

Abstract
The electronic characteristics of coupling partners in the transmetalation step for the cross-coupling reaction of arylsilanolates have been investigated. The ability to interrogate the transmetalation event by in situ preparation of the arylpalladium(II) silanolate intermediate has enabled a Hammett analysis for both the aryl electrophile and arylsilanolate to be conducted. These studies reveal that electron-donating groups on the silicon nucleophile and electron-withdrawing groups on the electrophile accelerate the transmetalation process.
Keywords: cross-coupling, mechanism, Hammett relationship, transmetalation, organosilicon
1. Introduction and Background
Transition-metal cross-coupling reactions have ascended to premier status in the panoply of strategy level carbon-carbon and carbon-heteroatom bond forming reactions. The great diversity of organometallic donors and organic electrophiles that participate in this process accounts, in large measure, for the widespread adoption of this technology.1 Despite the myriad permutations of donor and acceptor that engage in successful cross-coupling reactions, the basic catalytic cycle under palladium (or nickel) catalysis is remarkably uniform across various families and involves: (1) oxidative addition of an electrophilic halide or pseudo-halide to the M(0) catalyst, (2) transmetalation of the transferable group from an organometallic donor and (3) reductive elimination of the R1MLnR2 to generate a new C-C (or C-X) bond. Although the oxidative addition of an organic halide to a Pd(0) complex is common among most coupling reactions, the transmetalation step is distinctive to the organometallic donor employed. The transmetalation of organostannanes (Stille cross-coupling) has been extensively studied but is still a matter of considerable debate.2> These transfers have been suggested to follow an intermolecular SE2 mechanism by means of stereochemical models. Cross-coupling reactions of organoboranes (Suzuki cross-coupling) are highly base dependent and a pre-coordination of the transferring agent with the transition metal followed by delivery has been proposed for the transmetalation step but no concrete conclusions have been drawn to date.3,4> Similarly, organosilane transmetalations can proceed via a four-centered, closed transition structure (retentive), an acyclic, open process (invertive),5> and a six-centered closed transition structure (synfacial).6> Moreover, transmetalation via both neutral 8-Si-4 and anionic 10-Si-5 intermediates have been demonstrated.7,8 Most of the evidence available to support these pathways comes from kinetic analysis of the catalytic and, in some cases, stoichiometric reactions.
A more detailed understanding of the critical transmetalation step has been sought through the use of Hammett substituent effect analyses for a variety of organometallic donors.9 By establishing the influence of electronic perturbations in a catalytic reaction, the transition state structures can be further refined. The effects of substituents on arylstannanes10, -boranes11, silanes12 and -zincates13 have been studied for cross-coupling reactions, however the electronic influence by the organic acceptor for the transmetalation step is not well established. In a cleverly designed competition study, Hatanaka, Hiyama and co-workers established that electron-rich arenes undergo transmetalation from silicon faster than electron-deficient arenes (ρ = −1.5).12a A more recent study from Shukla and DeShong, however reaches a contradictory conclusion that electron-deficient arenes transfer faster in intermolecular coupling reactions of aryltriethoxysilanes (ρ = 1.4).12b The results described here will shed light on the origin of this disparity and provide the first unified insights into the electronic requirements of both the organic donor and acceptor in a palladium catalyzed cross-coupling reaction.
The general trends for various palladium-catalyzed cross-coupling reactions suggest that the transmetalation step is accelerated by more electron-rich nucleophiles (organic donors) and by electron-poor electrophiles (organic acceptors). However, these trends cannot be unambiguously ascribed to influences on the transmetalation step because the studies were conducted on fully operating catalytic systems wherein substituent effects are manifest in each of the stages of the catalytic cycle.14 Recent studies from these laboratories on the mechanism of the cross-coupling reaction of arylsilanolates with aryl halides in the presence of (t-Bu3P)2Pd documented the intermediacy of a three-coordinate arylpalladium(II) arylsilanolate complex as the pretransmetalation species.7 These studies included the isolation and spectroscopic, crystallographic and kinetic analysis of this intermediate.15 The ability to prepare the pretransmetalation intermediate enables a direct interrogation of the electronic characteristics of the critical step in the catalytic cycle. By systematic variation of the substituents on both the aryl halide and the arylsilanolate, the specific electronic demands can be elucidated. Herein, we describe the results of a Hammett substituent analysis for the transmetalation step in the cross-coupling of arylsilanolates through use of stoichiometrically generated arylpalladium(II) arylsilanolate complexes.
2. Results and Discussion
2.1. Preparation of tert-Butylphosphine Arylpalladium (II) Bromide Complexes
To facilitate the determination of the electronic influences of substituents on both components in the transmetalation step, a number of T-shaped arylpalladium(II) bromide complexes are needed. Fortunately, a number of methods are available to access a variety of electronically differentiated complexes, and the results of the preparation are compiled in Table 1.16 However, methods for the preparation and isolation of highly electron-deficient complexes are noticeably absent. Attempts to prepare the 4-trifluoromethylphenylpalladium bromide complex using neat aryl halide led to rapid decomposition after >1 h of heating at 70 °C.16a To avoid decomposition of the arylpalladium(II) bromide complex, a lower temperature (23 °C) was employed, but less than 20 % of (t-Bu3P)(4-CF3C6H4)PdBr (2e) was obtained. Ultimately, the preparation of 2e was accomplished by employing t-Bu3P•HBr as a catalyst, entry 5.17
Table 1.
Preparation of T-shaped Oxidative Addition Intermediates of Palladium
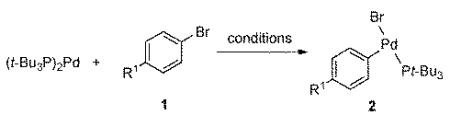 | |||||
|---|---|---|---|---|---|
| entry | R1 | time, min | solvent | product | yield, % |
| 1a | MeO | 45 | neat | 2a | 22 |
| 2b | H | 120 | neat | 2b | 50 |
| 3c | F | 45 | 2-butanone | 2c | 54 |
| 4c | CF3O | 50 | 2-butanone | 2d | 45 |
| 5c | CF3 | 45 | 2-butanone | 2e | 64 |
Conditions:
Pd(dba)2 (1.0 equiv), t-Bu3P (1.1 equiv) aryl bromide (45.0 equiv), 22 °C
(t-Bu3P)2Pd (1.0 equiv), aryl bromide (45.0 equiv), 70 °C
(t-Bu3P)2Pd (1.0 equiv), aryl bromide (40-45.0 equiv), t-Bu3P·HBr (0.03 equiv), 2-butanone, 70 °C (ref. 16)
2.2. Preliminary Investigation on the Electronic Influence of Substituents R1 and R2 on the Activated Transmetalation
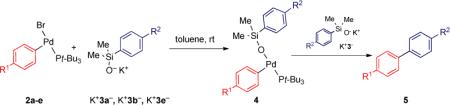
With a number of electronically disparate, three-coordinate, arylpalladium(II) bromide complexes in hand, the rate constants (kobs) for the cross-coupling were obtained. The influence of the electronic properties of R1 in the electrophilic partner (i.e. the arylpalladium(II) species derived from the aryl halide) was investigated for the activated transmetalation with potassium (4-methoxyphenyl)dimethylsilanolate K+3a−. Formation of the arylpalladium(II) silanolate complexes was accomplished in situ by treating the arylpalladium(II) bromides 2 with 0.95 equiv of a solution of K+3− in toluene at room temperature which resulted in the rapid formation of the desired T-shaped complexes 4. Addition of a second quantity of K+3− (1.05 equiv) initiated the transmetalation step and the rate constants were determined from a plot of the first order decay of 4(a-e)a.18 Each reaction was repeated in duplicate and the averaged first-order rate constants for each reaction and their relative rates are collected in Table 2. The arylpalladium(II) arylsilanolate complexes bearing electron-withdrawing substituents on the aryl electrophile ((4c-e)a) underwent transmetalation faster than those bearing either no substituent (4ba) or an electron-donating substituent (4aa) but the effect was small. For example the 4-CF3 and the 4-OCF3 substituted arylpalladium silanolate complexes 4da and 4ea reacted only 2.6 and 1.9 fold faster, respectively, than complex 4aa.
Table 2.
 | |||||
|---|---|---|---|---|---|
| Arylsilanolate K+3−, R2 | Arylpalladium Bromide 2, R1 |
||||
| OMe (2a) |
H (2b) |
F (2c) |
OCF3 (2d) |
CF3 (2e) |
|
| OMe (3a) | 2.80 | 3.27 | 4.64 | 5.56c | 7.20 |
| H (3b) | 1.53 | 1.82 | 2.39 | 2.28 | 3.18 |
| CF3 (3e) | 1.00d | 1.28 | 1.34 | 1.35 | 2.40 |
Reactions conditions: ca 25 mM in 2a-e, ca 50 mM in K+3− in toluene (0.8 mL).
The relative rate constants were calculated from the smallest rate constant which was for the transmetalation of 4ae with K+3e−.
Employed potassium (4-n-butoxyphenyl)dimethyl silanolate (see discussion).
kobs for reaction of 2a with 3e = 7.48 × 10-5 s−1
Given the matrix of data in Table 2, the electronic effect of substituents on the nucleophile (R2) in the transmetalation step could be secured by simply comparing the rate constants for a given electrophile (e. g. Table 2, R1 = H). This analysis reveals that the more electron-rich nucleophile (K+3a−) reacts at a faster rate compared to the other organosilicon donor groups (K+3b− (H) or K+3e− (4-CF3)). For example, the transmetalation of the 4-MeO-substituted arylsilanolate K+3a− was roughly 2.5-4 times faster compared to the 4-CF3-substituted arylsilanolate K+3e− independent of the substitution of the electrophilic partner. When the relative rates for the arylsilanolate transmetalation employing 4bx are plotted against the sigma values for the substituents on the nucleophile, a negative Hammett constant is obtained (Figure 1, ρ = −0.50).19 In addition, the Hammett value for the arylsilanolate seems to be independent of the substituent on the arylpalladium electrophile. Similar ρ values were determined for each arylpalladium electrophile investigated, suggesting that any synergistic effect between the two components for transmetalation was probably negligible.20
Figure 1.

Hammett plot for the transmetalation of various arylsilanolates in the phenylpalladium complexes 4bx.
To probe the contribution of the electronic properties of substituents on the electrophilic partner on the rate of cross-coupling of arylsilanolates, the relative rate constants for the coupling of both potassium (phenyl)dimethylsilanolate K+3b− and potassium (4-trifluoromethylphenyl)dimethylsilanolate K+3e− were investigated with each electrophilic partner, 4a-e (Table 2). Unsurprisingly, both of these nucleophiles displayed similar trends in the rate constants with the various T-shaped complexes. For each arylsilanolate employed, the more electron-rich electrophile (t-Bu3P)(4-MeOC6H4)PdX (4ax) provided the smaller rate constant (i.e. slower transmetalation) whereas the electron deficient complexes (t-Bu3P)(4-CF3OC6H4)PdX and (t-Bu3P)(4-CF3C6H4)PdBr (4dx and 4ex, resp.) provided the larger rate constants (i.e. faster transmetalation).
The Hammett analysis for the cross-coupling reaction of different electrophilic partners employing K+3a− provided a linear relationship (using simple σ values9) with a small ρ value (Figure 2, ρ = 0.49). The Hammett correlation for the effects of substituents on the arylpalladium electrophile on the rates of cross-coupling with arylsilanolate nucleophiles (K+3b− and K+3e−) also provided linear free-energy relationships with ρ = 0.35 and ρ = 0.40, respectively. The fact that different arylsilanolates generate plots with similar ρ values suggests that the influence that the substituent on the arylpalladium electrophile manifests in the transmetalation step is independent of the arylsilanolate nucleophile used. In addition, the small magnitude of the ρ values implies that the rate of cross-coupling of different bromoarenes should not vary widely. In fact, this trend was observed in the preparative cross-coupling of arylsilanolates using (t-Bu3P)2Pd wherein the reaction times for both electron-rich and electron-deficient bromoarenes were comparable within a given arylsilanolate set (3-4 h).21
Figure 2.

Hammett plot for electronic effect for the electrophilic partner employing K+3a−.
2.3. Preequilibrium Considerations
Previous studies on the mechanism of activated cross-coupling of arylsilanolates,7 revealed an interesting insight that potentially complicates the interpretation of the studies herein. Specifically, the rate of cross-coupling of stoichiometrically formed complexes 4 shows a first order dependence on the concentration of added silanolate, whereas under catalytic conditions, a zeroth-order dependence on the silanolate is seen. This behavior is interpreted to reflect a preequilibrium formation of the reactive 10-Si-5 silanolate complex i shown in Scheme 1. This preequilibrium complicates the determination of the electronic influence of substituents on the silanolate (R2) because these electronic perturbations can be manifested in two separate stages of the activated transmetalation step: (1) the association of the silanolate with the silicon atom in the 8-Si-4 arylpalladium silanolate to form the 10-Si-5 species and (2) the formation of the new carbon-palladium bond in the transmetalation step.22 From all of the foregoing studies, the rate of displacement of bromide in the initial formation of the 8-Si-4 species is extremely fast independent of the substituents on the silanolate or the arylpalladium bromide.7 However, the electronic properties of the silanolate will influence the activation of the silicon atom from the 8-Si-4 species to the 10-Si-5 siliconate and thus the rate of the transmetalation (Scheme 1). First, the electrophilicity of the silicon atom in complex 4, and the nucleophilicity of the oxygen atom in silanolate K+3− will affect the equilibrium concentration of complex i (step 1).23 Second, aryl substituents on both partners will influence the accumulation of negative charge on the 10-Si-5 moiety, and thus affect the intrinsic rate of transmetalation (step 2). To eliminate the contribution of the unknown and clearly substituent dependent preequilibrium (factor 1), the activated cross-coupling must be run under conditions where the rate constant for the transmetalation has reached a saturation point. If this kinetic regime could be attained, then the relative rate constants obtained from the transmetalation studies should reflect solely the formation of the palladium-carbon bond (factor 2).
Scheme 1.

2.4. Determination of Saturation Concentration
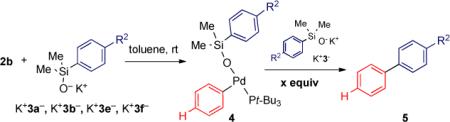
Experimentally, the rate constant at the saturation point can be established by increasing the concentration of the silanolate salts K+3a−, K+3b−, and K+3e− relative to silanolate complex 4 until no further rate enhancement is observed. Unfortunately, the solubility of K+3a− in toluene precluded the saturation point from being determined. Thus, a more soluble, electron-rich silanolate needed to be investigated.
Appending a more lipophilic alkoxy group at C(4) of the arene should improve organic solubility, but should not drastically impact the sigma value for the Hammett analysis.24 Thus, potassium (4-butoxyphenyl)dimethylsilanolate K+3f− was prepared. This reagent proved to be significantly more soluble and allowed the saturation point to be attained. When 2.0 equiv of K+3f− was added to the three-coordinate palladium silanolate complex 4bf to promote the transmetalation event, an increase in the rate constant was obtained (kobs = 3.88 × 10−4 s−1 versus kobs = 2.25 × 10−4 s−1 for 1.0 equiv). At this stage, additional quantities of silanolate were added until either a solubility threshold was reached or leveling of the rate constant was obtained. Gratifyingly, as the number of equivalents of K+3f− increased, the rate constant began to level-off (Table 3, entries 2-4). Finally, a ratio of 8:1 for K+3f−/4bf produced a maximum rate constant of 5.15 × 10−4 s−1.
Table 3.
 | ||||
|---|---|---|---|---|
| entry | R2 | silanolate | equiv of K+3− | kobs (s−1) |
| 1 | OMe | K+3a− | 1.0 | 2.25 × 10−4 |
| 2 | On-Bu | K+3f− | 2.0 | 3.88 × 10−4 |
| 3 | On-Bu | K+3f− | 4.0 | 5.00 × 10−4 |
| 4 | On-Bu | K+3f− | 8.0 | 5.15 × 10−4 |
| 5 | H | K+3b− | 1.0 | 1.25 × 10−4 |
| 6 | H | K+3b− | 2.0 | 1.67 × 10−4 |
| 7 | H | K+3b− | 4.0 | 1.98 × 10−4 |
| 8 | CF3 | K+3e− | 1.0 | 9.20 × 10−5 |
| 9 | CF3 | K+3e− | 2.0 | 9.00 × 10−5 |
| 10 | CF3 | K+3e− | 4.0 | 9.92 × 10−5 |
Reactions conditions: ca 25 mM in 2a-e, ca 50 mM in K+3− in toluene (0.8 mL).
Rate constants were determined by the first-order decay of the arylpalladium silanolate complex 4 using Ph3P(O) as an internal standard.
This same procedure was performed for both K+3b− and K+3e− and different saturation points were reached for each nucleophile (Table 3, entries 5-10). For example, the cross-coupling reaction of (t-Bu3P)(C6H5)Pd(OSiMe2C6H5) 4bb with 2.0 and 4.0 equiv of K+3b− resulted in an increase in rate constants by factors of 1.33 and 1.58, resp. clearly indicating a leveling off. Moreover, carrying out the cross-coupling of (t-Bu3P)(C6H5)Pd(OSiMe2C6H4-4-CF3) 4be with 2.0 and 4.0 equiv of K+3e− result in only a minor variation in the rate constant. Therefore, even at a 1:1 ratio of 4be/K+3e− the preequilibrium was nearly saturated.25,26
2.5. Implications of a Hammett Correlation in the Saturated Regime
The Hammett correlation for the electronic properties associated with the organosilanolate 3 yielded a negative ρ value thereby indicating that the more electron-rich substituents improve the reactivity. Using the saturated rate constants for each nucleophile, the Hammett correlation provides a value for only the palladium-carbon bond-forming event (Scheme 1, step 2). Although a modest change was observed (ρ = −0.80) compared to the values obtained employing only 1.0 equiv of arylsilanolate (ρ = −0.50),19 the slope remained negative, confirming that the transmetalation step is accelerated by electron-rich arylsilanolates (Figure 3). The increase in the slope arises from the greater equilibrium concentration of i(bf) compared to i(be) (see Scheme 1).
Figure 3.

Hammett plot employing kobs at the saturation point for the activated transmetalation step.
The negative Hammett value for the electronic effect of substituents on the arylsilanolate implies that, when saturated as the 10-Si-5 species, the greater electron density on the hypervalent siliconate moiety (in particular on the migrating aryl group) leads to a more facile transmetalation process. Nevertheless, the silicon atom in the 8-Si-4 species 2 plays an important role in establishing the preequilibrium, namely, the electrophilicity of this silicon atom impacted the saturation point of the activation step of the various arylsilanolate nucleophiles. As the concentration of K+3f− increased, the rate constant increased until the concentration dependence for K+3f− leveled off with an 8-fold excess of silanolate per complex 4. On the other hand, as the concentration of K+3e− increased, only minor variations in the rate constant were seen. If the nucleophilicity of the silanolate were the dominant factor, then the more nucleophilic silanolate is expected to reach saturation more quickly. Therefore, the observation that the least nucleophilic arylsilanolate reached a saturation point so readily (fewest equivalents) leads to the conclusion that the electrophilicity of the silicon atom is the major contributor to the activation preequilibrium.
The results presented herein are consistent with an (intramolecular)27 electrophilic aromatic substitution on an activated arylsilanolate moiety by an arylpalladium complex. The mechanism of the activated transmetalation proceeds by generation of an 8-Si-4 arylpalladium(II) silanolate complex (4, Scheme 2) from the first formed oxidative addition product of a bromoarene to (t-Bu3P)2Pd, compound 2 in combination with M+3−. In the presence of additional silanolate, a hypervalent 10-Si-5 siliconate i is formed from 4 in a preequilibrium, which renders the aryl group highly electron-rich. Because the palladium center is coordinatively unsaturated,7 the migrating aryl group can initiate the transmetalation by donation of electron density to the empty coordination site in the formation of a π-complex such as structure iii.28 The arene is then transferred in a rate- limiting electrophilic aromatic substitution process with concerted loss of a polysiloxane unit. This process may proceed via an intermediate σ-complex iv which places a partial positive charge on the arene ring.28,29 The negative ρ values for the arylsilanolates are consistent with an asynchronous generation of the σ-complex in which the formation of the palladium-carbon bond precedes the breaking of the carbon-silicon bond. Following reductive elimination, the monoligated palladium catalyst is regenerated to complete the catalytic cycle.30
Scheme 2.

3. Conclusion
The ability to prepare a pretransmetalation precursor has provided a refined insight into the electronic requirements for the critical transmetalation step in the cross-coupling reaction of organosilanolates. These results parallel other mechanistic studies on the transmetalation of, inter alia, organoboranes in the Suzuki cross-coupling reactions and indicate a common feature among the various cross-coupling reactions. The ability to isolate the pretransmetalation precursor provides a unique opportunity to interrogate other aspects of this critical step, the subject of these studies will reported in due course.
Supplementary Material
Acknowledgement
We are grateful to the National Institutes of Health (GM-63167) for financial support and to Johnson Matthey and Strem for gifts of palladium catalysts. R.C.S. thanks the Aldrich Chemical Company for a Graduate Student Innovation Award and Amgen for a Graduate Fellowship. W.-T.T.C acknowledges the University of Illinois for a graduate fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data: Full experimental procedures, kinetic analysis and characterization data of all new compounds.
References
- 1.(a) de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd Ed. Wiley-VCH; Weinhein: 2004. [Google Scholar]; (b) Negishi E, editor. Handbook of Organopalladium Chemistry. Wiley-Interscience; New York: 2002. [Google Scholar]
- 2.(a) Jutand A. Chem. Rev. 2008;108:2300–2347. doi: 10.1021/cr068072h. [DOI] [PubMed] [Google Scholar]; (b) Espinet P, Echavarren AM. Angew. Chem. Int. Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]
- 3.(a) Matos K, Soderquist JA. J. Org. Chem. 1998;63:461–470. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]; (b) Ridgway BH, Woerpel KA. J. Org. Chem. 1998;63:458–460. doi: 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]; (c) Braga AAC, Morgon NH, Ujaque G, Lledós A, Maseras F. J. Organomet. Chem. 2006;691:4459–4466. [Google Scholar]; (d) Braga AAC, Ujaque G, Maseras F. Organometallics. 2006;25:3647–3654. [Google Scholar]
- 4.Hartwig has demonstrated the intermediacy of an Rh-O-B bond in β-aryl eliminations to form Rh-aryl bonds. Zhao P, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2007;109:1876–1877. doi: 10.1021/ja068587q.
- 5.Hatanaka Y, Hiyama T. J. Am. Chem. Soc. 1990;112:7793–7794. [Google Scholar]
- 6.Denmark SE, Werner NS. J. Am. Chem. Soc. 2010;132:3612–3620. doi: 10.1021/ja910804u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denmark SE, Smith RC. J. Am. Chem. Soc. 2010;132:1243–1245. doi: 10.1021/ja907049y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The N-X-L descriptors were introduced by Martin et al. to designate the bonding about any atom (X) in a resonance structure in terms of the number of valence shell electrons (N) formally associated directly with that atom and the number of ligands (L) directly bonded to it. Perkins CW, Martin JC, Arduengo AJ, Lau W, Alegria A, Kochi J. J. Am. Chem. Soc. 1980;102:7753–7759.
- 9.Hansch C, Leo A, Taft R. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 10.(a) Labadie J, Stille JK. J. Am. Chem. Soc. 1983;105:6129–6137. [Google Scholar]; (b) Farina V, Krishnan B, Marshall D, Roth G. J. Org. Chem. 1993;58:5434–5444. [Google Scholar]
- 11.(a) Zim D, Lando VR, Dupont J, Monteiro AL. Org. Lett. 2001;3:3049–3051. doi: 10.1021/ol016526l. [DOI] [PubMed] [Google Scholar]; (b) Nunes C, Monteiro AL. J. Braz. Chem. Soc. 2007;18:1443–1447. [Google Scholar]; (c) Zim D, Nobre S, Monteiro AL. J. Mol. Cat. A: Chem. 2008;287:16–23. [Google Scholar]; (d) Liang L, Chien P, Huang M. Organometallics. 2005;24:353–357. [Google Scholar]; (e) Nishikata T, Yamamoto Y, Miyaura N. Organometallics. 2004;23:4317–4324. [Google Scholar]; (f) Lando VR, Monteiro AL. Org. Lett. 2003;5:2891–2894. doi: 10.1021/ol034948k. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hatanaka Y, Goda K, Okahara Y, Hiyama T. Tetrahedron. 1994;50:8301–8316. [Google Scholar]; (b) Shukla KH, DeShong P. J. Org. Chem. 2008;73:6283–6291. doi: 10.1021/jo8010254. [DOI] [PubMed] [Google Scholar]
- 13.Dong Z-B, Manolikakes G, Shi L, Knochel P, Mayr H. Chem. Eur. J. 2010;16:248–253. doi: 10.1002/chem.200902132. [DOI] [PubMed] [Google Scholar]
- 14.The study by Mayr and co-workers13 addressed this problem by circumventing the oxidative addition step. These authors provided convincing evidence that electron- donating groups on the arylzinc halide accelerate the transmetalation step but did not perform a detailed Hammett analysis for the organic electrophile in this step. They did clearly demonstrate the electronic demands of the organic halide in the oxidative addition step.
- 15.The conclusions from the preceding study (ref. 7) are as follows: (1) under catalytic conditions, the turnover limiting step is ligand dissociation from (t-Bu3P)2Pd so all subsequent steps are kinetically invisible, (2) arylpalladium(II) arylsilanolate complex 4ca could be fully characterized by solution NMR and X-ray crystallography, (3) 4ca undergoes thermal transmetalation to form biaryl 5ca, (4) the rate of transmetalation of 4ca is 10 fold faster in the presence of K+3a− (1.0 equiv), (5) the activated transmetalation of 4ca is first order in K+3a− but saturation could not be reached because of limited solubility, and (6) the rate of activated transmetalation of 4ca is 4.5 fold faster for Cs+3a− than for K+3a−.
- 16.(a) Stambuli J, Incarvito CD, Buhl M, Hartwig JF. J. Am. Chem. Soc. 2004;126:1184–1194. doi: 10.1021/ja037928m. [DOI] [PubMed] [Google Scholar]; (b) Yamashita M, Hartwig JF. J. Am. Chem. Soc. 2004;126:5344–5345. doi: 10.1021/ja0315107. [DOI] [PubMed] [Google Scholar]
- 17.Barrios-Landeros F, Carrow B, Hartwig JF. J. Am. Chem. Soc. 2008;130:5842–5843. doi: 10.1021/ja711159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The numbering scheme used throughout the paper is #xx where # is the compound number and the first letter refers to the residue from the aryl bromide and the second letter refers to the residue from the silanolate.
- 19.We recognize that the LFERs with three points and a modest R2 are not ideal, but they are sufficient for the purpose of qualitative analysis at this stage.
- 20.See the Supplementary Data for the remaining graphs.
- 21.Denmark SE, Smith RC, Chang W-TT, Muhuhi JM. J. Am. Chem. Soc. 2009;131:3104–3118. doi: 10.1021/ja8091449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The substituent R2 also influences the displacement step on 2 in the catalytic reactions, but that step need not be considered here because the formation of 4 is quantitative and irreversible (KBr precipitation) in the stoichiometric reactions herein.
- 23.Obviously, because the silanolate involved in these two aspects carries the same substituent, these two effects are at odds. Only experimentation can reveal which will dominate the preequilibrium.
- 24.σ (OMe) = −0.27, σ (On-Bu) = −0.32.
- 25.It should be mentioned in passing that our ability to reach a threshold value for the rate of these cross-coupling reactions provides strong support for the intermediacy of species i.
- 26.We have tried to confirm the establishment of a saturated equilibrium spectroscopically. In the saturation experiment employing 8 equiv of 4-n- butoxyaryldimethylsilanolate, the chemical shift change observed at the first data point compared to the unactivated silanolate-palladium complex is 0.206 ppm. This change is not significant enough to be interpreted as the 5-coordinate silicon. However, the perturbation of electron density on silicon from a tetravalent to a pentavalent silicate three bonds away from phosphorus is not likely to effect the 31P chemical shift. On the contrary, if the second silanolate is directly attached to a palladium atom to generate a tetracoordinate palladium species, a significant change in the 31P chemical shift is expected.
- 27.The results reported herein cannot be interpreted in terms of an intramolecular transmetalation, but the zeroth-order dependence on the concentration of silanolate under catalytic conditions supports this conclusion, see ref. 7.
- 28.(a) Ingold CK. Structure and Mechanism in Organic Chemistry. Cornell University Press; Ithaca: 1953. pp. 221–305. [Google Scholar]; (b) Pfeiffer P, Wizinger R. Justus Liebigs Ann. Chem. 1928;461:132–154. [Google Scholar]; (c) Wheland G. J. Am. Chem. Soc. 1942;64:900–908. [Google Scholar]
- 29.The depicted cis configuration of the diarylpalladium complex ii is not intended to imply that this is the immediate product of transmetalation, but rather the necessary configuration for reductive elimination.
- 30.This complete picture of the electronic demands of the two coupling partners together with the recognition that preequilibrium activation of the arylpalladium(II) arylsilanolate must be taken into consideration, provides the necessary backdrop for a discussion of the contradictory conclusions reached by Shukla and DeShong.12b In that study, the authors concluded, on the basis of a positive ρ value, that more electron deficient arylsiliconates undergo transmetalation faster. Although the authors noted that their conclusions contradicted those of Hiyama12a and Farina,10b they made no attempt to reconcile the disparity. Shukla and DeShong explain their observations by asserting that “Electron-withdrawing groups are better at stabilizing the developing negative charge on the ipso-carbon in (the) transition state through inductive effects.” This argument is clearly wrong because the negative charge at the ipso carbon in the transition state is less than in the ground state. Thus, the stabilizing effect of the substituent will be greater in the ground state of the fluorosilicate complex than in the transition state which will lead to a decrease in rate. An argument that reconciles their observations with all previous Hammett studies and with our own observations, focuses on the effect of the para substituent of the arylsiliconate on the activation preequilibrium with TBAF. Our studies have revealed a strong effect of the para substituent of an arylsilanolate on the activation preequilibrium by a second silanolate such that the more electron-deficient arylsilanolates reach the activated pentacoordinate state more readily (compare the saturation points for K+3e− vs. K+3f− (Table 3). Shukla and DeShong used only 1.0 equiv of TBAF for all of their competition studies and therefore likely had higher concentrations of the activated complexes from the electron-deficient arylsiliconates leading to an erroneously higher rate of reaction that they interpreted as a faster transmetalation. However, it must be noted that the reaction studied by Shukla and DeShong involves a different electrophile (π-allylpalladium cation) which could behave differently than the neutral palladium silanolate complexes studies herein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.