

Abstract
Hantaviruses primarily infect the endothelial cell lining of capillaries and cause two vascular permeability-based diseases. The ability of pathogenic hantaviruses to regulate the early induction of interferon determines whether hantaviruses replicate in endothelial cells. Tula virus (TULV) and Prospect Hill virus (PHV) are hantaviruses which infect human endothelial cells but fail to cause human disease. PHV is unable to inhibit early interferon (IFN) responses and fails to replicate within human endothelial cells. However, TULV replicates successfully in human endothelial cells, suggesting that TULV is capable of regulating cellular IFN responses. We observed a >300-fold reduction in the IFN-stimulated genes (ISGs) MxA and ISG56 following TULV versus PHV infection of endothelial cells 1 day postinfection. Similar to results with pathogenic hantaviruses, expressing the TULV Gn protein cytoplasmic tail (Gn-T) blocked RIG-I- and TBK1-directed transcription from IFN-stimulated response elements (ISREs) and IFN-β promoters (>90%) but not transcription directed by constitutively active IFN regulatory factor-3 (IRF3). In contrast, expressing the PHV Gn-T had no effect on TBK1-induced transcriptional responses. Analysis of Gn-T truncations demonstrated that the C-terminal 42 residues of the Gn-T (Gn-T-C42) from TULV, but not PHV, inhibited IFN induction >70%. These findings demonstrate that the TULV Gn-T inhibits IFN- and ISRE-directed responses upstream of IRF3 at the level of the TBK1 complex and further define a 42-residue domain of the TULV Gn-T that inhibits IFN induction. In contrast to pathogenic hantavirus Gn-Ts, the TULV Gn-T lacks a C-terminal degron domain and failed to bind tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3), a TBK1 complex component required for IRF3 activation. These findings indicate that the nonpathogenic TULV Gn-T regulates IFN induction but accomplishes this via unique interactions with cellular TBK1 complexes. These findings fundamentally distinguish nonpathogenic hantaviruses, PHV and TULV, and demonstrate that IFN regulation alone is insufficient for hantaviruses to cause disease. Yet regulating the early IFN response is necessary for hantaviruses to replicate within human endothelial cells and to be pathogenic. Thus, in addition to IFN regulation, hantaviruses contain discrete virulence determinants which permit them to be human pathogens.
INTRODUCTION
Hantaviruses are enveloped viruses which are transmitted to humans by discrete small mammal hosts (5, 40). Pathogenic hantaviruses are responsible for two human diseases, hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary syndrome (HPS), which are characterized by vascular permeability deficits and acute thrombocytopenia (24, 40). Eurasian hantaviruses, including Hantaan virus (HTNV), Dobrava virus (DOBV), and Puumala virus (PUUV), cause HFRS while hantaviruses identified throughout the Americas (i.e., Andes [ANDV], Sin Nombre virus ([SNV], and New York-1 virus ([NY-1V]) are associated with HPS (24, 40, 53). However, at least two nonpathogenic hantaviruses have been identified which are not associated with any human disease, Prospect Hill virus (PHV) and Tula virus (TULV) (24, 37, 40, 46). Contrasting functions of pathogenic and nonpathogenic hantaviruses has permitted analysis of virulence determinants which contribute to hantavirus pathogenesis. In contrast to PHV, pathogenic hantaviruses have been shown to regulate early interferon (IFN) induction (1, 14), and this finding is consistent with IFN regulation by the Gn protein of pathogenic hantaviruses but not nonpathogenic PHV (1, 2).
Interestingly, both pathogenic and nonpathogenic hantaviruses infect human endothelial cells (36, 50); however, PHV induces early cellular IFN responses (14) which block PHV replication in human endothelial cells and, as a result, the pathogenic potential of PHV (1, 2, 14, 40). Integrin receptor regulation further distinguishes pathogenic from nonpathogenic hantaviruses and is associated with increased vascular permeability (9, 10, 16, 39). Only pathogenic hantaviruses use αvβ3 integrin endothelial cell receptors, while TULV and PHV use α5β1 integrins for cell entry (7, 11, 39). β3 Receptors on platelets and endothelial cells regulate fluid barrier functions of the vasculature as well as permeability induced by vascular endothelial growth factor (VEGF) (8, 9, 16). Pathogenic hantaviruses dysregulate normal αvβ3 integrin functions through interactions with inactive conformations of the integrin and dramatically enhance endothelial cell permeability in response to VEGF (9, 33, 39). These responses clearly differentiate pathogenic hantaviruses from nonpathogenic PHV and TULV (9, 10) and are likely to contribute, at least in part, to hantavirus pathogenesis.
Hantaviruses contain a trisegmented (S, M, and L) negative-sense RNA genome and comprise a unique genus within the Bunyaviridae family (24, 40, 53). The Hantavirus L and S segments encode the viral polymerase and nucleocapsid proteins, respectively. The Hantavirus M segment encodes a precursor glycoprotein which is cotranslationally cleaved into two surface glycoproteins, Gn and Gc. The Gn protein contains a 142-amino-acid-long cytoplasmic tail (Gn-T) which mediates several functions, including interactions with the viral nucleocapsid protein that presumably nucleates viral budding into the cis-Golgi compartment (18, 48). In pathogenic hantaviruses the Gn-T has been shown to regulate RIG-I- and TBK1-directed IFN induction, disrupt TRAF3-TBK1 complexes, and mediate binding to tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3) and Src (1, 2, 12–14). However, consistent with the failure to replicate successfully in human endothelial cells, the Gn-T of nonpathogenic PHV fails to regulate RIG-I- and TBK1-directed IFN responses (1, 2, 14). In addition to Gn-T regulation of IFN responses, a nonstructural protein (90 residues) present in an alternate S segment (NSs) open reading frame (ORF) of TULV and PUUV has been suggested to regulate IFN responses by 30% (22, 23, 37, 46, 47). However, additional hantaviruses either do not encode NSs proteins or encode further truncated NSs ORFs (67 to 68 residues) that have not been identified following infection or functionally studied for their role in regulating IFN responses (22, 23).
IFN expression is part of the innate immune response and the first defense against invading viruses (42). Upon infection, viral double-stranded RNA (dsRNA) is detected by the C-terminal helicase domain of RIG-I, which initiates a signaling cascade resulting in the activation of TBK1/IKKε complexes (27, 42, 52). This, in turn, directs IFN regulatory factor 3 (IRF3) phosphorylation and NF-κB activation required for transcription from the IFN-β promoter (19, 21). IFN-β is secreted from endothelial cells and directs the induction of interferon-stimulated genes (ISGs) through the autocrine or paracrine activation of IFN-β receptors and downstream JAK/STAT pathway responses (38). IRF3 activation also induces transcription from a subset of promoters containing interferon-stimulated response elements (ISREs), and, collectively, ISG induction serves to limit viral replication and spread (42, 51). However, successful viral pathogens have evolved mechanisms to circumvent cellular IFN responses by regulating various IFN and ISG induction pathways (20, 26, 34, 42).
In contrast to PHV, pathogenic hantaviruses NY-1V, ANDV, and HTNV fail to induce early IFN responses (1, 14, 43), and previous findings have demonstrated that the Gn-Ts from pathogenic hantaviruses inhibit IFN-β induction (1, 2). NY-1V infection and expression of the NY-1V Gn-T disrupts the TRAF3-TBK1 complex formation required for IFN induction and IRF3 activation as well as TBK1- and TRAF2-directed NF-κB activation (1, 2). In contrast, the PHV Gn-T is unable to regulate IRF3 or NF-κB activation and IFN induction (1, 2, 14). These findings suggest that the Gn-Ts from only pathogenic hantaviruses inhibit TBK1-directed IFN induction and thereby block early IFN responses within infected endothelial cells (1, 2).
Interestingly, although TULV is nonpathogenic, we found that TULV replicates successfully in human endothelial cells and to titers similar to those of pathogenic hantaviruses (9). This indicated a fundamental difference in endothelial cell regulation by nonpathogenic TULV and PHV and suggested that TULV has the ability to regulate IFN induction within human endothelial cells. In this report, we show that, in contrast to PHV, TULV suppresses the IFN-induced genes MxA and ISG56 1 day postinfection. Further expressing the TULV Gn-T blocks TBK1-directed transcription from ISRE, IFN-β, and NF-κB promoters. These findings indicate that, similar to pathogenic hantaviruses, nonpathogenic TULV regulates IFN responses within human endothelial cells. We further demonstrated that the C-terminal 42 residues the Gn-T (Gn-T-C42) protein of TULV regulate TBK1-directed ISRE and IFN promoter transcriptional responses. These findings suggest that although IFN regulation is necessary for hantaviruses to be pathogenic, IFN regulation is not the only determinant of the pathogenic potential hantaviruses.
MATERIALS AND METHODS
Cells and virus.
Vero E6 (ATCC CRL-1586), Cos7, and HEK293 cells (ATCC CRL-1573) were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), penicillin (100 μg/ml), streptomycin sulfate (100 μg/ml), and amphotericin B (50 μg/ml) (Gibco). Human umbilical vein endothelial cells (HUVECs; passages 3 to 7) were purchased from Cambrex and grown in supplemented EBM-2 medium (Cambrex) in the presence of gentamicin (50 μg/ml), amphotericin B (50 μg/ml), and 10% fetal calf serum (Sigma). PHV and TULV were grown as previously described (11) in a biosafety level 2 facility and were determined to be mycoplasma free (Roche). Viral titers were determined by focus assay after immunoperoxidase staining of hantavirus nucleocapsid protein within Vero E6 cells as previously described (11).
Antibodies.
Monoclonal anti-Gal4 (RK5C1; catalog number sc-510) and monoclonal anti-myc (9E10; sc-40) were purchased from Santa Cruz Biotechnology; monoclonal anti-Flag M2 was purchased from Stratagene. Monoclonal anti-β-actin (AC-15) was purchased from Sigma. Antinucleocapsid rabbit polyclonal serum directed at the NY-1V nucleocapsid protein was used to detect nucleocapsid protein from both TULV and PHV as previously described (11). Horseradish peroxidase (HRP)-conjugated anti-mouse and goat anti-rabbit HRP conjugate were purchased from Kirkegaard and Perry Lab Laboratories.
Immunoperoxidase staining of hantavirus-infected cells.
Briefly, cell monolayers were fixed in ice-cold 100% methanol and incubated with a rabbit polyclonal antinucleocapsid serum (1:5,000) (11). Monolayers were washed with phosphate-buffered saline (PBS) and incubated with goat anti-rabbit horseradish peroxidase (HRP) conjugate (Kirkegaard and Perry Laboratories). Monolayers were washed and stained with 3-amino-9-ethyl-carbazole (0.026%) in 0.1 M sodium acetate, pH 5.2, and 0.03% H2O2 for 5 min, and nucleocapsid-containing cells were quantitated.
Quantitative real-time PCR.
Total RNA was extracted from PHV- and TULV-infected HUVECs using an RNeasy Kit (Qiagen) according to the manufacturer's protocol. RNA (1 μg) was reverse transcribed using oligo(dT)18 and a Transcriptor First Strand cDNA Synthesis Kit (Roche) (25°C at 10 min, 55°C at 30 min, and 85°C at 5 min). Specific primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), MxA, and ISG56 were previously described (1). Real-time PCR was performed with cDNA templates using the SYBR green PCR Master Mix (Applied Biosystems) in an Applied Biosystems 7300 Real Time PCR System with PCR conditions as follows: 50°C for 2 min and 95°C for 10 min, followed by 40 amplification cycles of 95°C for 15 s and 60°C for 1 min. All reaction mixtures were normalized to GAPDH mRNA levels, and experiments were performed at least three times with similar results.
Plasmids and transfections.
Plasmids expressing the PHV full-length cytoplasmic tail (pBIND PHV Gn-T) were generated by C-terminally fusing the Gn-T coding region to a Gal4 tag as previously described (1, 12, 13). TULV Gn-T was constructed by extracting total RNA from TULV-infected Vero E6 cells using an RNeasy Kit (Qiagen). RNA was reversed transcribed into cDNA as described above using specific primers containing sequences corresponding to TULV Gn-T (37, 46); amplified cDNA fragments were ligated into pCRII-TOPO (Invitrogen) and subcloned into the pBIND vector (Promega) as previously described (12). The constructs pBIND TULV Gn-T, pBIND TULV Gn-T-C42, and pBIND PHV Gn-T-C42 were generated by amplifying the coding regions of the cytoplasmic tail of TULV and the C-terminal 42 amino acids of TULV Gn-T and PHV Gn-T using PCR primers containing the BamHI and XbaI restriction sites and ligated directionally into pBIND. The pcDNA3-TBK1, pcDNA3-IRF3-5D (where IRF3-5D is IRF3 with five aspartic acid substitutions), pCAGGS (N)RIG-I (a plasmid harboring an N-terminal fragment of RIG-I), pRK-TRAF2, and pRK-TRAF3 N415 expression constructs were generated as previously described (1, 2).
For luciferase assays, transfections were performed using calcium phosphate and 60% confluent HEK293 cells as previously described (1). Cells were transfected with constant amounts of plasmid DNA for 16 h and subsequently maintained in DMEM with 10% FCS for 48 h prior to analysis. For coimmunoprecipitation experiments, Cos7 cells were transfected with 1 μg of pBIND NY-1V Gn-T, pBIND TULV Gn-T, or pBIND PHV Gn-T and 0.5 μg of pRK-TRAF3 N415 (a plasmid expressing C-terminally truncated TRAF3) using Fugene 6 (Roche) (2). Transfected cells were analyzed 48 h posttransfection.
Luciferase assays.
HEK293 cells were cotransfected with either pISRE-luciferase, IFN-β-luciferase, or κB-luciferase promoter reporter constructs (Clontech Laboratories, Inc.) as indicated and a Renilla luciferase construct (pRL-null; Promega) as previously described (1, 2). Cells were cotransfected with a plasmid expressing myc-tagged TBK1 (pcDNA-TBK1; 0.25 μg) or 0.5 μg of a plasmid expressing TRAF2 as previously described (1). Cells were cotransfected with constant amounts of total DNA using indicated amounts of PHV or TULV Gn-Ts or Gn-T-C42 expression vector and empty pBIND vector. Cells were lysed at 48 h posttransfection with 1× passive lysis buffer (Promega) for 15 min at room temperature. Luciferase assays were performed using a Dual Luciferase Assay Kit (Promega) according to the manufacturer's protocol and a Turner Designs TD 20/20 luminometer. Assays were performed in triplicate with similar results.
Immunoprecipitation assay and Western blot analysis.
Cos7 cells transfected with expression constructs expressing Gn-Ts and TRAF3 N415 were treated with MG132 (50 μM) as indicated 6 h before cell lysis. At 48 h posttransfection cells were lysed in coimmunoprecipitation lysis buffer (2, 17). Lysates were clarified by centrifugation, and the Gn-Ts were immunoprecipitated with anti-Gal4 monoclonal antibodies and protein A/G Plus-agarose beads (sc-2003; Santa Cruz Biotechnology). Coimmunoprecipitated proteins were analyzed by Western blotting. pBIND, pBIND Gn-Ts, pBIND-Gn-T-C42, pcDNA3-TBK1, pRK-TRAF2, and pRK-TRAF3 N415 expression was analyzed in cotransfected HEK293 cells and Cos7 cells. Cells were lysed in Laemmli buffer 48 h posttransfection and analyzed by Western blotting using anti-Gal4 (Gn-T) (1:1,000), anti-myc (TBK1) (1:1,000), and anti-Flag M2 (TRAF3 N415 and TRAF2) (1:1,000) (2). Blots were washed with Tris-buffered saline (50 mM Tris, 100 mM NaCl, pH 7.4, 0.05% Tween 20) and incubated with horseradish peroxidase-conjugated anti-mouse IgG (1:2,000). Where indicated, blots were stripped with stripping buffer (62.5 mM Tris-HCl, pH 6.8, 20% SDS, 100 mM β-mercaptoethanol) and incubated with monoclonal anti-β-actin (1:5,000). Blots were developed by fluorography with enhanced chemiluminescence reagent (Pierce).
RESULTS
TULV regulates early ISG induction in infected endothelial cells.
The pathogenic hantaviruses NY-1V and HTNV regulate early IFN responses and replicate successfully within human endothelial cells (1, 2, 14). In contrast, nonpathogenic PHV rapidly induces innate ISG responses which restrict PHV replication within human endothelial cells (1, 14). Expression of the cytoplasmic tail of pathogenic hantavirus Gn proteins (Gn-T), but not PHV Gn-T, reportedly inhibits RIG-I- and TBK1-directed ISRE and IFN-directed transcriptional responses (1, 2). This indicates that the Gn-T of pathogenic hantaviruses regulates IFN induction and that the regulation of early IFN responses is a determinant of hantavirus pathogenesis. However, we found that a discrete nonpathogenic hantavirus, TULV, also replicates successfully in human endothelial cells (Fig. 1). While nonpathogenic PHV fails to replicate in endothelial cells (1, 14) (Fig. 1), TULV titers increase 3 logs from 1 to 3 days postinfection of human endothelial cells, as well as following infection of IFN-deficient Vero E6 cells (Fig. 1A). Figure 1B demonstrates that both PHV and TULV similarly infect >95% of human endothelial cells at 1 day postinfection. However, 3 days postinfection nucleocapsid protein levels are reduced within PHV-infected (1) but not TULV-infected endothelial cells (Fig. 1B). Successful TULV replication within human endothelial cells fundamentally distinguishes nonpathogenic TULV and PHV interactions with human endothelial cells.
Fig. 1.

Hantavirus infection of endothelial cells. (A) Hantavirus replication in HUVECs and Vero E6 cells. HUVECS and Vero E6 cells were infected with PHV and TULV at a multiplicity of infection (MOI) of 1. Viral titers were analyzed 1 to 3 days postinfection by infectious focus assay as previously described (7). (B) HUVECs were either infected with PHV or TULV (MOI of 1) or mock infected. Cells were methanol fixed at 1 to 3 days postinfection, and infected cells were detected by immunostaining using an antinucleocapsid antibody as previously described (7).
PHV infection of human endothelial cells reportedly induces IFN-β and high levels of 24 ISGs 1 day postinfection (14), while antibodies to IFN-β block PHV-induced ISG induction (1, 14). In contrast, the successful replication of TULV within human endothelial cells suggests that TULV, similar to pathogenic hantaviruses, is capable of regulating early IFN responses (1, 2, 14). In order to determine whether TULV regulates the early induction of ISGs within human endothelial cells, we evaluated TULV induction of ISG56 (ISRE directed) and MxA (IFN induced), which are ISGs highly induced by PHV (14). Similar to previous reports (1, 2, 14), we found that PHV infection of human endothelial cells induced both ISG56 and MxA 400- and 800-fold, respectively, 1 day postinfection (Fig. 2). In contrast, TULV-infected endothelial cells resulted in only a 10- to 30-fold induction of MxA and ISG56 at 1 day postinfection (Fig. 2). However, 2 to 3 days postinfection both TULV and PHV (Fig. 2), like NY-1V and HTNV (1, 14), induced MxA and ISG56 >500-fold. Thus, nonpathogenic TULV, like pathogenic NY-1V and HTNV, transiently inhibits the early induction of ISGs within human endothelial cells (1, 14). These findings demonstrate a fundamental difference in early IFN regulation by nonpathogenic TULV and PHV that is consistent with the successful replication of only TULV within human endothelial cells.
Fig. 2.

Induction of MxA and ISG56 following TULV and PHV infection. Induction of the ISGs MxA and ISG56 was monitored by quantitative reverse transcription-PCR. Endothelial cells were infected with either PHV or TULV at an MOI of 1. One to 3 days postinfection, total RNAs were isolated, and MxA and ISG56 mRNA levels were determined in duplicate using quantitative reverse transcription-PCR normalized to GAPDH mRNA levels and relative to mock-infected controls.
TULV Gn-T blocks ISRE and IFN-β transcription at the level of the TBK1 complex.
The Gn-Ts from pathogenic NY-1V and nonpathogenic PHV reportedly differ in their abilities to regulate ISG induction (1, 2). Expressing the NY-1V, but not PHV, Gn-T inhibits RIG-I- and TBK1-directed ISRE and IFN promoter-directed transcriptional responses (1, 2). Since TULV, like NY-1V, replicates in human endothelial cells, we determined whether the Gn-T of TULV also regulates ISG induction. We expressed TULV and PHV Gn-Ts and comparatively evaluated their abilities to inhibit RIG-I- and TBK1-directed ISRE and IFN promoter-directed responses. We found that Gn-Ts from both PHV and TULV were stably expressed and present at comparable levels within transfected cells (Fig. 3A and B). Consistent with previous studies (1, 2) the PHV Gn-T failed to inhibit RIG-I- or TBK1-directed transcriptional responses (Fig. 3A and B). In contrast, expressing the TULV Gn-T inhibited both RIG-I-directed (Fig. 3A) and TBK1-directed (Fig. 3B) ISRE transcription in a dose-dependent manner (Fig. 3B). Cellular levels of TBK1 were unchanged by coexpression of either the TULV or PHV Gn-T (Fig. 3B), suggesting that TULV Gn-T expression does not inhibit ISRE-directed transcription by downregulating cellular TBK1 levels. Further, expressing the TULV, but not PHV, Gn-T dose dependently inhibited TBK1-directed transcription from the IFN-β promoter by >90% (Fig. 3C). These findings indicate that the TULV Gn-T inhibits transcriptional responses directed by ISRE and IFN-β promoters.
Fig. 3.

TULV Gn-T regulates RIG-I- and TBK1-directed ISRE and IFN-β transcription. HEK293 cells were transfected with an ISRE-driven luciferase reporter construct (A, B, and D) or an IFN-β-luciferase reporter construct (C) in the presence or absence of an activating (N)RIG-I (A), TBK1 (B and C), or IRF3-5D (D) expression vector. Cells were cotransfected with NY-1V Gn-T, PHV Gn-T, and TULV Gn-T (A and D) or increasing amounts of plasmid (0.5, 1, and 2 μg) expressing PHV Gn-T and TULV Gn-T (B and C) and the control empty vector (pBIND) to maintain constant DNA transfection levels. NY-1V Gn-T expression was used as a positive control. At 2 days posttransfection, cells were lysed, and luciferase activity was measured and normalized to Renilla luciferase levels. Luciferase activity is reported as the fold increase compared to that of controls lacking (N)RIG-I, TBK1, or IRF3-5D. Assays were performed in triplicate with similar results in at least three separate experiments. Western blot (WB) analysis indicates equal expression of TULV Gn-T and PHV Gn-T (A and B) and of TBK1 cotransfected with TULV Gn-T and PHV Gn-T (B). Cells were lysed at 48 h posttransfection, protein amounts were determined using bicinchoninic acid protein assay, and equal amounts were loaded onto a 10% SDS-PAGE gel. Proteins were detected by Western blotting using anti-Gal4 or anti-myc (Santa Cruz Biotechnology) and anti-mouse HRP-conjugated secondary antibody. Blots were stripped and reprobed with anti-β-actin (Sigma). IB, immunoblot.
TBK1 phosphorylates IRF3, which translocates to the nucleus and directs transcription from ISRE-containing promoters (20, 27, 42). A constitutively active form of IRF3 (IRF3-5D), containing five phospho-mimetic aspartic acid substitutions, induces ISRE transcription in the absence of TBK1 activation (1, 2). We cotransfected cells with IRF3-5D in addition to TULV or PHV Gn-T and evaluated ISRE-directed transcription. However, neither TULV nor PHV Gn-T expression significantly inhibited IRF3-5D-directed transcriptional responses (Fig. 3D). These findings indicate that the TULV Gn-T inhibits transcriptional responses at a point upstream of IRF3 activation at the level of the TBK1 complex.
The C-terminal 42 residues of the TULV Gn-T inhibit ISRE and IFN-β transcription.
Pathogenic hantavirus Gn-Ts contain degron domains within their C-terminal 42 residues (Gn-T-C42 constructs) that have been suggested to regulate IFN responses and contribute to pathogenesis (41). Interestingly, both the PHV and TULV Gn-T proteins are stably expressed in the absence of the proteasomal inhibitor MG132 (Fig. 3) (41). Thus, similar to PHV, the TULV Gn-T lacks apparent degron domains yet regulates IFN transcriptional responses, like pathogenic hantavirus Gn-Ts (1, 2). In order to determine whether the TULV Gn-T-C42 contains an IFN regulatory domain, we comparatively expressed the TULV and PHV Gn-T-C42 constructs and analyzed their effects on TBK1-induced transcription. Like the full-length Gn-T, we found that the C-terminal 42 residues of the TULV Gn protein were stably expressed and inhibited TBK1-directed ISRE and IFN-β transcription >80% and that inhibition was dose dependent (Fig. 4A and B). Although the PHV Gn-T-C42 was also stably expressed, it failed to regulate TBK1-directed transcriptional responses (Fig. 4A). These findings indicate that the C-terminal 42 residues of the TULV Gn-T protein are sufficient to regulate TBK1-directed signaling responses and inhibit IFN induction despite the lack of a degron domain.
Fig. 4.

TULV C42 inhibits TBK1-directed ISRE and IFN-β activation. (A) HEK293 cells were transfected with an ISRE-driven luciferase reporter construct in the presence or absence of an activating TBK1 expression vector. Cells were cotransfected with plasmids expressing PHV, TULV Gn-T-C42, or an empty vector control (pBIND expressing the Gal4 tag) to maintain DNA transfection levels. (B) HEK293 cells were transfected with an IFN-β-driven luciferase reporter construct and with or without an activating TBK1 expression vector. Cells were cotransfected with plasmids expressing PHV Gn-T-C42, increasing amounts of TULV Gn-T-C42, or an empty vector control (pBIND) to maintain DNA transfection levels. Two days posttransfection, luciferase activity was measured as described in the legends of previous figures, and values are reported as the fold increase compared to controls lacking TBK1. Assays were performed in triplicate with similar results in at least two separate experiments. Western blot (WB) analysis showing equal expression of TULV Gn-T-C42 and PHV Gn-T C42 (B) was performed. Cells were lysed at 48 h posttransfection, and equal amounts were loaded onto a 12% SDS-PAGE gel. Proteins were detected by Western blotting using anti-Gal4 (Santa Cruz Biotechnology) and anti-mouse HRP-conjugated secondary antibody. Blots were stripped and reprobed with anti-β-actin (Sigma).
TULV Gn-T blocks TBK1- and TRAF2-directed κB transcription.
IFN-β transcriptional responses require both ISRE and NF-κB activation directed by TBK1-TRAF3 and TBK1-TRAF2 complexes (19), respectively, and pathogenic hantavirus Gn-Ts have been shown to block TBK1-directed NF-κB activation (1, 2). Here, we determined whether the TULV Gn-T also blocks TBK1- and TRAF2-induced transcription from κB promoters. Figure 5 indicates that cells transfected with the TULV, but not PHV, Gn-T result in a 70% to 80% decrease in TBK1- or TRAF2-directed transcription from κB promoters (Fig. 5A and B). Similar to the Gn-T from pathogenic NY-1V (1, 2), these findings indicate that the TULV Gn-T blocks TBK1- and TRAF2-directed κB transcription. Collectively, our findings are consistent with the TULV Gn-T regulating ISRE and IFN-β transcriptional responses at the level of TBK1-TRAF2/TRAF3 (TRAF2/3) complex.
Fig. 5.
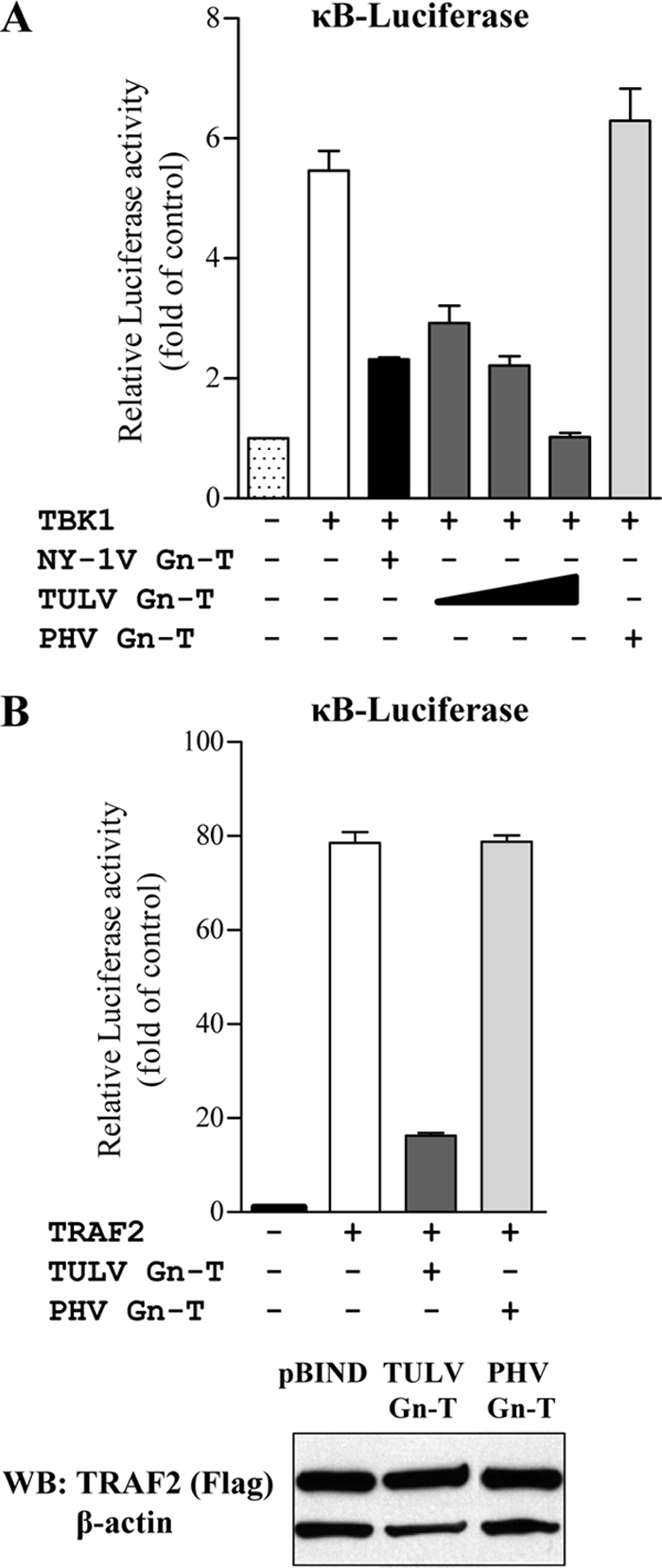
TULV Gn-T inhibition of TBK1- and TRAF2-directed NF-κB activation. HEK293 cells transfected with an NF-κB promoter-luciferase reporter construct in the presence or absence of activating TBK1 (A) or TRAF2 (B) expression vectors and plasmids expressing NY-1V (positive control) or PHV or TULV Gn-T. Luciferase activity within lysates was determined at 48 h posttransfection and normalized to Renilla luciferase activity, and values are reported as the fold increase compared to that of controls lacking TBK1 or TRAF2. Assays were performed in triplicate with similar results in at least three separate experiments. Western Blot (WB) analysis indicates equal expression of TRAF2 in the presence of pBIND, TULV Gn-T and PHV Gn-T (B). Cells were lysed at 48 h posttransfection, and equal amounts were loaded onto a 10% SDS-PAGE gel. Proteins were detected by Western blotting using anti-Flag M2 (Stratagene) and anti-mouse HRP-conjugated secondary antibody. Blots were stripped and reprobed with anti-β-actin (Sigma).
TULV and PHV Gn-T interactions with TRAF3.
A previous report indicates that the NY-1V Gn-T coprecipitates TRAF3 and that Gn-T expression or NY-1V infection disrupts TBK1-TRAF3 complex formation (2). The TULV Gn-T also inhibits transcriptional responses at the level of the TBK1 complex (Fig. 3 and 5), and thus we determined whether the TULV Gn-T inhibits TBK1 responses by binding TRAF3. Cells were cotransfected with Flag-tagged TRAF3 and the NY-1V, TULV, or PHV Gn-T construct and analyzed for the ability of immunoprecipitated Gn-T proteins to coprecipitate TRAF3. The NY-1V Gn-T contains a degron which directs its proteasomal degradation (Fig. 6) (41). Thus, the NY-1V Gn-T coprecipitated TRAF3 only in the presence of the proteasomal inhibitor MG132, while the stable PHV Gn-T (41) failed to coprecipitate TRAF3 in either the presence or absence of MG132. Contrary to our hypothesis, the TULV Gn-T also failed to coprecipitate TRAF3 in the presence or absence of MG132. Thus, the TULV Gn-T, similar to the NY-1V Gn-T, regulates ISG induction (1) but accomplishes this through a unique mechanism that is independent of degron domains and TRAF3 binding interactions.
Fig. 6.

The TULV Gn-T does not interact with TRAF3 N415. Cos7 cells were transfected with pBIND or pBIND-NY-1V Gn-T, -TULV Gn-T, and -PHV Gn-T and pRK-TRAF3 N415 as previously described (2). Briefly, at 6 h prior to lysis, transfected cells were treated with MG132 or mock treated as indicated, and proteins were analyzed at 48 h posttransfection. Hantavirus Gn-Ts were immunoprecipitated (IP) using an anti-Gal4 antibody (Santa Cruz Biotechnology) and protein A/G Plus-agarose beads (Santa Cruz Biotechnology). Coimmunoprecipitated TRAF3 N415 was detected by Western blotting using an anti-Flag M2 antibody (Stratagene). TRAF3 N415 expression was analyzed by anti-Flag Western blotting. IB, immunoblotting; *, IgG heavy chain.
DISCUSSION
Although both PHV and TULV are considered nonpathogenic, both hantaviruses enter and synthesize viral proteins within human endothelial cells (5, 32, 36, 40, 50). Interestingly, PHV titers are not amplified within human endothelial cells while TULV replicates to levels similar to those of pathogenic NY-1V and HTNV following infection of human endothelial cells (1, 9, 30, 46). While PHV replicates within IFN locus-deficient Vero E6 cells, PHV infection of endothelial cells results in high-level induction of ISGs, which restricts PHV replication (1, 14, 28, 43). In fact, both pathogenic and nonpathogenic hantaviruses are sensitive to IFN pretreatment or the addition of IFN at early times postinfection (1, 14). However, pathogenic NY-1V, HTNV, and ANDV hantaviruses successfully replicate within human endothelial cells by blocking early IFN responses (1, 14). In this report, we demonstrate that a TULV Gn-T protein regulates early cellular ISG responses similar to pathogenic hantaviruses, and thus nonpathogenic TULV differs from PHV at a fundamental level.
Our results demonstrate that in contrast to results with the PHV Gn-T, expressing the TULV Gn-T blocks IFN induction. These findings are similar to functions of Gn-Ts from pathogenic NY-1V, ANDV, and HTNV, which also regulate TBK1-directed transcriptional responses from ISRE, κB, and IFN-β promoters (1, 2). In this report we have further determined that the C-terminal 42 residues of the TULV Gn protein are sufficient to block TBK1-directed IFN responses, and this uniquely defines specific C-terminal residues that regulate IFN induction. These findings demonstrate for the first time that a nonpathogenic hantavirus Gn-T regulates cellular IFN responses. Thus, at one level, TULV has the potential to be pathogenic since it replicates within human endothelial cells. However, since TULV is nonpathogenic, this finding also demonstrates that inhibiting early IFN responses is necessary but insufficient for a hantavirus to be pathogenic.
Pathogenic hantavirus Gn-Ts contain a degron at their C termini which has been suggested be a determinant of hantavirus pathogenesis and IFN regulation (41, 49). However, the TULV Gn-T is not proteasomally degraded like pathogenic hantavirus Gn-T proteins yet regulates TBK1-directed transcriptional ISRE and IFN-β transcriptional responses. In contrast, the PHV Gn-T is stably expressed (41) but fails to regulate TBK1-directed transcription (1, 2). Although the degron may still be a virulence determinant of pathogenic hantaviruses, these findings demonstrate that the C-terminal degron is not a requirement for Gn-T regulation of IFN responses.
Like NY-1V, the TULV Gn-T regulates TBK1 responses; however, the TULV protein, unlike its pathogenic NY-1V Gn-T counterpart, is unable to bind TRAF3 (2). This suggests that the TULV Gn-T participates in interactions with components of the TBK1 complex that are discrete from those of the NY-1V protein. Our understanding of the TBK1 complex continues to evolve through the recent disclosure of unique pathway-specific regulatory functions of TRAF3 (15, 31, 54) and deubiquitinases which regulate TRAF2- and TRAF3-directed signaling pathways (15, 31). These findings suggest that the TULV Gn-T could interact with a number of TBK1 complex components or regulatory factors in order to inhibit TBK1-directed ISRE and NF-κB transcriptional responses and that inhibition may be restricted to specific pathway activators. These findings further suggest that the ubiquitination and degradation of pathogenic hantavirus Gn-Ts (41) may contribute to TRAF3 binding and TBK1 regulation (1, 2). The mechanism by which TULV and NY-1V Gn-Ts regulate TBK1 complexes remains to be investigated.
Hantaviruses have few cytoplasmic proteins that are capable of regulating IFN signaling pathways. The hantavirus nucleocapsid protein and polymerase are cytoplasmic proteins, and the hantavirus Gn protein contains a long, 142-residue cytoplasmic tail (Gn-T). N-terminal Gn-T domains contain zinc finger motifs which may serve the role of a viral matrix protein by recruiting nucleocapsid protein complexes and directing viral assembly (6, 18). However, expressing the complete Gn-T or the TULV Gn-T C terminus (Gn-T-C42) regulates RIG-I- and TBK1-directed ISG and IFN transcriptional responses (1, 2). Collectively, these findings demonstrate that IFN regulation is contained within C-terminal residues of the hantavirus Gn-T. However, residues required for IFN regulation and Gn-T binding interactions require further investigation.
Additional hantavirus proteins may also contribute to regulating IFN induction or NF-κB activation, both of which are required for IFN-β induction (19, 21). The nucleocapsid protein reportedly inhibits TNF-α-induced NF-κB activation by blocking importin α4-directed nuclear translocation (44). However, there are no data indicating that the nucleocapsid protein blocks IFN induction or that the PHV nucleocapsid protein differentially regulates IFN responses. One report demonstrates little or no change in NF-κB nuclear translocation in nucleocapsid-expressing cells, while another report indicates that the effects of the nucleocapsid protein are virus specific and limited to HTNV, DOBV, and Seoul virus (SEOV) (35, 45). However, the Gn-T, but not the nucleocapsid protein, was reported to inhibit TBK1- and TRAF2/3-directed ISRE-, IFN-, and NF-κB-directed transcriptional responses (1, 2). Yet another report suggests that expressing the SNV glycoproteins Gn and Gc, but not the nucleocapsid protein, reduces Sendai virus-induced IFN-β promoter responses (29). In contrast, neither the ANDV Gn and Gc glycoproteins nor the nucleocapsid protein inhibited IFN-β promoter responses alone, but they were suggested to reduce transcription 45% when coexpressed (29). Thus, viral proteins that regulate IFN responses may differ between hantaviruses. In addition to virus-specific differences, these discrepancies may reflect differences between assaying nuclear translocation or transcriptional responses or between different cell lines and pathway activators, which work through discrete receptors, pathways, and signaling intermediates.
The nonstructural protein NSs, which is expressed from an alternate ORF located within the S gene segment of some hantaviruses, has also been associated with IFN regulation (22, 23, 47). A recent paper suggests that TULV and PUUV NSs proteins localize to perinuclear domains and inhibit IFN-β and NF-κB activation (47). However, NSs inhibits at most 30% of IFN-β responses, and pathogenic ANDV, NY-1V, and HTNV have truncated NSs proteins which, according to previous reports, are neither expressed nor associated with IFN regulation. TULV strains with full-length or truncated NSs proteins replicate equally well for at least 10 passages in Vero E6 cells while TULV with the truncated NSs still replicates within IFN-competent MRC5 cells, but for fewer passages (22, 23). These findings make it unclear whether short NSs proteins regulate IFN responses, whether long NSs proteins are significant regulators of IFN responses, or whether NSs cooperates with Gn-T regulation of IFN responses.
Interestingly, hantaviruses are sensitive to the effects of IFN only at early times postinfection and strongly induce ISGs at later times when they appear to be resistant to IFN responses (1, 14, 25). In fact, high-level ISG induction is observed at late times after all hantavirus infections and occurs in the presence of both Gn and nucleocapsid protein expression. One report suggests that hantaviruses regulate downstream IFN receptor-directed STAT phosphorylation at late times postinfection (43). While all these findings conflict with the high-level ISG induction observed days after hantavirus infection (14), they demonstrate the transient ability of hantaviruses to regulate IFN responses and suggest that the ability of hantavirus proteins to regulate IFN responses is modulated by endothelial cell responses during the course of infection. This underscores the importance of understanding why hantaviruses regulate early but not late IFN responses and of defining the mechanism of hantavirus resistance to high-level IFN responses at late times postinfection.
TULV was previously shown to induce much lower levels of IFN-β than pathogenic HTNV at both transcriptional and secreted levels at all time points, yet MxA was detectable 16 h after TULV, but not HTNV, infection (28). In contrast, this report suggested that TULV replication was a poor relative to that of HTNV, based on titers determined by a chemiluminescence assay (28). Indeed, our results using an infectious focus assay indicate that TULV replicates to titers reported for pathogenic hantaviruses within human endothelial cells (1). In addition, successful TULV replication within human endothelial cells contrasts with that of PHV, which fails to increase in viral titers (1) and induced 300-fold more MxA than TULV 1 day postinfection. These findings indicate that the nonpathogenic nature of TULV is not the result of a failure to replicate within human endothelial cell targets.
Although replication within endothelial cells and evasion of early IFN responses are requirements for hantavirus pathogenesis, TULV replication within human endothelial cells demonstrates that blocking early IFN responses is not sufficient for a hantavirus to be pathogenic. Unique integrin usage may partially explain why both pathogenic and nonpathogenic hantaviruses enter human endothelial cells but have dramatically different effects on endothelial cell functions (7, 9–11). The function of β3 integrins is inhibited by pathogenic hantaviruses, and this integrin also plays an important role in stabilizing fluid barrier functions of the endothelium by regulating permeabilizing responses of VEGF and platelet activation (3, 4, 7, 8, 9–11, 16, 39). In contrast, nonpathogenic TULV and PHV have no effect on β3 integrin functions, and only pathogenic hantaviruses enhance VEGF-directed endothelial cell permeability, which may contribute to pathogenesis (7, 9–11, 16, 39). Findings presented here demonstrate that in addition to IFN regulation, other virulence determinants are required for TULV and PHV to be pathogenic. Conversely, these findings support the concept that altering IFN regulatory functions of pathogenic hantavirus Gn-Ts may be used as a mechanism for hantavirus attenuation.
ACKNOWLEDGMENTS
We thank Laura Cipp and Nicole Glennon for technical support and Nadine Dalrymple for helpful discussions and critical review of the manuscript.
This work was supported by National Institutes of Health grants R01AI47873, PO1AI055621, R21AI1080984, and U54AI57158 (Northeast Biodefense Center, [W. I. Lipkin, director]).
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Alff P. J., et al. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alff P. J., Sen N., Gorbunova E., Gavrilovskaya I. N., Mackow E. R. 2008. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 82:9115–9122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borges E., Jan Y., Ruoslahti E. 2000. Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J. Biol. Chem. 275:39867–39873 [DOI] [PubMed] [Google Scholar]
- 4. Brakenhielm E. 2007. Substrate matters: reciprocally stimulatory integrin and VEGF signaling in endothelial cells. Circ. Res. 101:536–538 [DOI] [PubMed] [Google Scholar]
- 5. Clement J. P. 2003. Hantavirus. Antiviral Res. 57:121–127 [DOI] [PubMed] [Google Scholar]
- 6. Estrada D. F., Boudreaux D. M., Zhong D., St Jeor S. C., De Guzman R. N. 2009. The hantavirus glycoprotein G1 tail contains dual CCHC-type classical zinc fingers. J. Biol. Chem. 284:8654–8660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gavrilovskaya I. N., Brown E. J., Ginsberg M. H., Mackow E. R. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by β3 integrins. J. Virol. 73:3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gavrilovskaya I. N., Gorbunova E. E., Mackow E. R. 2010. Pathogenic hantaviruses direct the adherence of quiescent platelets to infected endothelial cells. J. Virol. 84:4832–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gavrilovskaya I. N., Gorbunova E. E., Mackow N. A., Mackow E. R. 2008. Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor VEGF, while angiopoietin 1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82:5797–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gavrilovskaya I. N., Peresleni T., Geimonen E., Mackow E. R. 2002. Pathogenic hantaviruses selectively inhibit β3 integrin directed endothelial cell migration. Arch. Virol. 147:1913–1931 [DOI] [PubMed] [Google Scholar]
- 11. Gavrilovskaya I. N., Shepley M., Shaw R., Ginsberg M. H., Mackow E. R. 1998. β3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geimonen E., Fernandez I., Gavrilovskaya I. N., Mackow E. R. 2003. Tyrosine residues direct the ubiquitination and degradation of the NY-1 hantavirus G1 cytoplasmic tail. J. Virol. 77:10760–10868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geimonen E., et al. 2003. Hantavirus pulmonary syndrome-associated hantaviruses contain conserved and functional ITAM signaling elements. J. Virol. 77:1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geimonen E., et al. 2002. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. U. S. A. 99:13837–13842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez-Navajas, et al. 2010. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9-driven anti-inflammatory cytokine production. J. Exp. Med. 207:2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorbunova E., Gavrilovskaya I. N., Mackow E. R. 2010. Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 84:7405–7411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hacker H., et al. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207 [DOI] [PubMed] [Google Scholar]
- 18. Hepojoki J., et al. 2010. Cytoplasmic tails of hantavirus glycoproteins interact with the nucleocapsid protein. J. Gen. Virol. 91:2341–2350 [DOI] [PubMed] [Google Scholar]
- 19. Hiscott J. 2007. Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 18:483–490 [DOI] [PubMed] [Google Scholar]
- 20. Hiscott J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282:15325–15329 [DOI] [PubMed] [Google Scholar]
- 21. Hiscott J., et al. 2003. Convergence of the NF-κB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 1010:237–248 [DOI] [PubMed] [Google Scholar]
- 22. Jaaskelainen K. M., et al. 2007. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 79:1527–1536 [DOI] [PubMed] [Google Scholar]
- 23. Jaaskelainen K. M., Plyusnina A., Lundkvist A., Vaheri A., Plyusnin A. 2008. Tula hantavirus isolate with the full-length ORF for nonstructural protein NSs survives for more consequent passages in interferon-competent cells than the isolate having truncated NSs ORF. Virol. J. 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson K. M. 2001. Hantaviruses: history and overview. Curr. Top. Microbiol. Immunol. 256:1–14 [DOI] [PubMed] [Google Scholar]
- 25. Kanerva M., Melen K., Vaheri A., Julkunen I. 1996. Inhibition of Puumala and Tula hantaviruses in Vero cells by MxA protein. Virology 224:55–62 [DOI] [PubMed] [Google Scholar]
- 26. Kawai T., Akira S. 2005. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 17:338–344 [DOI] [PubMed] [Google Scholar]
- 27. Kawai T., et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 28. Kraus A. A., et al. 2004. Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. J. Virol. 78:6143–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levine J. R., et al. 2010. Antagonism of type I interferon responses by new world hantaviruses. J. Virol. 84:11790–11801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X. D., et al. 2004. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J. Gen. Virol. 85:3261–3268 [DOI] [PubMed] [Google Scholar]
- 31. Liang J., et al. 2010. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J. Exp. Med. 207:2959–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mackow E. R., Gavrilovskaya I. N. 2009. Hantavirus regulation of endothelial cell functions. Thromb. Haemost. 102:1030–1041 [DOI] [PubMed] [Google Scholar]
- 33. Matthys V. S., Gorbunova E. E., Gavrilovskaya I. N., Mackow E. R. 2010. Andes virus recognition of human and Syrian hamster β3 integrins is determined by an L33P substitution in the PSI domain. J. Virol. 84:352–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oganesyan G., et al. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211 [DOI] [PubMed] [Google Scholar]
- 35. Ontiveros S. J., Li Q., Jonsson C. B. 2010. Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 401:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pensiero M. N., Sharefkin J. B., Dieffenbach C. W., Hay J. 1992. Hantaan virus infection of human endothelial cells. J. Virol. 66:5929–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plyusnin A., et al. 1994. Tula virus: a newly detected hantavirus carried by European common voles. J. Virol. 68:7833–7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rani M. R., Ransohoff R. M. 2005. Alternative and accessory pathways in the regulation of IFN-beta-mediated gene expression. J. Interferon Cytokine Res. 25:788–798 [DOI] [PubMed] [Google Scholar]
- 39. Raymond T., Gorbunova E., Gavrilovskaya I. N., Mackow E. R. 2005. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent αvβ3 integrin conformers. Proc. Natl. Acad. Sci. U. S. A. 102:1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmaljohn C., Hjelle B. 1997. Hantaviruses: a global disease problem. Emerg. Infect. Dis. 3:95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sen N., Sen A., Mackow E. R. 2007. Degrons at the C terminus of the pathogenic but not the nonpathogenic hantavirus G1 tail direct proteasomal degradation. J. Virol. 81:4323–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seth R. B., Sun L., Chen Z. J. 2006. Antiviral innate immunity pathways. Cell Res. 16:141–147 [DOI] [PubMed] [Google Scholar]
- 43. Spiropoulou C. F., Albarino C. G., Ksiazek T. G., Rollin P. E. 2007. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 81:2769–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor S. L., Frias-Staheli N., Garcia-Sastre A., Schmaljohn C. S. 2009. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 83:1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor S. L., Krempel R. L., Schmaljohn C. S. 2009. Inhibition of TNF-alpha-induced activation of NF-κB by hantavirus nucleocapsid proteins. Ann. N. Y. Acad. Sci. 1171(Suppl. 1):E86–E93 [DOI] [PubMed] [Google Scholar]
- 46. Vapalahti O., et al. 1996. Isolation and characterization of Tula virus, a distinct serotype in the genus Hantavirus, family Bunyaviridae. J. Gen. Virol. 77:3063–3067 [DOI] [PubMed] [Google Scholar]
- 47. Virtanen J. O., Jaaskelainen K. M., Djupsjobacka J., Vaheri A., Plyusnin A. 2010. Tula hantavirus NSs protein accumulates in the perinuclear area in infected and transfected cells. Arch. Virol. 155:117–121 [DOI] [PubMed] [Google Scholar]
- 48. Wang H., Alminaite A., Vaheri A., Plyusnin A. 2010. Interaction between hantaviral nucleocapsid protein and the cytoplasmic tail of surface glycoprotein Gn. Virus Res. 151:205–212 [DOI] [PubMed] [Google Scholar]
- 49. Wang H., Strandin T., Hepojoki J., Lankinen H., Vaheri A. 2009. Degradation and aggresome formation of the Gn tail of the apathogenic Tula hantavirus. J. Gen. Virol. 90:2995–3001 [DOI] [PubMed] [Google Scholar]
- 50. Yanagihara R., Silverman D. J. 1990. Experimental infection of human vascular endothelial cells by pathogenic and nonpathogenic hantaviruses. Arch. Virol. 111:281–286 [DOI] [PubMed] [Google Scholar]
- 51. Yoneyama M., Fujita T. 2007. Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem. 282:15315–15318 [DOI] [PubMed] [Google Scholar]
- 52. Yoneyama M., et al. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737 [DOI] [PubMed] [Google Scholar]
- 53. Zaki S., et al. 1995. Hantavirus Pulmonary Syndrome: pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu S., et al. 2010. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J. Exp. Med. 207:2647–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]