

Abstract
The envelope (E) of dengue virus (DENV) is a determinant of tropism and virulence. At the C terminus of E protein, there is a stem region containing two amphipathic α-helical domains (EH1 and EH2) and a stretch of conserved sequences in between. The crystal structure of E protein at the postfusion state suggested the involvement of the stem during the fusion; however, the critical domains or residues involved remain unknown. Site-directed mutagenesis was carried out to replace each of the stem residues at the hydrophobic face with an alanine or proline in a DENV serotype 4 (DENV4) precursor membrane (prM)/E expression construct. Most of the 15 proline mutations at either EH1 or EH2 severely affected the assembly of virus-like particles (VLPs). Radioimmunoprecipitation and membrane flotation assays revealed that EH1 mutations primarily affect prM-E heterodimerization and EH2 mutations affect the membrane binding of the stem. Introducing four proline mutations at either EH1 or EH2 into a DENV2 replicon packaging system greatly affects assembly and entry. Moreover, introducing these mutations into a DENV2 infectious clone confirmed the impairment in assembly and infectivity. Sequencing analysis of adaptive mutations in passage 5 viruses revealed a change to a leucine or wild-type residue at the original site, suggesting the importance of maintaining the helical structure. Collectively, these findings suggest that the EH1 and EH2 domains are involved in both assembly and entry steps of the DENV replication cycle; this feature, together with the high degree of sequence conservation, suggests that the stem region is a potential target of antiviral strategies.
INTRODUCTION
Dengue viruses (DENVs) belong to the genus Flavivirus in the family Flaviviridae. There are four serotypes, DENV serotype 1 (DENV1), DENV2, DENV3, and DENV4. DENV is the leading cause of arboviral diseases in tropical and subtropical regions (10, 12). While most DENV infections are asymptomatic or result in a self-limited illness, known as dengue fever, some infected individuals develop severe and potentially life-threatening diseases, known as dengue hemorrhagic fever/dengue shock syndrome. Despite considerable efforts to develop therapeutic or prophylactic interventions, no antiviral or vaccine against DENV is currently available (9, 10, 12, 57). DENV is a positive-sense, single-stranded RNA virus with a genome of approximately 10.6 kb in length. Between the 5′ and 3′ untranslated regions, there is a single open reading frame encoding a polyprotein, which is subsequently cleaved by cellular and viral protease into three structural proteins, capsid (C), precursor membrane (prM), and envelope (E), at the N-terminal one-quarter, and seven nonstructural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, at the C-terminal three-quarters (26).
DENV enters the cell through receptor-mediated endocytosis (11, 26, 34, 44). The low-pH environment in the endosome triggers a series of conformational changes of the E protein and results in fusion between the viral membrane and endosomal membrane (3, 22, 26, 32, 34, 61). After uncoating, translation, and genome replication, assembly occurs in the membrane of the rough endoplasmic reticulum (ER), where the immature virions bud into the lumen of the ER and transport through the secretory pathway (26, 29, 34, 56). The mature virions are produced after the cleavage of prM protein by furin or furin-like protease in the trans-Golgi apparatus, though the cleavage is not efficient for DENV (20, 35, 45, 52, 54, 59). A common feature of flaviviral replication is the production of small and slowly sedimenting subviral particles (26, 46). Coexpression of prM and E proteins can produce recombinant virus-like particles (VLPs), on which the E proteins are structurally and antigenically similar to those on infectious virions (7, 47, 53). VLPs have been shown to be a model system to study the functions of prM/E proteins (7, 28, 47), as well as noninfectious serodiagnostic reagents and vaccine candidates (4, 6, 17, 23, 24, 30, 41). Recently, several packaging systems for flaviviral replicon particles, in which a flaviviral replicon-containing reporter gene is supplied in trans with structural proteins (CprME or prME) to generate replicon particles capable of completing one round of infection, have been shown to be convenient and useful tools to study different steps of flavivirus replication and screen for inhibitors (8, 18, 21, 27, 38, 40, 43, 49).
The E protein is a determinant of tropism and virulence, as well as the major target of neutralizing and enhancing antibodies to DENV (2, 9, 13, 26, 34). X-ray crystallographic studies of the N-terminal ectodomain of E protein revealed three domains (domains I, II, and III) consisting predominantly of β strands (31–33). At the C terminus of E protein, there is a stem region which contains two α helices (EH1 and EH2) and a stretch of conserved sequences (CSs) in between, followed by an anchor region, which contains two transmembrane domains (ET1 and ET2) crossing the two leaflets of the lipid bilayer (1, 60) (Fig. 1A). Previously, studies of E protein of the tick-borne encephalitis virus (TBEV) revealed that both ET2 and ET1 were required for assembly of E protein into particles. EH2 could stabilize the prM-E heterodimer, whereas EH1 was involved in the irreversible trimerization of soluble E protein in a low-pH environment (1, 37, 53). A study of the yellow fever virus reported that ET1 and ET2 were involved in the formation of VLPs (36). The possibility that the stem region is involved in the assembly was suggested by a recent report that substitutions of hydrophobic residues in the EH1 of DENV2 E protein increased the production of VLPs (42). Whether other domains of the stem also participate in the assembly and the mechanisms remain unknown. On the basis of the crystal structure of E protein at the postfusion state, the stem region was proposed to be involved in the fusion process, in which the folding back of the stem toward a channel along the intersubunit contact between domains II led to the formation of a fusion pore (3, 22, 32, 34, 61). The observation that peptides derived from the stem region as well as recombinant domain III, which bound to an intermediate following low-pH-induced trimerization, can block the fusion suggested that the stem region was exposed in the prefusion intermediate (14, 25, 48). Interestingly, a prefusion intermediate with outward extension of the stem region was recently captured by a cryo-electron microscopy (cryo-EM) study (19). However, the domains or critical residues of the stem region involved in virus entry and assembly remain largely unclear.
Fig. 1.

Schematic drawing of the stem region of DENV4 E protein, expression, production of VLPs, and prM-E heterodimerization of proline mutants. (A) The C terminus of E protein contains two α-helical domains (EH1 and EH2) and a stretch of CSs in between, followed by two transmembrane domains (ET1 and ET2) (60). Single-letter designations of amino acids are shown, with numbers above indicating the positions of residues at the a or d position of the helical wheel. (B) Helical wheel analysis of the EH1 and EH2 domains revealed an amphipathic helix with hydrophobic residues at the a or d position facing one side and charged or polar residues facing the other side. (C) At 48 h posttransfection, 293T cell lysates and pellets derived from culture supernatants of WT pCB-D4 or mutants were subjected to Western blot analysis using serum from a human dengue case (16, 55) and anticalnexin MAb for cell lysates (lower gel, left). The long-exposure gels for prM bands in pellets are shown (lower gel, right). One representative experiment of three is shown. (D) Dot blot binding assay by using different mouse anti-E MAbs, including flavivirus group-reactive MAbs recognizing domain II (4G2, FL0231), complex-reactive MAb (DD18-5), and type-specific MAb (1H10-6-7), as well as mixed human sera to recognize WT and mutant E proteins (from panel C) in 1% NP-40 lysis buffer and mixtures containing decreasing amounts of native WT E protein in 1% NP-40 lysis buffer and increasing amounts of denatured E protein in reducing (R) buffer. (E) 293T cells transfected with WT or mutants were labeled with [35S]Met, immunoprecipitated with anti-E MAb FL0232, and subjected to SDS-12% PAGE. The prM/E index of a mutant is equal to (intensity of mutant prM band/intensity of WT prM band)/(intensity of mutant E band/intensity of WT E band). Data are means and standard errors from two experiments. The sizes of the molecular mass markers are shown in kDa.
In this study, we investigated the roles of different stem domains in the assembly and entry of DENV by proline- and alanine-scanning mutagenesis in the prM/E expression constructs of DENV4 and found that most of the 15 proline mutations at either EH1 or EH2 severely impair the assembly of VLPs. Examining four mutants with such mutations in a packaging system of DENV2 replicon particles and a full-length infectious clone revealed that in all four mutants the mutations greatly affect assembly and entry, suggesting that EH1 and EH2 are involved in both assembly and entry steps of the DENV replication cycle.
MATERIALS AND METHODS
Plasmid constructs of VLPs.
The prM/E expression construct of DENV4 (pCB-CD4) was described previously (15, 16). To generate mutants of the stem region, a two-step PCR mutagenesis was performed using pCB-D4 as template and primers d4EI398P-A2, d4EI398P-B1, d4EM401P-A2, d4EM401P-B1, d4ET405P-A2, d4ET405P-B1, d4EG408P-A2, d4EG408P-B1, d4EM412P-A2, d4EM412P-B1, d4EL415P-A2, d4EL415P-B1, d4ET418P-A2, d4ET418P-B1, d4EF422P-A2, d4EF422P-B1, d4EV425P-A2, d4EV425P-B1, d4EF429P-A2, d4EF429P-B1, d4EL432P-A2, d4EL432P-B1, d4EV436P-A2, d4EV436P-B1, d4EV439P-A2, d4EV439P-B1, d4EV443P-A2, d4EV443P-B1, d4ET446P-A2, and d4ET446P-B1, as described previously (16). After the second-round PCR, the 596-bp product containing the stem mutation was digested with DraIII-NotI and cloned into pCB-D4. All constructs were confirmed by sequencing the entire insert in each construct to rule out a second-site mutation. The sequences of all primers used will be provided upon request.
Cell lysates, VLPs, Western blot analysis, and dot blot assay.
293T cells, prepared in a 10-cm culture dish at 5 × 105 cells per dish 1 day earlier, were transfected with 10 μg of plasmid DNA by the calcium phosphate method. At 48 h posttransfection, culture supernatants were collected (see below) and cells were washed with phosphate-buffered saline (PBS) and treated with 1% NP-40 lysis buffer (100 mM Tris [pH 7.5], 150 mM NaCl, 20 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, and protease inhibitors [Roche Diagnostics]), followed by centrifugation at 20,000 × g and 4°C for 30 min to obtain cell lysates (15). Culture supernatants were clarified by centrifugation at 1,250 × g for 20 min, filtered through a 0.22-μm-pore-sized membrane (Sartorius), layered over a 20% sucrose buffer, and ultracentrifuged at 65,000 × g and 4°C for 5 h to obtain pellets, which were resuspended in 30 μl TNE (Tris-NaCl-EDTA) buffer (15). For Western blot analysis, cell lysates or pellets were added to nonreducing buffer (2% SDS, 0.5 M Tris [pH 6.8], 20% glycerol, 0.001% bromophenol blue [final concentrations]) and subjected to 12% polyacrylamide gel electrophoresis (PAGE), followed by transfer to a nitrocellulose membrane, blocking, and incubation with primary antibody, serum from a confirmed human dengue case or anticalnexin mouse monoclonal antibody (MAb) E-10 (Santa Cruz), and secondary antibody (15, 55). After the final wash, the signals were detected by enhanced chemiluminescence reagents (Perkin Elmer Life Sciences). For dot blot assay, cell lysates in 1% NP-40 lysis buffer were diluted in bromophenol blue containing PBS and blotted to a nitrocellulose membrane by using a 96-dot formatted dot blotter (Labrepco), followed by blocking, incubation with primary antibodies (mixed human dengue sera or mouse MAbs 4G2, FL0231, DD18-5, and 1H10-6-7) and secondary antibodies, and detection, as described above (15, 55). For dots containing mixtures of native E protein in 1% NP-40 lysis buffer and denatured E protein in reducing buffer (nonreducing buffer with 0.71 M β-mercaptoethanol [final concentration]), denatured E protein was first blotted, followed by washing with PBS three times and blotting with native E protein.
Radioimmunoprecipitation.
293T cells prepared in a 6-well plate were transfected with plasmid DNA by the calcium phosphate method. At 20 h, cells were washed, incubated with methionine-free medium, followed by 50 μCi [35S]methionine (Amersham Biosciences) at 37°C for 6 h, and collected to obtain cell lysates (15). Cell lysates were precleared and then, a incubated with anti-E mouse MAb FL0232 (Chance Biotechnology) at 4°C overnight and then with protein A-Sepharose beads (Amersham Biosciences) at 4°C for 6 h. After the beads were washed, they were mixed with 2× sample buffer and heated, and the solubilized fraction was subjected to 12% PAGE (15). The intensities of the prM and E bands were analyzed by a UVP Biochemi image system (15), and the prM/E index of a mutant equals (intensity of mutant prM band/intensity of wild-type [WT] prM band)/(intensity of mutant E band/intensity of WT E band).
Subcellular fractionation and enzyme digestion.
293T cells transfected with plasmid DNA were washed 3 times with PBS at 48 h, resuspended in modified buffer B (10% sucrose, 20 mM Tris, 150 mM NaCl, 10 mM magnesium acetate, 1 mM EGTA [pH 7.6]), and subjected to freeze-thaw cycles 8 times, as described previously (58). After the nuclei and debris were cleared by centrifugation at 1,000 × g for 5 min, the membrane fraction was obtained by centrifugation at 20,000 × g for 30 min at 4°C. The resulting supernatants were layered over a 20% sucrose buffer and ultracentrifuged at 246,000 × g and 4°C for 1 h to obtain the pellets of the soluble fraction, which were resuspended in 30 μl TNE buffer. The membrane fraction and pellets of the soluble fraction were subjected to Western blot analysis (15). Aliquots from the suspension described above were treated with 500 U of endo-β-N-acetylglucosaminidase H (endo H) or peptide N-glycosidase F (PNGase F) at 37°C for 1 h, according to the manufacturer's instructions (New England BioLabs), and subjected to Western blot analysis.
Plasmid constructs for membrane flotation assay.
Plasmids pCDNA3-β-Gal/gp41 and pCDNA3-β-Gal, which contained the β-galactosidase gene with or without fusion to the cytoplasmic tail (residues 706 to 856) of gp41 of human immunodeficiency virus type 1 (HIV-1), respectively, were described previously (5). The stem regions of WT pCB-D4 and mutants were amplified by PCR and cloned into pCDNA3-β-Gal by use of the EcoRI and XbaI sites.
Membrane flotation assay.
293T cells (1 × 105 cells) in a 10-cm dish were transfected with 10 μg DNA by the calcium phosphate method. The transfected cells were washed twice with buffer containing 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 1 mM EGTA and sonicated twice on ice in 0.5 ml hypotonic TE buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, complete protease inhibitor cocktail [Roche]) supplemented with 10% sucrose. After centrifugation at 1,000 × g and 4°C for 10 min, 0.25 ml postnuclear supernatants was mixed with 1.25 ml of 85.5% sucrose and overlaid with 7 ml of 65% sucrose and then 3.25 ml of 6% sucrose, followed by ultracentrifugation at 98,768 × g and 4°C for 18 h (5). A total of 12 fractions were collected; each fraction was concentrated by trichloroacetic acid (TCA) precipitation and subjected to Western blot analysis using anti-β-galactosidase MAb (Promega) (5).
Infectious clone, replicon, and CprME constructs.
An infectious cDNA clone of DENV2 (pACYC FLTSV; strain TSV01) and subclone pACYC TSV-E were used to engineer mutations in the EH1 and EH2 domains in the context of genome-length RNA. The subclone pACYC TSV-E contains the SacII-XhoI fragment of pACYC FLTSV (spanning from the T7 promoter [for the RNA transcription of genome-length RNA] to nucleotide position 5426 of the viral genome; GenBank accession number AY037116). A QuikChange II XL site-directed mutagenesis kit (Stratagene) was used to engineer EH1 and EH2 mutations into pACYC TSV-E. The mutated DNA fragment was cut and cloned back into pACYC FLTSV with BsrGI and MluI (representing nucleotide positions 1840 to 3851 of the genome).
The DENV2 TSV01 Rluc2A replicon was constructed by replacing the structural genes (CprME) with the Renilla luciferase gene, followed by the foot-and-mouth disease virus (FMDV) 2A protease-coding sequence; the coding sequences of 38 amino acids at the N terminus of C protein and 31 amino acids at the C terminus of E protein were retained to preserve the correct topology of the nonstructural polyprotein across the membrane of the ER. For replicon particle packaging experiments, the complete CprME-coding fragment, with a Kozak sequence at the 5′ end and a stop codon at the 3′ end, was cloned into pCR2.1-TOPO (Invitrogen); the resulting plasmid was used to engineer EH1 and EH2 mutations by use of a QuikChange II XL site-directed mutagenesis kit; the mutated fragments were then cloned into a Semliki Forest virus (SFV) expression vector (40) through a unique XmaI site, resulting in the SFV-DENV2-CprME cDNA plasmid. All the constructs were verified by DNA sequencing.
In vitro transcription, RNA transfection, and immunofluorescence assay (IFA).
The genome-length RNA and replicon RNA were in vitro transcribed from corresponding cDNA plasmids that were linearized with ClaI. A T7 mMESSAGE mMACHINE kit (Ambion) was used for RNA synthesis as described previously (50). The SFV-DENV2-CprME RNA was in vitro transcribed from a SapI-linealized DNA using an SP6 mMESSAGE mMACHINE kit (Ambion). Both genome-length and replicon RNAs were electroporated into BHK-21 cells (50). After transfection of genome-length RNA, the cells were cultured at 37°C for 24 h and then at 30°C; culture supernatants containing viruses were collected every 24 h until day 5 and were aliquoted and stored at −80°C.
For IFA, BHK-21 cells transfected with genome-length RNA were seeded on an 8-well chamber slide (Nalgene Nunc). At the indicated time points, the cells were fixed in 100% methanol at 4°C for 30 min, washed with PBS, and incubated with mouse anti-E MAb 4G2 at room temperature for 1 h, followed by three washings with PBS and incubation with Alexa Fluor 488 goat anti-mouse IgG at room temperature for 1 h. After three washings with PBS and mounting with 4′,6-diamidino-2-phenylindole, the slides were analyzed under a fluorescence microscope (Leica).
Replicon particle assay.
Replicon particles of DENV2 were prepared by supplying viral CprME proteins to replicon RNA in trans as reported previously (43). Briefly, 8 × 106 BHK-21 cells were electroporated with 10 μg of DENV2 luciferase-reporting replicon RNA in a 0.4-cm cuvette with a GenePulser apparatus (Bio-Rad) using the settings of 0.85 kV, 25 μF, and three pulsings at 3-s intervals. The transfected cells were resuspended in Dulbecco modified Eagle medium with 10% fetal bovine serum, incubated at 37°C for 24 h, and electroporated again with 10 μg of SFV replicon RNA expressing WT or mutant CprME using the settings described above. After incubation at 30°C for 24 h and 48 h after the second transfection, culture supernatants were centrifuged to remove cellular debris and the supernatants containing replicon particles were aliquoted and stored at −80°C. To determine the RNA copy numbers in the replicon particles, replicon RNA was extracted from 140 μl supernatants and dissolved in 50 μl RNase-free water, of which 5 μl was quantified by a real-time reverse transcription-PCR (RT-PCR) assay using primers targeting the NS5 gene (5′-GAAGGAGAAGGGCTGCACAAAC-3′ and 5′-GCACACGCACCACCTTGTTTTG-3′) and an iScriptTM one-step RT-PCR kit with SYBR green (Bio-Rad). For replicon particle infection assay, Vero cells (4 × 104 cells per well in a 96-well plate) were infected with equal amounts of WT or mutant replicon particles (normalized by viral RNA copy numbers). At 48 h postinfection, the cells were washed once with cold PBS, lysed in 20 μl of 1× Renilla luciferase lysis buffer for 20 min, and assayed for luciferase activities by using a Renilla luciferase assay kit (Promega) and a Clarity luminescence microplate reader (BioTek).
Virion production from infectious clone.
Western blot analysis and real-time RT-PCR were used to evaluate the effects of mutations on virion production. Briefly, 20 ml of culture supernatants from genome-length RNA-transfected BHK-21 cells at day 5 was centrifuged at 4,000 × g and 4°C for 30 min to remove the cell debris. The supernatants (containing virions) were transferred to sterile Sorvall SS-34 tubes and ultracentrifuged at 45,000 × g and 4°C for 1 h. After careful removal of the supernatants, the pellets containing virions were resuspended in 500 μl PBS; aliquots were dissolved in SDS sample buffer, boiled for 5 min, and subjected to SDS-5 to 12% PAGE and Western blot analysis using mouse anti-E MAb 4G2 as described above. Viral RNA in the pellets was quantified by real-time RT-PCR as described above.
Plaque assay, continuous passaging of mutants, and adaptive mutations.
Plaque assay in BHK-21 cells was performed as described previously (39). Viruses collected on day 5 posttransfection (passage 0) of each mutant were continuously cultured on Vero cells for 5 rounds (5 days per round) to obtain the passage 5 viruses, which could reach a similar peak titer (about 1 × 107 PFU/ml) as the WT. Viral RNA extracted from culture supernatants of passage 0 and 5 viruses was subjected to RT-PCR using SuperScript III one-step RT-PCR kits (Invitrogen) and sequencing of the entire CprME gene to examine the adaptive mutations.
RESULTS
EH1 and EH2 domains are involved in production of VLPs.
Helical wheel analysis of the stem region revealed two amphipathic helices (EH1 and EH2), in which hydrophobic residues at the a or d position of the wheel faced one side and charged or polar residues faced the other side (Fig. 1B). To examine the roles of the EH1 and EH2 helical domains in the assembly of VLPs, site-directed mutagenesis was carried out to replace each of the residues at the a or d position with a proline or alanine in a DENV4 prM/E expression construct, pCB-D4 (Fig. 1A) (16). Two CS domain residues extending from the EH2 helix, phenylalanine at position 422 and valine at position 425, were also included in the analysis. Substitutions with proline, an α-helix breaker, and alanine are commonly employed to study the effect of disrupting the helical structure and the effect of removing a side chain with the helical structure preserved, respectively.
As shown in Fig. 1C, the amounts of prM/E proteins of the 15 proline mutants in cell lysates were generally comparable to those of WT pCB-D4, suggesting that substitutions of proline do not greatly affect the expression of prM/E proteins. In contrast, the amounts of prM/E proteins in pellets compared to those of the WT were greatly reduced in 14 mutants except the mutant with the V425P mutation, which was located in the CS domain between EH1 and EH2. These findings suggest that proline substitutions introduced to residues at either the EH1 or EH2 domain greatly affect the production of VLPs. To exclude the possibility that these proline mutations affect the folding and conformation of E protein, we employed a panel of conformation-sensitive mouse anti-E MAbs of different classes in the dot blot binding assay, in which cell lysates were prepared in 1% NP-40 lysis buffer. As shown in Fig. 1D, native E protein (in 1% NP-40 lysis buffer) but not denatured E protein (in reducing buffer) can be recognized by human sera or the MAbs. In addition, a gradual decrease in the intensity of dots was observed as the amount of native E protein decreased and that of denatured E protein increased, suggesting that the assay signal is sensitive to increasing proportions of denatured E protein. Compared with the WT E protein, the binding of mutant E proteins to flavivirus group-reactive MAbs recognizing domain II (4G2, FL0231), a complex-reactive MAb (DD18-5), or a type-specific MAb recognizing domain III (1H10-6-7) was not reduced (Fig. 1D), suggesting that the overall conformation of E protein was not affected by these mutations.
Alanine substitutions were also introduced into each of these 15 residues, and the production of VLPs for most mutants in the pellets was either mildly or not reduced compared to that for the WT (data not shown), suggesting that the helical structure of EH1 and EH2 is important for the assembly of VLPs. To further investigate whether particular amino acids are required for VLP production, substitutions with different types of amino acids, including hydrophobic, polar, and charged residues, were introduced into the first 1 of these 15 residues, isoleucine at position 398. As shown in Fig. 2A, the amounts of prM/E proteins in pellets relative to those in the cell lysates were greatly reduced by substitutions to aromatic (I398W), polar (I398S, I398Q), and charged (I398E) residues but not by substitution to another hydrophobic residue (I398L), suggesting the importance of a hydrophobic residue at this position to maintain the assembly function of VLPs.
Fig. 2.

Expression, production of VLPs, and prM-E heterodimerization of different mutants with mutations at residue 398 of the stem of DENV4 E protein. (A) At 48 h posttransfection, 293T cell lysates and pellets derived from culture supernatants of WT pCB-D4 or mutants were subjected to Western blot analysis using serum from a human dengue case (16, 55). (B) 293T cells transfected with WT or mutants were labeled with [35S]Met, immunoprecipitated with anti-E MAb FL0232, and subjected to SDS-12% PAGE. The prM/E index of each mutant is calculated as described in the legend to Fig. 1E. The sizes of the molecular mass markers are shown in kDa. One representative experiment of at least two experiments is shown.
EH1 and EH2 domains are involved in the release of VLPs from the membrane to the lumen of the ER.
To further investigate whether the intracellular prM/E proteins of these proline mutants are retained in the membrane-bound fraction or form VLPs in the soluble fraction of ER or other compartments, 293T cells transfected with two proline mutants with mutations of the EH1 domain (I398P, T405P), the first two mutations located at the a position of the EH1 wheel (Fig. 1B), were subjected to a previously described subcellular fractionation experiment (15, 58). As shown in Fig. 3A, the amounts of prM/E proteins in pellets of the soluble fraction relative to those in the membrane fraction were reduced for these mutants compared with those for the WT. As a control, calnexin, an integral ER membrane protein, was found in the membrane fraction but not in the soluble fraction. Digestion of the membrane fraction and pellets of the soluble fraction with endo H or PNGase F enzyme revealed a predominantly endo H-sensitive pattern of glycosylation for both the WT and mutants (Fig. 3B), suggesting that intracellular prM/E proteins of the WT and mutants, either in the membrane-bound fraction (before assembly into VLPs) or in the soluble fraction (after assembly), were mainly present in a compartment prior to the trans-Golgi apparatus, most likely ER (15, 51). It is worth noting that more than 2-fold the amount of soluble fraction pellets of mutants I398P and T405P compared with that for the WT was added to the enzyme digestion experiment to see the deglycosylated E protein bands because of severe reduction of the prM/E proteins in the soluble fractions of these mutants. Subcellular fractionation experiments carried out for other proline mutants with mutations in the EH2 domain (F429P, V436P, V439P) revealed similar results (data not shown). Together, these findings suggest that proline substitutions introduced into the EH1 or EH2 domain result in impairment in the release of VLPs from the membrane to the lumen of the ER and thus reduce the production of VLPs in the culture supernatants.
Fig. 3.

Subcellular fractionation experiment of the EH1 mutants of DENV4. (A) 293T cells mock transfected or transfected with WT or mutant (I398P, T405P) plasmid DNA were washed with PBS, resuspended in modified buffer B, and subjected to freeze-thaw cycles 8 times (15). After the nuclei and debris were cleared, the membrane fraction and the pellets derived from the soluble fraction by ultracentrifugation were subjected to Western blot analysis using serum from a human dengue case (upper gels) (55) and then reprobing with anticalnexin MAb (lower gels). The long-exposure gels for prM bands in pellets are shown (middle gels). (B) The membrane fraction and pellets of the soluble fraction of WT and mutants were treated with endo H (H) or PNGase F (F) and subjected to Western blot analysis as described above (55). Arrowheads indicate E or deglycosylated E (dE) protein. The sizes of the molecular mass markers are shown in kDa. One representative experiment of two is shown.
Stem mutants affected prM-E heterodimerization or membrane binding ability.
Previous studies of TBEV suggested that a proper prM-E heterodimeric interaction was important for assembly of VLPs (1). To investigate whether these proline mutations affect the prM-E interaction, lysates of 293T cells transfected with each of these mutants were subjected to a radioimmunoprecipitation assay using anti-E MAb (15). As shown in Fig. 1E, the amounts of prM protein relative to those of E protein were reduced for mutants with EH1 mutations, greatly reduced for those with mutations at the center (M401P, T405P, G408P, M412P), and slightly reduced for those with mutations at both ends (I398P, L415P T418P) but were not reduced for those with CS and EH2 mutations. Notably, since there were 18 and 10 methionine residues in the E and prM proteins of pCB-D4, respectively, the prM/E indices for mutants M401P and M412P, whose E proteins contained 17 rather than 18 methionine residues, were corrected by a factor of 17/18 in Fig. 1E. For mutants with different mutations of the isoleucine residue at position 398, the amounts of prM protein relative to the amounts of E protein were slightly reduced (Fig. 2B). Together, these findings suggest that the proline substitutions introduced into several residues at the center of the EH1 domain rather than those introduced into the CS or EH2 domain greatly affect the prM-E heterodimeric interaction, which may account for the impairment in the assembly of VLPs by these EH1 mutants.
Cryo-EM study of DENV2 virions at high resolution revealed that the EH1 and EH2 domains are partially buried in the outer leaflet of the viral membrane (60). The possibility that mutations in these domains might affect the association of the stem with the membrane in the ER after biosynthesis and thus affect the assembly of particles was investigated by examining the membrane binding ability of the WT or mutant stem. After cloning of the WT or mutant stem into a plasmid (pCDNA3-β-Gal) encoding β-galactosidase, a cytosol protein, a previously described membrane flotation assay was performed (Fig. 4A) (5). As shown in Fig. 4B, β-galactosidase fused to the WT stem was found mainly in fraction 9, whereas β-galactosidase alone was present in fractions 1 and 2 at the bottom, suggesting that the stem region can bind to the membrane. As a control, β-galactosidase fused to the cytoplasmic tail of HIV-1 gp41, which has been shown to bind to the membrane, was found in fraction 9 (5). Notably, β-galactosidase fused to WT EH1 alone was found in fractions 1 and 2, whereas β-galactosidase fused to WT EH2 alone was found mainly in fraction 9, suggesting that EH2 contributed primarily to the membrane binding ability of the stem (Fig. 4B), though its membrane binding was not as good as that of β-galactosidase fused to the entire stem. Therefore, the stems of each of the EH2 and CS proline mutants as well as two of the EH1 mutants (I398P and T405P) were cloned into pCDNA3-β-Gal and analyzed. The two EH1 mutants (I398P and T405P) were included in the analysis to further test whether EH1 mutations affect membrane binding. As shown in Fig. 4B, β-galactosidase fused to the stem of an EH1 mutant stem (T405P) was found predominantly in fraction 9, suggesting that it does not affect the membrane binding ability of the stem. In contrast, β-galactosidase fusion proteins containing the stems of the EH2 mutants (F429P, L432P, V436P, V439P, V443P, T446P), as well as those of the EH1 mutant (I398P) and CS mutant (F422P), that affected the VLP assembly shifted from fraction 9 to multiple fractions, including fractions 1 and 2 at the bottom. Chimeric β-galactosidase containing the stem of another CS mutant (V425P) that did not affect VLP assembly was found mainly in fraction 9. Together, these findings suggest that the mutations in all proline mutants with mutations of EH2, as well as one mutant with an EH1 mutation (I398P) and one mutant with a CS mutation (F422P), affected the membrane binding ability, which may account for the impairment in assembly of VLPs by these mutants.
Fig. 4.

Chimeric constructs and membrane flotation assay to measure the membrane binding ability of the stem region. (A) Schematic drawing of constructs expressing β-galactosidase alone (pCDNA3-β-Gal) or β-galactosidase fused to the stem, EH1, EH2, or cytoplasmic tail (CT) of HIV-1 gp41 (pCDNA3-β-Gal/fusion) (5). At 48 h posttransfection, 293T cells were washed, resuspended in hypotonic TE buffer, and sonicated twice. After the nuclei and debris were cleared, the postnuclear fraction was loaded onto a 6 to 85.5% (wt/wt) sucrose gradient and ultracentrifuged; each of the 12 fractions was collected, concentrated by TCA precipitation, and subjected to Western blot analysis using anti-β-galactosidase MAb (5).
EH1 and EH2 domains are involved in the assembly and entry of replicon particles.
To investigate the effects of EH1 and EH2 mutations on the assembly of replicon particles and infectivity, we utilized a packaging system for replicon particles, in which a DENV2 luciferase-reporting replicon, TSV01 Rluc2A, was supplemented in trans with WT or mutant DENV2 structural proteins (CprME) by a previously described sequential transfection protocol (43). Four DENV2 CprME constructs containing each of the proline mutations in either the EH1 domain (I398P, T405P) or EH2 domain (F429P, L436P) were generated. After transfection of both replicon and CprME RNA to BHK-21 cells, Western blot analysis revealed comparable amounts of E proteins in the WT and mutant packaging cells, suggesting that these mutations did not affect the expression of structural proteins (Fig. 5A). Similar levels of luciferase activity were detected in the WT and mutant packaging cells, suggesting that these mutations did not affect the replication efficiency of replicon RNA (Fig. 5B). To examine the amounts of replicon particles released in the culture supernatants, a real-time RT-PCR assay was carried out to quantify the extracellular replicon RNA posttransfection. As shown in Fig. 5C, the amounts of replicon RNA in the culture supernatants of all four mutants were drastically reduced. Compared with the WT packaging cells, the four mutant packaging cells produced less than 3% of replicon particles in the supernatants at 72 h posttransfection. These findings suggest that EH1 and EH2 mutations greatly affect the assembly of replicon particles and release.
Fig. 5.
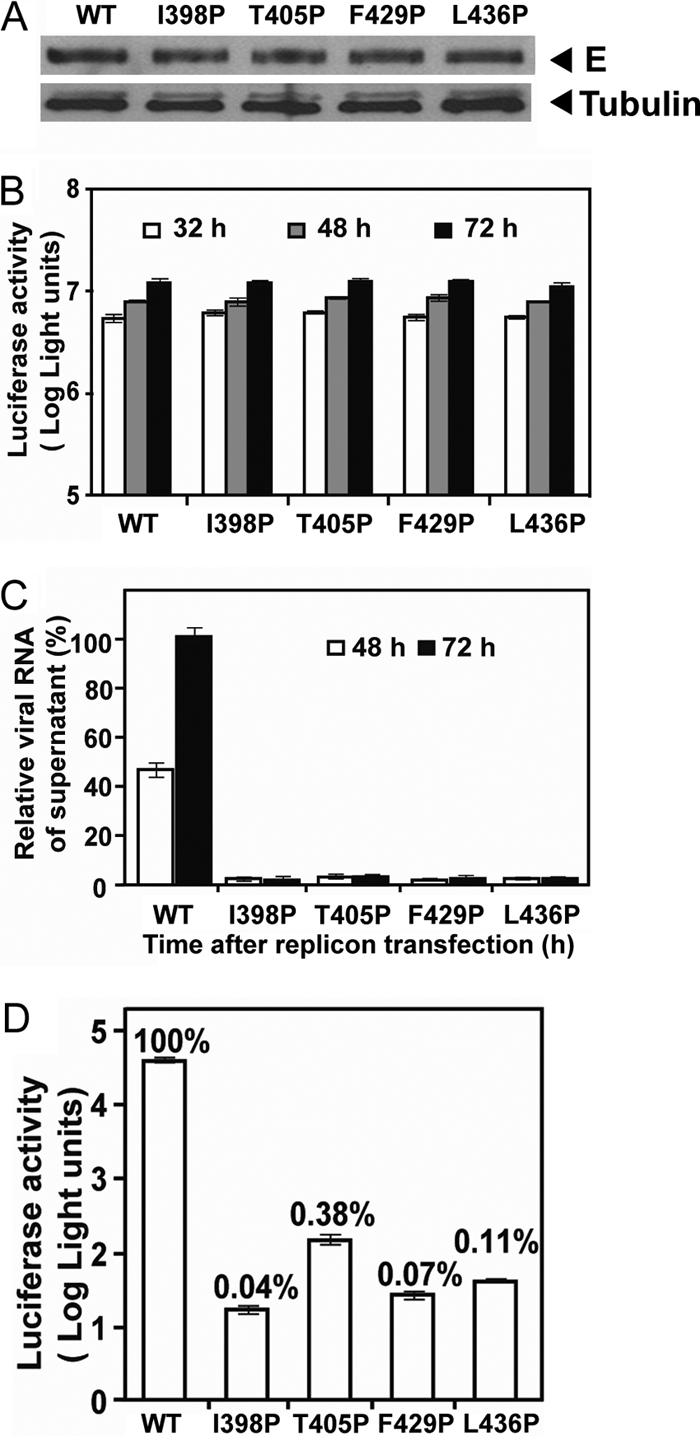
Effects of E protein stem mutations on the assembly and infectivity of replicon particles. (A) DENV2 replicon particles were prepared by sequential transfections of BHK-21 cells with replicon RNA and WT or mutant CprME expression vector. At 30 h after the first transfection, the cells were examined for E protein expression by Western blot analysis using anti-E MAb 4G2. Host γ-tubulin protein was used as a loading control. (B) At 32, 48, and 72 h posttransfection, the luciferase activities in the transfected cells were measured. (C) Viral RNA extracted from the culture supernatants (containing replicon particles) was quantified by real-time RT-PCR; the relative amounts of replicon RNA to the amount of the WT replicon RNA at 72 h posttransfection (100%) are presented. (D) Equal amounts of WT and mutant replicon particles (normalized by RNA copy numbers) were inoculated into Vero cells. At 48 h, the luciferase activities in the infected cells were measured; the luciferase signals relative to those of WT replicon particle-infected cells (100%) are shown. Data are means and standard errors from duplicate experiments.
To further examine the effect of these mutations on the infectivity of replicon particles, Vero cells were infected with equal amounts of WT or mutant replicon particles normalized by replicon RNA copy number, and the luciferase activities were measured at 48 h postinfection. As shown in Fig. 5D, the luciferase signals in the mutant replicon particle-infected cells were more than 2 to 3 log units lower than those in the WT replicon particle-infected cells. Notably, the mutations were present on the surface of replicon particles and did not affect the replication of replicon RNA (Fig. 5B); thus, the reduced luciferase signals in the target cells were most likely due to a defect in the entry of these mutants.
EH1 and EH2 domains are involved in the assembly and infectivity of virions.
To further examine the involvement of the EH1 and EH2 domains in the assembly and infectivity of virions, each of the four proline mutations in EH1 (I398P, T405P) or EH2 (F429P, L436P) was engineered to a DENV2 infectious clone, pACYC FLTSV. After transfection of BHK-21 cells with equal amounts of RNA, the numbers of IFA-positive cells (expressing E protein) increased rapidly from 24 to 72 h in the WT RNA-transfected cells (Fig. 6A). In contrast, the numbers of IFA-positive cells remained similar from 24 to 72 h in three mutant RNA-transfected cells (I398P, F429P, L436P) and increased slightly in mutant T405P-transfected cells, suggesting no or limited spreading of virus in the cultures. At 72 h, the proportions of IFA-positive cells among the cells transfected with WT, I398P, T405P, F429P, and L436P RNA were 80%, 15%, 19%, 15%, and 15%, respectively (calculated from three random views under a microscope).
Fig. 6.

Effects of E protein stem mutations on assembly and infectivity of virions. (A) BHK-21 cells were transfected with equal amounts (10 μg) of WT and mutant genome-length RNA derived from a DENV2 infectious clone, and viral protein synthesis was analyzed by IFA at 24, 48, and 72 h posttransfection. Anti-E MAb 4G2 and Alexa Fluor 488 goat anti-mouse IgG were used as primary and secondary antibodies, respectively. (B) Pellets (containing virions) derived from ultracentrifugation of culture supernatants on day 5 posttransfection were examined for E protein by Western blot analysis using anti-E MAb 4G2 (upper panel). The sizes of the molecular mass markers are shown in kDa. Viral RNA extracted from the pellets (containing virions) were quantified by real-time RT-PCR; the relative amount of viral RNA of each mutant to that of WT virus (100%) is shown (lower panel). Data are means of duplicate experiments.
To investigate the production of virions, pellets derived from culture supernatants posttransfection were examined by Western blot analysis using anti-E MAb. As shown in Fig. 6B, upper panel, the amounts of E protein in pellets derived from these mutants were greatly reduced compared with those in pellets derived from the WT; a very faint band was detected for F429P. In addition, the amounts of viral RNA in the pellets (containing virions) of mutants I398P, T405P, F429P, and L436P were only 0.006%, 0.013%, 0.016%, and 0.022%, respectively, of that of the WT (Fig. 6B, lower panel). Since viral RNA was extracted from the pellets (containing virions) after ultracentrifugation rather than directly from culture supernatants and original transfected RNA degraded to the background level on day 5 posttransfection (data not shown), the viral RNA detected was most likely from new virion RNA. Collectively, these findings suggest that mutations in both EH1 and EH2 affect the assembly and release of virions.
Characterization of the adaptive changes of EH1 and EH2 mutants.
As an alternative approach to investigate the functional roles of the EH1 and EH2 domains in the replication cycle, we selected adaptive viruses from these four mutants with EH1 and EH2 mutations by continuous passage on Vero cells. Apparent cytopathic effects with peak viral titers similar to those of the WT were observed in the passage 5 viruses for all four mutants (data not shown), suggesting that the passage 5 viruses have restored the infectivity through adaptive mutations. Consistent with this, the plaque sizes of the passage 5 viruses of all four mutants were similar to those of the WT, whereas the passage 0 viruses of mutants I398P, T405P, and F429P exhibited slightly smaller plaques than the WT virus (Fig. 7). The entire CprME genes of the passage 5 viruses of each mutant were completely sequenced, and the sequences were compared to the WT and engineered sequences at each site, which was originally designed to contain 2 nucleotide changes to avoid rapid reversion to WT. Interestingly, all the adaptive changes in the passage 0 and 5 viruses were found only in the engineered mutational sites. As shown in Fig. 7, the passage 0 viruses of mutants T405P, F429P, and L436P retained the originally engineered mutations, whereas passage 0 viruses of mutant I398P displayed a mixed population of 398P and 398L, suggesting that the I398P mutation could not be tolerated and adaptation from proline to leucine at position 398 (398L) via a single nucleotide change occurred quickly. In the passage 5 viruses of mutants I398P, F429P, and L436P, the original proline mutation was completely reverted to a leucine residue, also through a single nucleotide change (398L, 429L, 436L), suggesting the importance of a hydrophobic residue at these positions. Notably, the passage 5 virus of mutant T405P reverted the proline mutation back to the WT threonine residue rather than to a leucine residue, of which both required a single nucleotide change, suggesting that a threonine residue at position 405 probably resulted in more efficient replication than a leucine residue. Taken together, the observation that these proline mutations of the EH1 or EH2 domain completely restore viral replication by adaptation to a hydrophobic residue or reversion to the WT residue suggested the importance of the EH1 and EH2 α-helical structure for productive viral replication.
Fig. 7.

Characterization of adaptive viruses after continuous passage of four viruses with stem mutations. Plaque morphologies on BHK-21 cells and sequencing chromatograms of WT and mutant viruses of passage 0 (collected on day 5 posttransfection) and passage 5 (continuously cultured on Vero cells for 5 rounds) viruses (39). The plaque sizes (means ± standard errors) of the passage 0 WT, I398P, T405P, F429P, and L436P viruses were 1.61 ± 0.09 mm, 0.97 ± 0.11 mm, 1.20 ± 0.16 mm, 0.91 ± 0.05 mm, and 1.60 ± 0.09 mm, respectively. The nucleotides and corresponding amino acids are indicated above the sequence chromatogram peaks.
Validation of I398L adaptation.
Since our sequence analysis of the adaptive mutations involved the structural genes only, the possibility that additional mutations outside the structural genes also contribute to the restored viral replication could not be ruled out. Therefore, we engineered a single adaptive mutation into the WT infectious clone for one of the four mutants and examined its replication in comparison with that of the WT virus. The adaptive mutation of 398L from mutant I398P was chosen, because I398P was the least-tolerated mutation with a rapid adaptive change (Fig. 7). After transfection of BHK-21 cells with equal amounts of RNA, the numbers of IFA-positive cells increased from 24 to 72 h for both WT and I398L RNA-transfected cells (Fig. 8A), suggesting rapid virus spread in both cultures; this was in contrast to no virus spread in the I398P RNA-transfected cells (Fig. 6A). The plaque size and morphology of mutant I398L were similar to those of the WT virus (Fig. 8B). Sequencing results revealed that the mutant I398L retained the engineered mutation (data not shown). In addition, the viral titers of mutant I398L were comparable to those of the WT at various time points posttransfection (Fig. 8C). These findings suggest that a single substitution from proline to leucine at position 398 of E protein can restore viral replication of mutant I398P.
Fig. 8.

Characterization of I398L adaptation. (A) Analysis of viral protein synthesis in BHK-21 cells, which were transfected with equal amounts (10 μg) of WT and mutant I398L genome-length RNA, by IFA at 24, 48, and 72 h posttransfection. Data are presented as described in the legend to Fig. 6A. (B) Plaque morphologies of the WT and mutant I398L viruses collected on day 5 posttransfection (39). (C) Production of the WT and mutant I398L viruses after RNA transfection. Viral titers (PFU/ml) in the culture supernatants collected every 24 h from day 1 to 5 posttransfection were determined by plaque assay on BHK-21 cells (39).
To further validate the adaptation mutation involving a nonconservative amino acid substitution, we have engineered another adaptive mutation, F429L, into a WT infectious clone. As shown in Fig. 9A, the numbers of IFA-positive cells increased from 24 to 72 h in F429L RNA-transfected cells. The plaque size, morphology, and replication kinetics of mutant F429L were comparable to those of the WT virus (Fig. 9B and C). These findings suggest that a single substitution from proline to leucine, which has a side chain very different from the WT phenylalanine residue, at position of 429 of E protein can restore viral replication of mutant F429P.
Fig. 9.

Characterization of F429L adaptation. (A) Analysis of viral protein synthesis in BHK-21 cells, which were transfected with equal amounts (10 μg) of WT and mutant F429L genome-length RNA, by IFA at 24, 48, and 72 h posttransfection; (B) plaque morphologies of WT and mutant F429L viruses collected on day 5 posttransfection (39); (C) production of WT and mutant F429L viruses after RNA transfection. Data are presented as described in the legend to Fig. 8.
DISCUSSION
In this study, we investigated the roles of the EH1 and EH2 domains of the E protein stem region in the replication cycle of DENV by the approach of site-directed mutagenesis and different functional assays. Analysis of VLPs revealed that most of the proline mutations in either EH1 or EH2 greatly affected assembly, whereas alanine mutations in general had a mild or no effect, suggesting the importance of the helical structures of EH1 and EH2 in the assembly of VLPs. A subcellular fractionation experiment suggested that mutations in EH1 or EH2 affect the release of VLPs from the membrane to the lumen of the ER and thus the production of VLPs. Biochemical analyses revealed that EH1 mutations primarily affected prM-E heterodimerization, whereas EH2 mutations mainly affected the membrane binding ability of the stem. Introducing four proline mutations in either the EH1 or EH2 domain into a DENV2 replicon packaging system revealed that all four mutations affect the assembly and entry of replicon particles. The impairment in the assembly and infectivity were further verified in a DENV2 infectious clone. To our knowledge, this is the first report demonstrating that the EH1 and EH2 domains of the DENV E protein are involved in both assembly and entry, two important steps of the replication cycle.
Previous studies of TBEV E protein by using C-terminal truncational constructs revealed that the prM-E interaction was reduced by truncation of ET1 and abolished by truncation of EH2, whereas assembly of VLPs was abolished by truncation of ET1 (1, 53). It was concluded that EH2 and ET1 are required for prM-E interaction and assembly, respectively, and that proper prM-E heterodimerization is important for assembly. However, the possibility that EH1 is also involved in assembly or the prM-E interaction could not be addressed by such an approach. Our mutational analysis revealed that several of the proline substitutions introduced into EH1 (such as M401P, T405P, G408P, and M412P) greatly affected the assembly of VLPs, presumably through their effect on prM-E heterodimerization (Fig. 1E). Moreover, proline substitutions introduced into EH2 did not affect the prM-E interaction but greatly affected the assembly of VLPs. To explore other plausible mechanisms involved in assembly, we employed a membrane flotation assay to examine a series of chimeric β-galactosidase constructs containing WT or mutant stem and demonstrated that the stem region of DENV E protein alone can bind to the membrane and that impairment in membrane binding correlated with impaired assembly of VLPs for those mutants that did not affect the prM-E interaction. The observation that all EH2 mutations affected the membrane binding ability of the stem suggests that the EH2 domain is mainly involved in the membrane binding of the stem (Fig. 4B; Table 1). Consistent with this, a chimeric construct containing EH2 alone but not EH1 alone was able to bind to the membrane (Fig. 4B). One EH1 mutation (I398P) at the N terminus of the EH1 domain also affected membrane binding (Table 1). These observations correlated nicely with the high-resolution structure of DENV2 virions depicted by cryo-EM, in which the N terminus of the EH1 domain and the entire EH2 domain are partially buried in the outer leaflet of the viral membrane (60). It is conceivable that substitutions introduced into the EH2 domain or N-terminal EH1 domain (I398P) might affect the association of the stem region with the membrane and thus interfere with the curving and bending of the membrane lipid bilayer during assembly in the ER. Although in the cryo-EM study the CS domain was not found to associate with the membrane, one of the CS mutations (F422P) which affected assembly also affected the membrane binding of the stem (Table 1). Taken together, our findings suggest that the stem region is involved in the assembly of VLPs by two different mechanisms, contributing to the prM-E heterodimeric interaction and the association with the membrane.
Table 1.
Summary of effects of stem mutations at different domains on production of VLPs, prM-E heterodimerization, and membrane bindinga

After DENV enters the cell through receptor-mediated endocytosis, a series of conformational changes of the E protein and fusion process are triggered by the low-pH environment in the endosome, including the hinge outward of domain II and lateral rearrangement of the E protein, insertion of the exposed fusion loop of domain II into the endosomal membrane, trimerization of E protein, folding back of domain III and the stem-anchor toward domain II, approximation of the viral membrane and endosomal membrane, and formation of the fusion pore (3, 22, 26, 32, 34, 61). A recent cryo-EM study revealed that the stem region extended outward in the prefusion intermediate (19). It is likely that our proline mutations of the stem (I398P, T405P, F429P, L436P) may affect the EH1 or EH2 helical structure required for extension of the stem region during this process. Using different peptides covering the stem region, a study reported recent that peptides spanning the CS plus EH2 domains (residues 419 to 447) can block fusion, whereas peptides spanning an individual domain alone (EH1, CS, or EH2) or the EH1 plus CS domains (residues 396 to 429) cannot, suggesting the importance of both the CS and EH2 domains as potential targets during the fusion process of entry (14, 48). In agreement with this, two mutations in the EH2 domain (F429P, L436P) were found to affect entry and virus spread (Fig. 5D and Fig. 6A). Notably, the mutations in the EH1 domain (I398P, R405P) also affected virus entry and spread, even though the peptide spanning the EH1 domain could not block fusion. This could be due to poor accessibility of the EH1 domain compared to that of the EH2 domain during fusion or a nonoptimal conformation or folding of the peptide spanning EH1. Together, our findings suggest that both EH1 and EH2 are involved in the entry steps of the DENV replication cycle.
Investigation of the adaptive changes after continuous passage of these mutant viruses provided interesting and important information regarding the structure and function of the EH1 and EH2 domains. The observation that the plaques of passage 0 viruses were slightly smaller than (I398P, T405P, F429P) or comparable to (L436P) those of WT virus (Fig. 7) suggests that these mutations did not completely abrogate virus infectivity. Since the plaques of our viruses did not show clear morphology in double-layer agar, which was required for plaque purification, we sequenced the adaptive viruses after continuous passage rather than individually purified plaques. After complete sequencing of the CprME gene of passage 0 and 5 viruses, all the adaptive changes were found in the original mutational sites rather than in other residues of the stem or C-prM-E proteins, suggesting that the EH1 and EH2 domains are probably structurally distinct from other domains of the structural proteins. Except for reversion to the original threonine residue by mutant T405P, the observation that adaptation in three other mutants (I398P, F429P, L436P) that contained substitutions to a hydrophobic residue but not a nonhydrophobic residue suggests the importance of hydrophobic residues at the a and d positions to maintain the helical structure of EH1 and EH2. Interestingly, the adaptation involved substitutions to leucine rather than to other hydrophobic residues, such as isoleucine, valine, methionine, and phenylalanine. Analysis of the codons of proline, leucine, and all other hydrophobic residues revealed that the substitutions from proline to leucine (CCT to CTT of residue 398, CCC to CTC of residue 429, and CCT to CTT of residue 436) observed involved only one nucleotide change, presumably the easiest and quickest way to acquire viable adaptation, whereas substitutions to other hydrophobic residues generally involve two or more nucleotide changes (Fig. 7 and data not shown). The observation that introducing one substitution (I398L or F429L) into the WT infectious clone completely restores replication of I398P or F429P virus (Fig. 8 and 9) indicates that a single amino acid adaptation from proline to leucine at position 398 or 429 is sufficient. Consistent with this, introducing a leucine substitution into residue 398 (I398L) in the DENV4 prM/E construct completely restores the impairment in assembly of VLPs by mutant I398P (Fig. 2A).
Comparing the amino acid sequences of the stem region among different flaviviruses revealed that residues at the a and d positions of the EH2 domain are not absolutely conserved by particular amino acids but are conserved by hydrophobic amino acids (Table 2). This feature is consistent with the phenotype of EH2 mutants, in which substitutions to alanine resulted in mild or no impairment in the assembly of VLPs (data not shown), as well as the adaptation of EH2 mutant viruses (F429P and L436P), in which substitutions to a leucine (rather than the original residue) were tolerated (Fig. 7). In contrast, most residues at the a and d positions of the EH1 and CS domains are highly conserved by all four DENV serotypes or by all flaviviruses, the exception being residue 425. Importantly, several residues located adjacent to residues at the a and d positions examined in this study are highly conserved by all four DENV serotypes or all flaviviruses. The high degree of sequence conservation of the stem together with its involvement in two steps of the replication cycle suggests that the stem is an attractive target of antivirals. Due to the helix-breaker effect of the proline substitution and the focus on residues at the hydrophobic face in this study, the phenotypes of stem mutants were attributed to the effect on the helical structure rather than the side chains of particular amino acids. Nonetheless, the overall conformation of E protein was not affected by these mutations. Further delineation of the functional roles of the highly conserved stem residues by substitutions to different groups of amino acids is needed and would lead to the identification of potential targets of antivirals in the future.
Table 2.
Comparison of amino acid sequences of the stem region of E proteins of different flavivirusesa

The stem region of E protein contains two α-helical domains (EH1 and EH2) and a stretch of CSs in between (60). Sequences of the stem regions of multiple strains of flaviviruses were obtained from GenBank and analyzed. Single-letter amino acid designations are shown, with the numbers above indicating amino acid positions and the numbers beneath indicating the numbers of strains containing such a residue. The residues at the a or d position of the helical wheel are in boldface. Asterisks indicate residues highly conserved among the four DENV serotypes (*) or flaviviruses (**) analyzed. JEV, Japanese encephalitis virus; YFV, yellow fever virus; WNV, West Nile virus.
ACKNOWLEDGMENTS
This work was supported by the National Science Council Taiwan (NSC95-2320-B-002-084-MY3), the start-up fund from JABSOM, and award 2P20RR018727 from the National Center for Research Resources of the National Institutes of Health.
We thank Han-Chung Wu and Steve S. L. Chen at the Academia Sinica, Taiwan, for kindly providing MAb DD18-5 and plasmids (pCDNA3-β-Gal/gp41, pCDNA3-β-Gal), respectively, and Duane Gubler at the Duke-National University of Singapore Graduate Medical School for providing MAb 1H10-6-7.
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Allison S. L., Stiasny K., Stadler K., Mandl C. W., Heinz F. X. 1999. Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J. Virol. 73:5605–5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bray M., Men R., Tokimatsu I., Lai C. J. 1998. Genetic determinants responsible for acquisition of dengue type 2 virus mouse neurovirulence. J. Virol. 72:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bressanelli S., et al. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 23:728–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang G. J., et al. 2003. Enhancing biosynthesis and secretion of premembrane and envelope proteins by the chimeric plasmid of dengue virus type 2 and Japanese encephalitis virus. Virology 306:170–180 [DOI] [PubMed] [Google Scholar]
- 5. Chen S. S., Lee S. F., Wang C. T. 2001. Cellular membrane-binding ability of the C-terminal cytoplasmic domain of human immunodeficiency virus type 1 envelope transmembrane protein gp41. J. Virol. 75:9925–9938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis B. S., et al. 2001. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J. Virol. 75:4040–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferlenghi I., et al. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol. Cell 7:593–602 [DOI] [PubMed] [Google Scholar]
- 8. Gehrke R., et al. 2003. Incorporation of tick-borne encephalitis virus replicons into virus-like particles by a packaging cell line. J. Virol. 77:8924–8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Green S., Rothman A. 2006. Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr. Opin. Infect. Dis. 19:429–436 [DOI] [PubMed] [Google Scholar]
- 10. Gubler D. J. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100–103 [DOI] [PubMed] [Google Scholar]
- 11. Guirakhoo F., Hunt A. R., Lewis J. G., Roehrig J. T. 1993. Selection and partial characterization of dengue 2 virus mutants that induce fusion at elevated pH. Virology 194:219–223 [DOI] [PubMed] [Google Scholar]
- 12. Guzman M. G., Kouri G. 2002. Dengue: an update. Lancet Infect. Dis. 2:33–42 [DOI] [PubMed] [Google Scholar]
- 13. Halstead S. B. 1988. Pathogenesis of dengue: challenges to molecular biology. Science 239:476–481 [DOI] [PubMed] [Google Scholar]
- 14. Hrobowski Y. M., Garry R. F., Michael S. F. 2005. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol. J. 2:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsieh S. C., Liu I. J., King C. C., Chang G. J., Wang W. K. 2008. A strong endoplasmic reticulum retention signal in the stem-anchor region of envelope glycoprotein of dengue virus type 2 affects the production of virus-like particles. Virology 374:338–350 [DOI] [PubMed] [Google Scholar]
- 16. Hu H. P., Hsieh S. C., King C. C., Wang W. K. 2007. Characterization of retrovirus-based reporter viruses pseudotyped with the precursor membrane and envelope glycoproteins of four serotypes of dengue viruses. Virology 368:376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hunt A. R., Cropp C. B., Chang G. J. 2001. A recombinant particulate antigen of Japanese encephalitis virus produced in stably-transformed cells is an effective noninfectious antigen and subunit immunogen. J. Virol. Methods 97:133–149 [DOI] [PubMed] [Google Scholar]
- 18. Jones C. T., Patkar C. G., Kuhn R. J. 2005. Construction and applications of yellow fever virus replicons. Virology 331:247–259 [DOI] [PubMed] [Google Scholar]
- 19. Kaufmann B., et al. 2009. Capturing a flavivirus pre-fusion intermediate. PLoS Pathog. 5:e1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keelapang P., et al. 2004. Alterations of pr-M cleavage and virus export in pr-M junction chimeric dengue viruses. J. Virol. 78:2367–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khromykh A. A., Varnavski A. N., Westaway E. G. 1998. Encapsidation of the flavivirus Kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 72:5967–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kielian M., Rey F. A. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 4:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konishi E., Fujii A. 2002. Dengue type 2 virus subviral extracellular particles produced by a stably transfected mammalian cell line and their evaluation for a subunit vaccine. Vaccine 20:1058–1067 [DOI] [PubMed] [Google Scholar]
- 24. Kroeger M. A., McMinn P. C. 2002. Murray Valley encephalitis virus recombinant subviral particles protect mice from lethal challenge with virulent wild-type virus. Arch. Virol. 147:1155–1172 [DOI] [PubMed] [Google Scholar]
- 25. Liao M., Kielian M. 2005. Domain III from class II fusion proteins function as a dominant-negative inhibitor of virus membrane fusion. J. Cell Biol. 171:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindenbach B. D., Thiel H. J., Rice C. M. 2007. Flaviviridae: the viruses and their replication, p. 1101–1152In Knipe D. M., Howley P. M. (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 27. Lo M. K., Tilgner M., Bernard K. A., Shi P. Y. 2003. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3′ untranslated region of West Nile virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 77:10004–10014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lorenz I. C., et al. 2003. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J. Virol. 77:4370–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackenzie J. M., Westaway E. G. 2001. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 75:10787–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin J. E., et al. 2007. A West Nile virus DNA vaccine induces neutralizing antibody in healthy adults during a phase 1 clinical trial. J. Infect. Dis. 196:1732–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Modis Y., Ogata S., Clements D., Harrison S. C. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100:6986–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Modis Y., Ogata S., Clements D., Harrison S. C. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319 [DOI] [PubMed] [Google Scholar]
- 33. Modis Y., Ogata S., Clements D., Harrison S. C. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 79:1223–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mukhopadhyay S., Kuhn R. J., Rossmann M. G. 2005. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3:13–22 [DOI] [PubMed] [Google Scholar]
- 35. Murray J. M., Aaskov J. G., Wright P. J. 1993. Processing of the dengue virus type 2 proteins prM and C-prM. J. Gen. Virol. 74(Pt 2):175–182 [DOI] [PubMed] [Google Scholar]
- 36. Op De Beeck A., et al. 2003. Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J. Virol. 77:813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orlinger K. K., Hoenninger V. M., Kofler R. M., Mandl C. W. 2006. Construction and mutagenesis of an artificial bicistronic tick-borne encephalitis virus genome reveals an essential function of the second transmembrane region of protein E in flavivirus assembly. J. Virol. 80:12197–12208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pierson T. C., et al. 2006. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346:53–65 [DOI] [PubMed] [Google Scholar]
- 39. Poh M. K., et al. 2009. A small molecule fusion inhibitor of dengue virus. Antiviral Res. 84:260–266 [DOI] [PubMed] [Google Scholar]
- 40. Puig-Basagoiti F., et al. 2005. High-throughput assays using luciferase-expressing replicon, virus-like particle, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 49:4980–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Purdy D. E., Noga A. J., Chang G. J. 2004. Noninfectious recombinant antigen for detection of St. Louis encephalitis virus-specific antibodies in serum by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 42:4709–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Purdy D. E., Chang G. J. 2005. Secretion of noninfectious dengue virus-like particles and identification of amino acids in the stem region involved in intracellular retention of envelope protein. Virology 333:239–250 [DOI] [PubMed] [Google Scholar]
- 43. Qing M., Liu W., Yuan Z., Gu F., Shi P. Y. 2010. A high-throughput assay using dengue-1 virus-like particles for drug discovery. Antiviral Res. 86:163–171 [DOI] [PubMed] [Google Scholar]
- 44. Randolph V. B., Stollar V. 1990. Low pH-induced cell fusion in flavivirus-infected Aedes albopictus cell cultures. J. Gen. Virol. 71:1845–1850 [DOI] [PubMed] [Google Scholar]
- 45. Randolph V. B., Winkler G., Stollar V. 1990. Acidotropic amines inhibit proteolytic processing of flavivirus prM protein. Virology 174:450–458 [DOI] [PubMed] [Google Scholar]
- 46. Russell P. K., Brandt W. E., Dalrymple J. M. 1980. Chemical and antigenic structure of flaviviruses, p. 503–529In Schlesinger R. W. (ed.), The togaviruses. Academic Press, New York, NY [Google Scholar]
- 47. Schalich J., et al. 1996. Recombinant subviral particles from tick-borne encephalitis virus are fusogenic and provide a model system for studying flavivirus envelope glycoprotein functions. J. Virol. 70:4549–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt A. G., Yang P. L., Harrison S. C. 2010. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 6:e1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scholle F., Girard Y. A., Zhao Q., Higgs S., Mason P. W. 2004. Trans-packaged West Nile virus-like particles: infectious properties in vitro and in infected mosquito vectors. J. Virol. 78:11605–11614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shi P. Y., Tilgner M., Lo M. K., Kent K. A., Bernard K. A. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shin J., Dunbrack R. L., Jr., Lee S., Strominger J. L. 1991. Signals for retention of transmembrane proteins in the endoplasmic reticulum studied with CD4 truncation mutants. Proc. Natl. Acad. Sci. U. S. A. 88:1918–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stadler K., Allison S. L., Schalich J., Heinz F. X. 1997. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 71:8475–8481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stiasny K., Allison S. L., Marchler-Bauer A., Kunz C., Heinz F. X. 1996. Structural requirements for low-pH-induced rearrangements in the envelope glycoprotein of tick-borne encephalitis virus. J. Virol. 70:8142–8147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang S., He R., Anderson R. 1999. PrM- and cell-binding domains of the dengue virus E protein. J. Virol. 73:2547–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang W. K., et al. 2006. Slower rates of clearance of viral load and virus-containing immune complexes in patients with dengue hemorrhagic fever. Clin. Infect. Dis. 43:1023–1030 [DOI] [PubMed] [Google Scholar]
- 56. Welsch S., et al. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. World Health Organization 2009. Dengue and dengue hemorrhagic fever. Fact sheet no. 117. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs117/en [Google Scholar]
- 58. Xu Z., Bruss V., Yen T. S. 1997. Formation of intracellular particles by hepatitis B virus large surface protein. J. Virol. 71:5487–5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu I. M., et al. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837 [DOI] [PubMed] [Google Scholar]
- 60. Zhang W., et al. 2003. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 10:907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Y., et al. 2004. Conformational changes of the flavivirus E glycoprotein. Structure 12:1607–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]