PubMed] [Google Scholar]
- 30. Pollack P., Groothuis J. R. 2002. Development and use of palivizumab (Synagis): a passive immunoprophylactic agent for RSV. J. Infect. Chemother. 8:201–206 [DOI] [PubMed] [Google Scholar]
- 31. Rao J. K., Argos P. 1981. Structural stability of halophilic proteins. Biochemistry 20:6536–6543 [DOI] [PubMed] [Google Scholar]
- 32. Record M. T., Jr., Zhang W., Anderson C. F. 1998. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 51:281–353 [DOI] [PubMed] [Google Scholar]
- 33. Ruiz-Arguello M. B., et al. 2004. Thermostability of the human respiratory syncytial virus fusion protein before and after activation: implications for the membrane-fusion mechanism. J. Gen. Virol. 85:3677–3687 [DOI] [PubMed] [Google Scholar]
- 34. Russell C. J., Jardetzky T. S., Lamb R. A. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanchez-San Martin C., Sosa H., Kielian M. 2008. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4:600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shay D. K., et al. 1999. Bronchiolitis-associated hospitalizations among US children, 1980–1996. JAMA 282:1440–1446 [DOI] [PubMed] [Google Scholar]
- 37. Tanabayashi K., Compans R. W. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J. Virol. 70:6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Techaarpornkul S., Barretto N., Peeples M. E. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 75:6825–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ternette N., Stefanou D., Kuate S., Uberla K., Grunwald T. 2007. Expression of RNA virus proteins by RNA polymerase II dependent expression plasmids is hindered at multiple steps. Virol. J. 4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ugwu S. O., Apte S. P. 2004. The effect of buffers on protein conformational stability. Pharm. Technol. 2004:86–113 [Google Scholar]
- 41. Vonhippel P. H., Wong K. Y. 1964. Neutral salts: the generality of their effects on the stability of macromolecular conformations. Science 145:577–580 [DOI] [PubMed] [Google Scholar]
- 42. Wharton S. A., Skehel J. J., Wiley D. C. 2000. Temperature dependence of fusion by Sendai virus. Virology 271:71–78 [DOI] [PubMed] [Google Scholar]
- 43. Yin H. S., Paterson R. G., Wen X., Lamb R. A., Jardetzky T. S. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yin H. S., Wen X., Paterson R. G., Lamb R. A., Jardetzky T. S. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yunus A. S., et al. 2010. Elevated temperature triggers human respiratory syncytial virus F protein six-helix bundle formation. Virology 396:226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao X., Singh M., Malashkevich V. N., Kim P. S. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. U. S. A. 97:14172–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zimmer G., Budz L., Herrler G. 2001. Proteolytic activation of respiratory syncytial virus fusion protein. Cleavage at two furin consensus sequences. J. Biol. Chem. 276:31642–31650 [DOI] [PubMed] [Google Scholar]
- 48. Zimmer G., Trotz I., Herrler G. 2001. N-glycans of F protein differentially affect fusion activity of human respiratory syncytial virus. J. Virol. 75:4744–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]