

Abstract
The untranslated regions (UTRs) located at the 5′ and 3′ ends of the Japanese encephalitis virus (JEV) genome, a positive-sense RNA, are involved in viral translation, the initiation of RNA synthesis, and the packaging of nascent virions. The cellular and viral proteins that participate in these processes are expected to interact with the UTRs. In this study, we used biotinylated RNA-protein pulldown and liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) analyses to identify that the far upstream element (FUSE) binding protein 1 (FBP1) binds with JEV 5′ and 3′ UTRs. The impact of FBP1 on JEV infection was determined in cells with altered FBP1 expression. JEV replication was enhanced by knockdown and reduced by the overexpression of FBP1, indicating a negative role for FBP1 in JEV infection. FBP1, a nuclear protein, was redistributed to the perinuclear region and appeared as cytoplasmic foci that partially colocalized with JEV RNA in the early stage of JEV infection. By using a JEV replicon reporter assay, FBP1 appeared to suppress JEV protein expression mediated by the 5′ and 3′ UTRs. Thus, we suggest that FBP1 binds with the JEV UTR RNA and functions as a host anti-JEV defense molecule by repressing viral protein expression.
INTRODUCTION
Japanese encephalitis virus (JEV), a member of the family Flaviviridae, is the causative agent of Japanese encephalitis (JE), the most prevalent viral encephalitis in Asia. The JEV genome is a single-stranded, positive-sense RNA of about 11 kb in length, encoding a large polyprotein of about 3,400 amino acids. After translation, the polyprotein is processed by host and viral proteases into three structural proteins (core [C], precursor membrane [prM], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The JEV genome, with a type I cap at the 5′ end but no poly(A) tract at the 3′ end, is flanked by 5′ and 3′ untranslated regions (UTRs), which are 95 and 585 nucleotides (nt) long, respectively (46). The flavivirus UTRs contain highly conserved sequences and secondary structures (10, 24, 41) that are important for the regulation of viral protein translation (21, 27–28) and RNA synthesis (1, 48, 52, 58–59).
Positive-sense RNA viruses rely on host cell proteins for various aspects of their life cycle. Following the first round of viral translation in the cytoplasm, genomic RNA replicates to convert the positive-sense RNA into a replication-intermediate negative-sense RNA. The negative-sense RNA then acts as a template for the synthesis of progeny virion RNAs, and the assembly of viral RNA and viral proteins is coordinated to generate an infectious virus. The cellular and viral proteins that participate in these replication processes are expected to bind with the 5′ and 3′ UTRs of viral RNA to regulate viral translation and replication (11). For instance, elongation factor-1α (EF-1α) interacts with the 3′ UTR of West Nile virus (WNV) and dengue virus serotype 4 (DEN-4) (7, 9, 16–17), and EF-1α binding to the 3′ stem-loop (SL) of WNV positive-sense RNA facilitates viral negative-sense RNA synthesis (16). T-cell intracellular antigen 1 (TIA-1) and TIA-1-related (TIAR) proteins also bind to the 3′ SL of WNV negative-sense RNA and play a positive role in virus replication (34). The La autoantigen interacts with the 3′ UTR of JEV, and the short interfering RNA (siRNA)-mediated downregulation of the La protein results in the repression of JEV replication in cultured cells (51). The Y-box binding protein 1 (YB-1) is an antiviral protein for dengue virus serotype 2 (DEN-2) through binding to its 3′ SL and repressing viral translation (42).
To identify the cellular proteins that interact with JEV UTRs, we used streptavidin beads to capture biotinylated JEV UTR RNA. We then analyzed the copurified cellular proteins by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) to reveal the identity of cellular proteins binding with the JEV UTRs. Far upstream element (FUSE) binding protein 1 (FBP1) was one of the cellular proteins that interacted with JEV UTR RNA. FBP1 originally was identified as a single-stranded DNA (ssDNA) binding protein that binds to the FUSE, an A/T-rich element located in the c-myc promoter, and it modulates c-myc mRNA levels (5, 15, 19–20, 26). It also has been demonstrated to be a member of the AU-rich element (ARE) binding protein family (18) and was reported to bind with the 3′ UTR of growth-associated protein 43 (29) and cyclo-oxygenase-2 mRNA (45) to influence their stability. Three variants of this protein, FBP1, FBP2 (also known as KSRP, for K-homology splicing regulator protein), and FBP3, currently are known. These FBP proteins contain four KH domains that are involved in DNA and RNA binding and several Arg-Gly-Gly motifs, which are present in many RNA binding proteins and helicases (53). Thus, by binding to DNA and RNA, FBP1 modulates gene expression, regulates the stability of mRNA, and functions as an ATP-dependent DNA helicase. Recently, FBP1 also has been reported to interact with the poly(U) tract of the hepatitis C virus (HCV) 3′ UTR and is required for efficient HCV replication (60).
In this study, we characterized the role of FBP1 in JEV infection by the knockdown and overexpression of FBP1. We found that FBP1 functions as an anti-JEV protein, because JEV replication was enhanced by knockdown and reduced by the overexpression of FBP1. The cellular distribution of FBP1 was traced during the course of JEV infection, and the impact of FBP1 on JEV translation and replication was investigated by using a JEV replicon system. The novel function of FBP1 as a negative regulator in JEV infection is reported and discussed.
MATERIALS AND METHODS
Cell lines, virus, and chemicals.
N18, a mouse neuroblastoma cell line (2), and BHK-21, a baby hamster kidney cell line, were cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS). HeLa and 293FT (Invitrogen) were grown in Dulbecco's modified essential medium (DMEM) supplemented with 10% FBS. NT2, a human neuronal precursor cell line, was cultured in DMEM supplemented with 4% FBS. JEV strain RP-9 (13) and dengue virus serotype 2 (DEN-2) PL046 strain (37) were propagated in mosquito C6/36 cells. For viral infection, cells were absorbed with virus at 37°C for 1 h. Virus titers were determined by plaque-forming assays on BHK-21 cells. Puromycin was obtained from Sigma. Lipofectamine 2000 and G418 were purchased from Invitrogen.
In vitro transcription.
The plasmid JEV UTR-Luc/pBR322 contains an SP6 promoter upstream of a Renilla luciferase gene flanked by the JEV 5′ UTR plus a portion of the C gene (nt 96 to 158) in its 5′ end and portions of the E (nt 2388 to 2477), NS1 (nt 2478 to 2692), and NS5 (nt 10207 to 10391) genes and 3′ UTRs in its 3′ end. The plasmid UTR-GAPDH/pBR322 contains an SP6 promoter upstream of a mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene flanked by the 5′ and 3′ UTRs of GAPDH. The JEV 5′ UTR and 3′ UTR, used as RNA transcription templates, were PCR amplified from JEV UTR-Luc/pBR322 by using the following primer pairs: JEV 5′ UTR forward (5′-ACGCGTCGACATTTAGGTGACACTATAGAGAAGTTTATCTGTGTGAACTTCTTG-3′) and JEV 5′ UTR reverse (5′-GGTTATCTTCCGTTCTAAAAAACTG-3′), JEV 3′ UTR forward (5′-ACGCGTCGACATTTAGGTGACACTATAGTGTGATTTAAGGTAGAAAAGTAG-3′) and JEV 3′ UTR reverse(5′-AGATCCTGTGTTCTTCCTCAC-3′). Underlined nucleotides represent the SP6 promoter. RNA transcripts were synthesized using the RiboMax SP6 RNA polymerase system kit (Promega), and a 7-methyl-GpppA nucleotide (Ambion) was incorporated at the initial adenosine residue of the 5′ UTR. Biotinylated RNAs were synthesized by adding 1.25 μl of 20 mM biotinylated UTP, biotin-16-UTP (Roche), into a final volume of 20 μl of an in vitro transcription mixture for RNA labeling. After incubation at 37°C for 2 h, template DNA was removed with RNase-free DNase I. Synthesized RNAs were purified using the RNeasy minikit (Qiagen) and analyzed on 1% agarose gels.
Isolation of proteins that bound with biotinylated JEV UTR-Luc by pulldown assay.
The pulldown assay was performed based on a previously published method (25, 36). Briefly, 200 μg of N18 cell extract {10 mM Tris-HCl (pH 7.4), 1 mM MgCl2, 1 mM EGTA, 0.5% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 10% glycerol, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 5 mM 2-mercaptoethanol} was incubated with 6.5 μg of JEV UTR-Luc RNA with or without biotin label in a final volume of 100 μl of RNA mobility shift buffer (5 mM HEPES [pH 7.1], 40 mM KCl, 0.1 mM EDTA, 2 mM MgCl2, 2 mM dithiothreitol, 1 unit RNasin, and 0.25 mg/ml heparin) for 15 min at 30°C. After incubation, 400 μl of Streptavidin MagneSphere paramagnetic particles (Promega) was added for 10 min at room temperature. The protein-RNA complexes were washed three times with RNA mobility shift buffer without heparin. After the samples were washed, 30 μl of 2× SDS-PAGE sample buffer was added to the beads, and they were incubated for 10 min at room temperature. The bound proteins were eluted and resolved by 12% SDS-PAGE and stained with SYPRO Ruby (Invitrogen). Images were taken with a Typhoon 9410 variable-mode imager (Amersham Biosciences). The extra protein bands, which bound to biotin-UTR-Luc RNA but not to the unbiotinylated control, were excised for in-gel trypsin digestion and analyzed by LC-MS/MS.
Tryptic digestion and mass spectrometry.
Each gel band was cut into small pieces and washed three times with 25 mM NH4HCO3 in 50% acetonitrile (ACN). Gel pieces then were dehydrated with 100% ACN at room temperature for 5 min and were subjected to trypsin digestion by adding a solution containing 25 mM NH4HCO3 (pH 8.4) and 10 μg/ml trypsin (sequencing grade modified; Promega). Following 16 to 24 h of incubation at 37°C, reaction mixtures were adjusted to 50% ACN-5% trifluoroacetic acid (TFA) for the extraction of peptides from the gel. The peptide mixtures were identified by using nano-LC-MS/MS on a Finnigan LCQ Deca XP ion trap mass spectrometer (ThermoFisher, San Jose, CA). The samples were loaded onto a 75-μm-inner-diameter (360-μm-outer-diameter, 15-μm tip, 11-cm-length) capillary column/spray needle packed in house with MAGIC C18AQ reverse-phase material (5 μm, 100 Å) from Michrom Bioresources (Auburn, CA). The LCQ was run in a top-three configuration with one mass spectrometry scan followed by three MS/MS scans. Spectra were acquired with the instrument operating in the data-dependent mode. Sequence analyses were performed using Sequest through the Bioworks Browser, version 3.1 (Thermo Scientific). Sequential database searches were made using the NCBI nonredundant protein database. Modifications specified in the search were that cysteine was differentially carbamidomethylated and that methionine was differentially oxidized.
Plasmid constructs.
Human and mouse FBP1 cDNA from HeLa cells and mouse brain were PCR amplified using the primers human FBP1 forward (5′-AACCATGGCAGACTATTCAACAGTGC-3′), mouse FBP1 forward (5′-TGGGTATGGCCGACTACTCCAC-3′), and FBP1 reverse (5′-TTGGCCCTGAGGTGCTGGAG-3′) and were cloned into pcDNA3.1/V5-His using the TOPO TA expression kit (Invitrogen). The cloned human and mouse FBP1 cDNAs were verified by sequencing and were the same as those reported in GenBank (NM_003902 and NM_057172, respectively). A single-primer mutagenesis protocol (39) was used to generate a human shFBP1-resistant FBP1/pcDNA3.1/V5-His construct by using the primer 5′-CAAAAAAGACCTTTAGAAGATGGTGACCAACCAGATGCTAAGAAAGTTG-3′, annealing to nt 187 to 235 of the human FBP1 gene; the mutated sequences are underlined. For lentivirus expression, human FBP1 with a V5-His tag at the C terminus was subcloned into the self-inactivating lentiviral vector (pSIN) in which the expression of an inserted gene is under the control of a constitutive spleen focus-forming virus promoter (23). The mouse GAPDH cDNA containing its 5′ and 3′ UTRs was PCR amplified using the primers 5′-ACGCGTCGACATTTAGGTGACACTATAGAGAGACGGCCGC-3′ and 5′-CCATCGATTTTTTTTTTTTTTTTTT-3′ and cloned into the SalI and ClaI sites of the pBR322 vector. Underlined nucleotides represent the SP6 promoter. The cloned mouse GAPDH cDNA was verified by sequencing and was the same as that reported as GenBank NM_008084.2. For reporter assays, Renilla luciferase flanked by GAPDH 5′ and 3′ UTRs was cloned into pBR322 and named GAPDH-Luc.
Two JEV replicons were used in this study. The first, containing a neomycin resistance gene, has been reported previously (33). The second, J-R2A, as outlined in Fig. 7A, contains a Renilla luciferase reporter and was generated by cloning JEV RP-9 cDNA into pBR322 under the control of an SP6 promoter. The Renilla luciferase gene was placed after the first 102 nt of the C gene, followed by the foot-and-mouth disease virus 2A self-cleaving protease (FMDV 2A), to enable the cleavage of the luciferase away from downstream nonstructural proteins. FMDV 2A was fused to the last 90 nt of the E gene, which contained the leader sequence required for the proper topology of the remaining viral polyprotein. To ensure RNA stability and processing, a hepatitis delta virus ribozyme was placed immediately adjacent to the 3′ end of the JEV cDNA, followed by a simian virus 40 (SV40) poly(A) sequence, as previously described (35). The replication-dead J-R2A-NS5mt replicon construct containing a mutation (GDD→AAG) in the RNA-dependent RNA polymerase motif was generated by single-primer mutagenesis with the primer 5′-CCAGGATGGCGATCAGCGCAGCCGGCTGTGTCGTCAAGCCGCTG-3′, annealing to nt 9658 to 9701 of the NS5 gene; the mutated sequences are underlined. For the replicon luciferase reporter assay, cells were seeded in 12-well plates and cotransfected with 0.6 μg of in vitro-transcribed J-R2A replicon or J-R2A-NS5mt replicon RNA, plus 0.1 μg of a firefly luciferase RNA in vitro transcribed from a luciferase SP6 control (Promega) as an internal control using Lipofectamine 2000. The control experiment using GAPDH-Luc, a GAPDH 5′ and 3′ UTR-flanking luciferase reporter, was performed as described for the JEV replicon. At the indicated times posttransfection, cell lysates were collected for the dual-luciferase assay (Promega).
Fig. 7.

FBP1 negatively regulated JEV UTR-dependent protein expression measured by a JEV reporter replicon. (A) Schematic representation of the J-R2A replicon. The construct contains an SP6 promoter upstream of the JEV 5′ UTR, followed by the first 34 amino acids of JEV C protein (C96-197), Renilla luciferase, the foot-and-mouth disease virus 2A self-cleaving protease (FMDV 2A), the last 30 amino acids of JEV E protein (E2388-2477), NS1-NS5 of JEV nonstructural proteins, and the JEV 3′ UTR. To ensure RNA stability and processing, a hepatitis delta virus ribozyme was placed immediately adjacent to the 3′ end of the JEV cDNA, followed by an SV40 poly(A) sequence. (B) Human neuronal NT2 cells transduced with lentivirus carrying shRNA-targeting FBP1 (shFBP1; TRCN0000013293) or LacZ (shLacZ) were selected by puromycin (5 μg/ml). To verify the knockdown effect, cell lysates were harvested for immunoblotting with anti-FBP1 and anti-actin antibodies. NT2-shFBP1 or NT2-shLacZ cells were cotransfected with a Renilla luciferase JEV replicon J-R2A (C), a replication-dead replicon J-R2A-NS5mt (D), or a Renilla luciferase reporter flanked by GAPDH 5′ and 3′ UTRs (E) plus firefly luciferase RNA as a control. At various times posttransfection, cell lysates were collected for dual-luciferase assays. Renilla luciferase activity was normalized to that of firefly luciferase. The results are expressed as averages and standard deviations from two independent samples. The data of shFBP1 and shLacZ at the same time points were compared by two-tailed Student's t test. *, P < 0.05.
Lentiviral vector preparation.
The lentivirus vector pLKO.1, carrying the short hairpin RNA (shRNA) targeting human FBP1 (TRCN0000013293, 5′-CGACTTGATGAAGATCTTAAT-3′, targeting the 3′ UTR of the human FBP1 mRNA; and TRCN0000013297, 5′-CCTTTAGAAGATGGAGATCAA-3′, targeting nt 196 to 216 of the human FBP1 mRNA) and the negative control targeting LacZ (TRCN0000072223, 5′-TGTTCGCATTATCCGAACCAT-3′) were obtained from the Taiwan National RNAi Core Facility. For lentivirus preparation, 293FT cells were cotransfected with pLKO.1-shRNA or a lentivirus expression construct (pSIN-human FBP1-V5-His) and two helper plasmids, pMD.G and pCMVΔR8.91, with Lipofectamine 2000 reagent (Invitrogen). The culture supernatants containing the viral particles were harvested and stored at −80°C. To knock down human FBP1 expression, the cells were transduced with shFBP1 lentivirus for 24 h and selected with puromycin (5 μg/ml).
Immunoprecipitation and RT-PCR.
We used coimmunoprecipitation and reverse transcription-PCR (RT-PCR) to detect the association of FBP1 with JEV UTR in JEV-infected cells. Briefly, 200 μg of JEV-infected HeLa cell extracts (at 4 h postinfection) were mixed with 5 μg of mouse anti-FBP1 antibody (BD Biosciences), mouse anti-Bst2 (H00000684-B02; Abnova), or control mouse IgG isolated from pooled normal serum (I 8765; Sigma) and incubated at 4°C for 4 h. Prewashed protein A/G-agarose beads (GE Healthcare) were added to each sample and incubated overnight at 4°C. Immune complexes then were washed three times with RNA mobility shift buffer, and the RNA was extracted from the immunoprecipitated complex with an RNeasy kit (Qiagen) according to the manufacturer's instructions. cDNA was synthesized, and RT-PCR was performed by using the primers JEV 3′ UTR forward (5′-TAGTGTGATTTAAGGTAGAAAAGTAGACT-3′, annealing to nt 10392 to 10421 of JEV) and JEV 3′ UTR reverse (5′-AGATCCTGTGTTCTTCCTCAC-3′, annealing to nt 10976 to 10956 of JEV). To check for an FBP1 knockdown effect, total RNA was extracted from cells with an RNeasy kit and RT-PCR was performed by using primers FBP1 forward (5′-ATGATTCCAGCCAGCAAAGCAG-3′) and FBP1 reverse (5′-TTGGCCCTGAGGTGCTGGAG-3′). Actin serves as an internal control with primers actin forward (5′-TCCTGTGGCATCCACGAAACT-3′) and actin reverse (5′-GAAGCATTTGCGGTGGACGAT).
Western immunoblotting.
For Western immunoblotting, cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% TritonX-100, and 1% sodium deoxycholate) containing a cocktail of protease inhibitors (Roche). The protein concentrations were determined by using the Bio-Rad Dc protein assay (Bio-Rad). Equal amounts of proteins were loaded and separated by SDS-PAGE and then transferred to a nitrocellulose membrane (Hybond-C Super; Amersham/GE Healthcare). The nitrocellulose membrane was blocked with skim milk in phosphate-buffered saline (PBS) with 0.1% Tween 20 (PBST) and subsequently incubated with primary antibody against various proteins, including mouse anti-FBP1 (1:1,000; BD Biosciences), mouse anti-JEV NS1, NS3 (1:1,000) (12), mouse anti-DEN-2 NS3 (37), mouse anti-V5 (1:5,000; Sigma), and mouse anti-actin (1:10,000; Chemicon). The blots then were reacted with a horseradish peroxidase-conjugated secondary antibody (1:2,500; Jackson ImmunoResearch) and developed using an enhanced chemiluminescence system (ECL; Pierce).
IFA.
For immunofluorescence assay (IFA), the cells were fixed with 4% formaldehyde in PBS and permeabilized with 0.5% Triton X-100. After being blocked with skim milk in PBS, JEV protein expression was detected using a mouse anti-JEV NS3 antibody (12), and viral RNA was detected using a mouse anti-double-stranded RNA (dsRNA) antibody (1:1,000; J2 MAb; English & Scientific Consulting). The expression of V5-tagged human FBP1 was detected with a rabbit anti-V5 antibody (1:5,000; Sigma). The secondary antibodies, Alexa Fluor 568-conjugated anti-mouse antibody (1:1,000; Molecular Probes) and Alexa Fluor 488-conjugated anti-rabbit antibody (1:1,000; Molecular Probes), were added sequentially for 1 h at room temperature. The nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI) (1:3,500; Molecular Probes), and cells were examined with a Leica fluorescent microscope.
Confocal imaging.
Cells were seeded on coverslips in 12-well plates for 16 h and then infected with JEV (multiplicity of infection [MOI], 5) for the indicated times. Cells were fixed with 4% paraformaldehyde, followed by permeabilization with 0.5% Triton X-100 for 10 min. Human FBP1 proteins were stained with a rabbit anti-V5 antibody, and viral RNA was detected using a mouse anti-dsRNA antibody (J2 MAb; English & Scientific Consulting). For stress granules (SGs) and P body induction, cells were stimulated by heat shock (44°C for 30 min) or sodium arsenite (0.5 mM for 1 h) as previously described (30). Human V5-tagged FBP1 proteins were detected with anti-V5 antibody, and SG markers were stained with anti-eIF4E (1:500; Cell Signaling), anti-PABP1 (1:500; Sigma), anti-G3BP (1:500; BD Biosciences), and anti-p54/RCK (1:500; Bethyl Laboratories) antibodies. Subsequently, secondary antibodies conjugated with Alexa Fluor 568 and Alexa Fluor 488 were added for 1 h at room temperature. The nuclei were stained with DAPI, and specimens were observed using confocal microscopy (Zeiss LSM 510 META).
RESULTS
Identification of FBP1 interacting with JEV UTRs.
We used a biotinylated RNA-protein pulldown assay (25, 36) followed by LC-MS/MS protein identification to investigate the mammalian host factors interacting with JEV UTRs. Because long-range viral 5′-to-3′ UTR RNA interactions are involved in viral translation and RNA synthesis (1, 21), we in vitro synthesized a biotin-labeled RNA containing both of the 5′ and 3′ JEV UTRs flanking a luciferase gene and named it JEV UTR-Luc RNA. Protein extracts from mouse neuroblastoma N18 cells that support JEV replication (44) were incubated with the biotinylated JEV UTR-Luc RNA or control RNA without biotin-16-UTP labeling. Streptavidin beads then were used to capture the biotin-UTR-Luc RNA and its associated proteins. After being washed extensively, proteins were eluted from the beads and subjected to SDS-PAGE followed by SYPRO Ruby staining. The protein bands of interest were excised and submitted for identification by LC-MS/MS analysis. By using this pull-down strategy, several host proteins were found to bind with the JEV UTR-Luc RNA (Fig. 1A). One of them was the heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP A2/B1), which has been reported to interact with the 3′ UTR of DEN-2 (42), validating of our RNA-protein pulldown assay.
Fig. 1.

Interaction of FBP1 with JEV UTRs. (A) Identification of the cellular proteins interacting with JEV UTRs by pulldown assay as described in Materials and Methods. Mouse N18 cell extracts (200 μg) were incubated with UTR-Luc or biotin-UTR-Luc RNA (6.5 μg), and then streptavidin beads were used to capture the biotinylated RNA. After the beads were washed, the bound proteins were eluted and resolved using 12% SDS-PAGE and stained with SYPRO Ruby. The extra protein bands, which bound to biotin-UTR-Luc but not to the unbiotinylated control (UTR-Luc), were excised for in-gel trypsin digestion and analyzed by LC-MS/MS. The protein bands identified as FBP1 and hnRNP A2/B1 are indicated by arrows. (B) The interaction of JEV UTR with FBP1 was confirmed by anti-FBP1 antibody. The pulldown assay was performed as described above, and then the proteins were separated on SDS-PAGE and subjected to immunoblotting with anti-FBP1 antibody. An unrelated RNA, UTR-GAPDH, with biotin label, was included as a negative control (lane 2). (C) Human FBP1 from HeLa cells also interacted with JEV UTRs. HeLa cell extracts (200 μg) were incubated with 6.5 μg of unbiotinylated or biotinylated UTR-Luc RNA (lanes 1 and 2) or with 4 μg of biotinylated 5′ UTR or 3′ UTR (lanes 3 and 4). The pulldown proteins then were analyzed by immunoblotting with anti-FBP1 antibody, with the HeLa cell lysate as the protein control (lane 5). (D) JEV RNA was pulled down with FBP1 from JEV-infected cell extracts. HeLa cells were infected by JEV (MOI, 10) for 4 h before cell extract collection. The mouse anti-FBP1 (αFBP1; lane 1), mouse anti-Bst2 (αBst2; lane 2), or normal mouse IgG control (lane 3) was incubated with 200 μg of JEV-infected cell extracts, and then protein G/A-agarose beads were added to each sample to pull down the immune complexes. Following the washing step, the RNA was extracted and subjected to RT-PCR with JEV 3′ UTR-specific primers. The cDNA was separated by agarose gel electrophoresis, and the expected band of 585 bp is indicated by an arrow.
A protein band with a molecular mass of around 72 kDa was pulled down by the biotin-UTR-Luc RNA (Fig. 1A). LC-MS/MS data indicated that 11 of the identified peptides had an amino acid sequence matching different regions of the sequence of a protein named far upstream element (FUSE) binding protein 1 (FBP1; GenBank accession no. NP_476513). The interaction between FBP1 and JEV UTR RNA was further verified by immunoblotting with anti-FBP1 antibody. The 72-kDa protein pulled down by biotin-UTR-Luc reacted strongly with the anti-FBP1 antibody (Fig. 1B). We also included a biotinylated cellular GAPDH RNA, including its 5′ and 3′ UTRs that has been reported not to bind with FBP1 (40), as a negative control, and FBP1 was not pulled down by this RNA in our assay (Fig. 1B). The interaction of JEV UTR RNA and FBP1 was not restricted to one species or one cell type, as FBP1 from human HeLa cells also was pulled down by biotin-UTR-Luc (Fig. 1C). To further determine the region of JEV UTR RNA interacting with FBP1, in vitro-transcribed biotinylated JEV 5′ UTR or 3′ UTR RNA was used in the pulldown assay. The JEV 3′ UTR seems to be the major contributor for FBP1 binding; however, 5′ UTR also may bind weakly to FBP1 (Fig. 1C). To further demonstrate the association of FBP1 with JEV UTR RNA in JEV-infected cells, HeLa cell extracts collected at 4 h postinfection with JEV (MOI, 10) were subjected to immunoprecipitation with antibody against FBP1. Two negative controls, a control antibody against an unrelated Bst2 protein and a normal mouse IgG, were included in this experiment. The RNA pulled down by the immunocomplexes was extracted and amplified by RT-PCR by using the primers specific to the JEV 3′ UTR. A cDNA band with the expected size (585 bp) was noted in immunoprecipitates brought down by anti-FBP1 but not by anti-Bst2 or IgG control (Fig. 1D), indicating that JEV 3′ UTR RNA associates with FBP1 in JEV-infected cells. Thus, using this RNA-protein pulldown and proteomic assays, we identified FBP1 as a binding partner for JEV UTR RNA for mouse and human cells.
FBP1 is a negative regulator against JEV and DEN-2 replication in human cells.
To address the role of FBP1 binding to JEV RNA, HeLa cells were deprived of their FBP1 expression by transduction with a lentivirus expressing an shRNA targeting the human FBP1 (shFBP1; TRCN0000013297), resulting in the HeLa-shFBP1 cell line. FBP1 expression was greatly decreased by shFBP1 at protein and RNA levels (Fig. 2 A). These cells then were tested for their ability to support JEV replication. Virus production measured by plaque assays at several time points postinfection with JEV was significantly enhanced in cells with reduced FBP1 expression compared to that of shLacZ control cells (Fig. 2B). We also observed accelerated viral protein expression in cells deprived of FBP1 expression (Fig. 2C). Similarly, DEN-2 infection also was enhanced in cells with reduced FBP1 expression, as measured for viral protein expression by immunoblotting (Fig. 3A) and viral progeny production by plaque assays (Fig. 3B).
Fig. 2.

Reduction of FBP1 expression enhanced JEV protein expression and viral production. (A) HeLa cells transduced with lentivirus carrying shRNA-targeting FBP1 (shFBP1; TRCN0000013297) or LacZ (shLacZ) were selected by puromycin (5 μg/ml). The FBP1 knockdown effect was verified by immunoblotting with anti-FBP1 antibody at the protein level (left) and by RT-PCR with FBP1-specific primers at the RNA level (right). The band intensities were quantified with Adobe Photoshop, and the relative ratios of FBP1 to actin were calculated and are shown at the bottom. (B) HeLa cells carrying shFBP1 or control shLacZ were infected with JEV (MOI, 0.1), and at 24, 30, 48, and 72 h postinfection (p.i.) culture supernatants were harvested for plaque assays. The virus titers (PFU/ml) at the same time points from three independent samples, shown here as averages and standard deviations (SD), were compared using a two-tailed Student's t test (n = 3), and the results are shown (**, P < 0.001; ***, P < 0.0005). (C) Extracts from HeLa-shFBP1 or control shLacZ cells infected with JEV (MOI, 5) were collected at 8, 10, 12, and 24 h p.i. and subjected to immunoblotting using antibodies against JEV NS3 and actin as indicated.
Fig. 3.
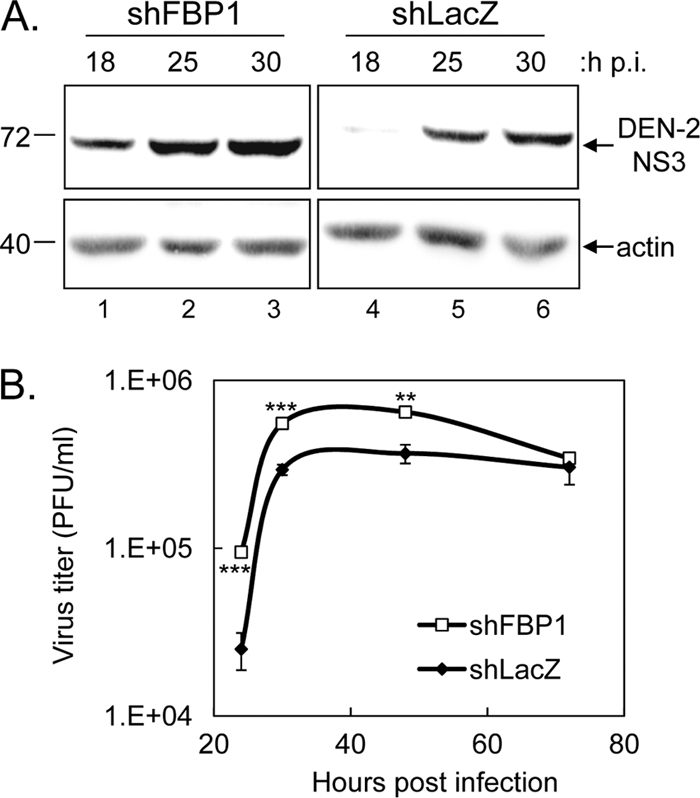
DEN-2 protein expression and viral production were increased in cells with reduced FBP1 expression. (A) Extracts from HeLa-shFBP1 or control shLacZ cells infected with DEN-2 (MOI, 5) were collected at 18, 25, and 30 h p.i. and subjected to immunoblotting using antibodies against DEN-2 NS3 and actin as indicated. (B) HeLa cells carrying shFBP1 or control shLacZ were infected with DEN-2 (MOI, 0.1), and at 24, 30, 48, and 72 h p.i. culture supernatants were harvested for plaque assays. The virus titers at the same time points from three independent samples were compared using a two-tailed Student's t test (n = 3), and the results are shown (**, P < 0.001; ***, P < 0.0005).
To validate the specificity of FBP1 knockdown on JEV infection, we augmented the FBP1 level of HeLa-shFBP1 cells by ectopic expression with a mouse FBP1-expressing construct, which is resistant to the shRNA targeting human FBP1 (Fig. 4A). HeLa-shFBP1 cells transfected with a vector (pcDNA3.1) or a construct expressing V5-tagged mouse FBP1 (M-FBP1/pcDNA3.1) were challenged with JEV (MOI, 5). After reconstitution with mouse FBP1, the higher level of JEV NS3 protein expression no longer was noted in HeLa-shFBP1 cells (Fig. 4B). We further established a JEV replicon cell line by using a JEV replicon RNA carrying a neomycin resistance gene (33) in HeLa-shFBP1 cells. To rescue human FBP1 expression, we also created a human FBP1-expressing construct that carried two wobble mutations, rendering it resistant to shFBP1 targeting but not changing its amino acid sequence (Hr-FBP1/pcDNA3.1) (Fig. 4A). The JEV NS3 protein levels were reduced by transfection with Hr-FBP1/pcDNA3.1 compared to that with the vector (pcDNA3.1) and untransfected cell control (Fig. 4C). These results strongly suggest that the deprivation of FBP1 expression enhances JEV replication, and that FBP1 acts as a host defense protein during JEV infection.
Fig. 4.

Rescue of FBP1 expression in HeLa-shFBP1 cells repressed JEV production. (A) The sequences of shRNA targeting human FBP1 (TRCN0000013297), the relative region of human and mouse FBP1, and shFBP1-resistant human FBP1 are shown. The different nucleotides are underlined and presented as italic letters. The amino acids encoded by this region also are indicated at the top. (B) HeLa-shFBP1 cells transfected with a vector control (pcDNA3.1) or a V5-tagged mouse FBP1-expressing plasmid (M-FBP1) were infected with JEV (MOI, 5). Cells lysates were harvested at 10, 12, and 24 h p.i. and subjected to immunoblotting with antibodies against JEV NS3, V5-tag (for FBP1), and actin as indicated. (C) HeLa-shFBP1 cells carrying a neomycin-resistant gene-containing JEV replicon (HeLa-shFBP1/JEV replicon) were transfected with pcDNA3.1 (vector) or a V5-tagged human FBP1 wobble mutant that is resistant to the targeting of shFBP1 (Hr-FBP1). At 10, 18, and 24 h posttransfection (p.t.), cells lysates were harvested for immunoblotting with antibodies against JEV NS3, V5-tag (for FBP1), and actin as indicated.
Overexpression of FBP1 suppresses JEV replication.
The notion that FBP1 is an anti-JEV protein was further verified by FBP1 overexpression. We transduced HeLa cells with recombinant lentiviruses overexpressing C-terminal V5-tagged human FBP1 (HeLa-FBP1) or enhanced green fluorescent protein (EGFP) as a control (Fig. 5A). The cells then were infected with JEV (MOI, 5) and, consistently with the knockdown data, the overexpression of FBP1 resulted in 8.83- and 10.27-fold decreases in infectious JEV progeny production compared to that of the vector and EGFP control, respectively (Fig. 5D). The JEV viral proteins, such as NS1 and NS3, also were reduced in HeLa-FBP1 cells compared to that in the vector and EGFP control cells, as measured by immunofluorescence assay and Western blotting (Fig. 5A and C). The level of viral RNA indicated by an anti-dsRNA antibody also was lower in cells with FBP1 overexpression (Fig. 5B). These results demonstrate that FBP1 negatively regulates JEV replication in infected cells.
Fig. 5.

Overexpression of FBP1 reduced JEV protein expression and viral production. (A) HeLa cells transduced with lentivirus expressing EGFP, V5 tagged-FBP1, or vector control were infected with JEV (MOI, 5). After 24 h of infection, cells were fixed for immunofluorescence assays with anti-JEV NS3 (red) (A) or anti-dsRNA antibody (red) (B). FBP1 expression was detected by anti-V5 tag antibody (green), and nuclei were stained with DAPI (blue). The signals were photographed using a fluorescence microscope. (C) Cells were infected with JEV (MOI, 5), and cell lysates were collected at 8, 10, 12, and 24 h p.i. for immunoblotting with antibodies against JEV NS3, JEV NS1, V5-tag (for FBP1), EGFP, and actin as indicated. (D) At 24 h p.i., culture supernatants were harvested for virus titration by plaque-forming assays. The virus titers (PFU/ml), shown as averages and standard deviations from three independent samples, were compared by two-tailed Student's t test (n = 3).
Subcellular localization of FBP1 and viral RNA in JEV-infected cells.
FBP1 is a transcription activator localized in the nucleus, whereas JEV replication occurs mainly in the cytoplasm. To address the discrepancy in the cellular localization of FBP1 and JEV, we determined the cellular distribution of FBP1 during the course of JEV infection. FBP1 protein and JEV RNA in HeLa-FBP1 cells were detected by anti-V5 and anti-dsRNA antibodies, respectively. The anti-dsRNA signals have been used as a marker of flavivirus replication complex (55), as the nascent viral RNA labeled by bromouridine incorporation in the presence of actinomycin D was located in the same subcellular site as dsRNA in Kunjin virus-infected cells (54). In the absence of JEV infection, FBP1 protein was localized in the cell nucleus (Fig. 6A), whereas FBP1 was redistributed to the perinuclear and cytoplasmic areas of JEV-infected cells in the early stage of viral infection, such as 2, 4, and 10 h postinfection (p.i.) (Fig. 6B to D). Furthermore, some cytoplasmic FBP1 signals colocalized with those of viral RNA, especially at 4 h p.i. (Fig. 6C). However, in the late stage of JEV infection (24 h p.i.), the colocalization of FBP1 and dsRNA was no longer noted (Fig. 6E). Thus, our results suggest that in the early stages of JEV infection, when viral translation and RNA replication take place, FBP1 is relocated from the nucleus to the cytoplasm and partially interacts with viral RNA; however, in the late stage of the JEV life cycle, when viral RNA is involved mainly in assembly and packaging, FBP1 no longer interacts with JEV RNA. We noted that FBP1 relocated to the perinuclear region of JEV-infected cells and formed cytoplasmic foci that were similar to stress granules (SGs). We thus further determined whether SG markers colocalized with FBP1 under stress conditions. FBP1 was located predominantly in the nuclei of HeLa-FBP1 cells without stimulus. Under heat shock conditions, some FBP1 signals relocalized to cytoplasmic foci and colocalized with SG markers, such as eIF4E (31), PABP1 (31), and Ras-GAP SH3 domain binding protein (G3BP) (49) (data not shown). Because SGs and processing bodies (P bodies) often are physically associated (3), we further tested whether FBP1 also colocalized with P bodies. FBP1 proteins in HeLa-FBP1 cells treated with sodium arsenite did not colocalize with those of p54/RCK, a marker for P bodies (14); however, these two proteins were found in nearby areas (data not shown). These results suggest that FBP1 is a component protein of SGs during stress conditions.
Fig. 6.

Cellular localization of FBP1 and dsRNA. HeLa cells with V5-tagged FBP1 stable expression (HeLa-FBP1) were infected with JEV (MOI, 5) for 2, 4, 10, and 24 h and stained with antibodies against dsRNA (red), V5-tag (green), and DAPI (blue). The samples were observed and photographed by using a confocal microscope (Zeiss LSM 510 META).
FBP1 is a negative regulator for JEV UTR-mediated protein expression.
Since FBP1 interacts with JEV RNA in the early stage of the JEV life cycle, we used a JEV replicon reporter system to test whether FBP1 modulated viral translation and/or RNA replication. We constructed a luciferase-containing JEV replicon, J-R2A (Fig. 7A), which was comprised of JEV 5′ and 3′ UTRs, a Renilla luciferase reporter gene followed by FMDV 2A self-cleaving protease, and JEV nonstructural regions as described for other flaviviruses (38, 50). Besides HeLa cells, we also generated FBP1 knockdown cells in NT2, a human neuroblastoma cell line, by transduction with an shFBP1-expressing lentivirus (TRCN0000013293). FBP1 protein expression was greatly decreased in these NT2 cells (Fig. 7B). NT2 cells deprived of FBP1 or the LacZ control were cotransfected with in vitro-transcribed J-R2A RNA and firefly luciferase RNA as the transfection control. Cells then were harvested for dual luciferase assays at 3, 6, 9, 24, 48, and 72 h posttransfection. As expected, two peaks of replicon luciferase activity were noted (Fig. 7C), which represented the first translation and the second replication peak, as reported for other flaviviral replicon systems. Higher luciferase activities were noted for cells deprived of FBP1 expression for all of the time points (Fig. 7C), indicating that viral protein translation and viral RNA replication were hampered by the presence of FBP1. Because JEV RNA replication depends on viral proteins, we suspect FBP1 mainly targets viral translation, which then leads to the suppression of RNA replication. We thus generated a replication-dead replicon, J-R2A-NS5mt, which contained a GDD→AAG mutation in NS5 polymerase (8), enabling us to analyze the role of FBP1 in viral translation. Higher levels of luciferase activity derived from replication-dead J-R2A-NS5mt RNA also were noted in cells with FBP1 knockdown (Fig. 7D). In contrast, the luciferase activities derived from a control reporter, Renilla luciferase flanked by cellular GAPDH 5′ and 3′ UTRs, were not significantly different between the control shLacZ and shFBP1 cells (Fig. 7E). Since the reporter activity of JEV replicon is dependent on the 5′ and 3′ UTRs, our results strongly suggest that FBP1 targets JEV UTRs and represses JEV protein expression.
DISCUSSION
By using a biotinylated RNA-protein pulldown system followed by proteomic analysis, we identified FBP1 as a JEV UTR RNA binding protein that negatively regulates the JEV and DEN-2 life cycle by blocking viral protein expression. Recently, FBP1 also has been found to bind with the 3′ UTR of HCV, a member of the Flaviviridae family; however, binding with FBP1 is required for efficient HCV replication (60). Thus, FBP1 may function as a negative or positive host factor for different members of the Flaviviridae family, such as JEV/DEN-2 and HCV, and it would be interesting to study further the roles of FBP1 in the life cycles of other members of this virus family.
FBP1 was first identified as a single-stranded DNA binding protein that regulated c-myc gene expression by binding to FUSE of the c-myc promoter (5, 15, 19–20, 26). Subsequently, FBP1 and FBP2 have been found to bind to AU-rich elements (AREs) in several mRNA species and have been implicated in various aspects of RNA metabolism (18, 29, 45). More recently, FBP1 and FBP2 have been found to interact with TIA-1 and TIAR and relocate to SGs in cells subjected to arsenite-induced oxidative stress (3, 43). SGs are dynamic aggregates of nontranslating mRNAs and a subset of proteins that form in cells upon exposure to various stresses, e.g., heat, oxidative conditions, viral infection, and hypoxia (4). Notable RNA binding proteins in SGs include TIA-1, TIAR, and G3BP, and when these proteins are overexpressed they promote SG formation (22, 49). SGs often snuggle up against P bodies, which are associated with mRNA decay, suggesting that mRNAs move between these two compartments for dynamic sorting between translation repression and RNA degradation.
Components of SGs and P bodies have been found to affect virus life cycles in a negative or positive manner by repressing viral transcripts or promoting viral replication (6). For example, mouse embryo fibroblasts lacking the TIA-1 protein show increased virus production from a wide range of different viruses, including vesicular stomatitis virus (a negative-sense RNA virus), Sindbis virus (a positive-sense RNA virus), and herpes simplex virus (a DNA virus replicating in the nucleus); in contrast, WNV (a positive-sense RNA flavivirus) grows less efficiently in these TIA-1 knockout cells (34). Our finding that FBP1 functions as a negative regulator for JEV translation and replication is concordant with the results for another TIA-interacting SG-associated protein, YB-1 (57), that represses DEN-2 translation (42). Taking these findings together, we add FBP1 to the growing list of SG/P body-associated proteins that may function in the host antiviral defense response by targeting viral RNA for translation repression and RNA decay.
For a positive-sense RNA virus, such as JEV, the incoming viral RNA undergoes a first round of translation after entry and uncoating. The viral RNA then serves as a template for RNA replication, and further translation may occur on the newly synthesized viral RNA. Finally, the viral RNA will be packaged within a capsid and assembled for mature virus production. The mechanisms by which JEV segregates translation from replication and assembly, and the host factors involved in these processes, are not well understood. Our results that FBP1 colocalizes with JEV RNA, as indicated by anti-dsRNA antibody, especially at 4 h after viral infection (Fig. 6), and that FBP1 reduces JEV translation (Fig. 7) suggest that FBP1 interacts mainly with the incoming RNA undergoing translation and not with the viral RNA template for RNA replication. In a recent study, FBP1 was found to interact with the 3′ UTR of nucleophosmin, a multifunctional oncoprotein, and to repress its translation through the exclusion of nucleophosmin mRNA from actively translating polysomes (40). It would be interesting to further study whether FBP1 also affects JEV RNA translation in a similar fashion. The finding that at 24 h postinfection FBP1 no longer colocalized with dsRNA may reflect that in the late stage of viral infection, viral RNA is packaged with the capsid proteins and is not accessible by FBP1.
JEV is the leading cause of viral encephalitis in Asia, and it is estimated that there are more than 50,000 cases of JE and 10,000 deaths from JE annually. Thus, JE is a regionally important public health issue; however, research on JEV lags behind that for related flaviviruses, such as WNV and DEN. Regarding the cellular factors involved in JEV infection, only a few host proteins have been found to bind with JEV RNA, and their roles in JEV infection are largely unclear. The Mov34 protein from mouse brain tissue binds with the positive-sense JEV 3′ SL RNA (47), whereas polypyrimidine tract binding protein interacts with the negative-sense JEV 3′ SL RNA (32). GAPDH interacts with the 3′ end of JEV UTR RNA and colocalizes with the viral RNA-dependent RNA polymerase NS5 protein (56). La protein interaction with the 3′ UTR of JEV may play an important role in JEV replication; however, the mechanisms for this remain unclear (51). Most of these published results were conducted by using traditional experimental strategies, such as UV cross-linking and electrophoretic mobility shift assays, to fish out the cellular proteins that interact with JEV RNA. In this study, we used biotin-labeled RNA comprising both 5′ and 3′ UTRs of JEV to pull down the interacting cellular proteins and identified FBP1 as a candidate. More importantly, the role FBP1 plays in JEV infection was elucidated by the overexpression and knockdown of FBP1, and our results indicate a negative role for FBP1 in JEV infection. The step in the virus life cycle targeted by FBP1 likely is at viral protein translation, because FBP1 colocalized with viral RNA at SGs during the early stage of infection and the reporter activities derived from a JEV replicon increased in cells deprived of FBP1 expression. Overall, we recapitulate how FBP1 participates in JEV infection and pave the way for the better understanding of the regulatory mechanisms of cellular proteins in JEV replication.
ACKNOWLEDGMENTS
We thank the Core Facility of the Institute of Biomedical Sciences, Academia Sinica, Taiwan, for performing the LC-MS/MS analysis and the National RNAi Core Facility, Taiwan (supported by the National Research Program for Genomic Medicine Grants of National Science Council), for shRNA constructs.
This work was supported by grants awarded to Y.-L.L. from the National Science Council (NSC98-2320-B-001-011-MY3 and NSC99-3112-B-001-016) and from Academia Sinica, Taiwan.
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Alvarez D. E., Lodeiro M. F., Luduena S. J., Pietrasanta L. I., Gamarnik A. V. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 79:6631–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amano T., Richelson E., Nirenberg M. 1972. Neurotransmitter synthesis by neuroblastoma clones (neuroblast differentiation-cell culture-choline acetyltransferase-acetylcholinesterase-tyrosine hydroxylase-axons-dendrites). Proc. Natl. Acad. Sci. U. S. A. 69:258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson P., Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141–150 [DOI] [PubMed] [Google Scholar]
- 4. Anderson P., Kedersha N. 2002. Stressful initiations. J. Cell Sci. 115:3227–3234 [DOI] [PubMed] [Google Scholar]
- 5. Avigan M. I., Strober B., Levens D. 1990. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J. Biol. Chem. 265:18538–18545 [PubMed] [Google Scholar]
- 6. Beckham C. J., Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackwell J. L., Brinton M. A. 1997. Translation elongation factor-1 alpha interacts with the 3′ stem-loop region of West Nile virus genomic RNA. J. Virol. 71:6433–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blight K. J., McKeating J. A., Marcotrigiano J., Rice C. M. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brinton M. A. 2001. Host factors involved in West Nile virus replication. Ann. N. Y. Acad. Sci. 951:207–219 [DOI] [PubMed] [Google Scholar]
- 10. Brinton M. A., Dispoto J. H. 1988. Sequence and secondary structure analysis of the 5′-terminal region of flavivirus genome RNA. Virology 162:290–299 [DOI] [PubMed] [Google Scholar]
- 11. Chen C. J., et al. 1997. RNA-protein interactions: involvement of NS3, NS5, and 3′ noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 71:3466–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen L. K., et al. 1996. Persistence of Japanese encephalitis virus is associated with abnormal expression of the nonstructural protein NS1 in host cells. Virology 217:220–229 [DOI] [PubMed] [Google Scholar]
- 13. Chen L. K., et al. 1996. Generation and characterization of organ-tropism mutants of Japanese encephalitis virus in vivo and in vitro. Virology 223:79–88 [DOI] [PubMed] [Google Scholar]
- 14. Cougot N., Babajko S., Seraphin B. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165:31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis-Smyth T., Duncan R. C., Zheng T., Michelotti G., Levens D. 1996. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J. Biol. Chem. 271:31679–31687 [DOI] [PubMed] [Google Scholar]
- 16. Davis W. G., Blackwell J. L., Shi P. Y., Brinton M. A. 2007. Interaction between the cellular protein eEF1A and the 3′-terminal stem-loop of West Nile virus genomic RNA facilitates viral minus-strand RNA synthesis. J. Virol. 81:10172–10187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Nova-Ocampo M., Villegas-Sepulveda N., del Angel R. M. 2002. Translation elongation factor-1alpha, La, and PTB interact with the 3′ untranslated region of dengue 4 virus RNA. Virology 295:337–347 [DOI] [PubMed] [Google Scholar]
- 18. Dean J. L., Sully G., Clark A. R., Saklatvala J. 2004. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 16:1113–1121 [DOI] [PubMed] [Google Scholar]
- 19. Duncan R., et al. 1994. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8:465–480 [DOI] [PubMed] [Google Scholar]
- 20. Duncan R., Collins I., Tomonaga T., Zhang T., Levens D. 1996. A unique transactivation sequence motif is found in the carboxyl-terminal domain of the single-strand-binding protein FBP. Mol. Cell. Biol. 16:2274–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edgil D., Harris E. 2006. End-to-end communication in the modulation of translation by mammalian RNA viruses. Virus Res. 119:43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilks N., et al. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15:5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Godfrey A., Anderson J., Papanastasiou A., Takeuchi Y., Boshoff C. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510–2518 [DOI] [PubMed] [Google Scholar]
- 24. Hahn C. S., et al. 1987. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 198:33–41 [DOI] [PubMed] [Google Scholar]
- 25. Harris D., Zhang Z., Chaubey B., Pandey V. N. 2006. Identification of cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome. Mol. Cell Proteomics 5:1006–1018 [DOI] [PubMed] [Google Scholar]
- 26. He L., et al. 2000. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 19:1034–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holden K. L., Harris E. 2004. Enhancement of dengue virus translation: role of the 3′ untranslated region and the terminal 3′ stem-loop domain. Virology 329:119–133 [DOI] [PubMed] [Google Scholar]
- 28. Holden K. L., et al. 2006. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3′ stem-loop structure. Virology 344:439–452 [DOI] [PubMed] [Google Scholar]
- 29. Irwin N., Baekelandt V., Goritchenko L., Benowitz L. I. 1997. Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 25:1281–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kedersha N., Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61–81 [DOI] [PubMed] [Google Scholar]
- 31. Kedersha N., et al. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i) (Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell 13:195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim S. M., Jeong Y. S. 2006. Polypyrimidine tract-binding protein interacts with the 3′ stem-loop region of Japanese encephalitis virus negative-strand RNA. Virus Res. 115:131–140 [DOI] [PubMed] [Google Scholar]
- 33. Lee C. J., Lin H. R., Liao C. L., Lin Y. L. 2008. Cholesterol effectively blocks entry of flavivirus. J. Virol. 82:6470–6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W., et al. 2002. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 76:11989–12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang J. J., Liao C. L., Liao J. T., Lee Y. L., Lin Y. L. 2009. A Japanese encephalitis virus vaccine candidate strain is attenuated by decreasing its interferon antagonistic ability. Vaccine 27:2746–2754 [DOI] [PubMed] [Google Scholar]
- 36. Lin J. Y., Li M. L., Shih S. R. 2009. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 37:47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin Y. L., et al. 1998. Study of dengue virus infection in SCID mice engrafted with human K562 cells. J. Virol. 72:9729–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lo M. K., Tilgner M., Bernard K. A., Shi P. Y. 2003. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3′ untranslated region of West Nile virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 77:10004–10014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Makarova O., Kamberov E., Margolis B. 2000. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 29:970–972 [DOI] [PubMed] [Google Scholar]
- 40. Olanich M. E., Moss B. L., Piwnica-Worms D., Townsend R. R., Weber J. D. 2011. Identification of FUSE-binding protein 1 as a regulatory mRNA-binding protein that represses nucleophosmin translation. Oncogene 30:77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olsthoorn R. C., Bol J. F. 2001. Sequence comparison and secondary structure analysis of the 3′ noncoding region of flavivirus genomes reveals multiple pseudoknots. RNA 7:1370–1377 [PMC free article] [PubMed] [Google Scholar]
- 42. Paranjape S. M., Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3′-untranslated region and mediates antiviral effects. J. Biol. Chem. 282:30497–30508 [DOI] [PubMed] [Google Scholar]
- 43. Rothé F., Gueydan C., Bellefroid E., Huez G., Kruys V. 2006. Identification of FUSE-binding proteins as interacting partners of TIA proteins. Biochem. Biophys. Res. Commun. 343:57–68 [DOI] [PubMed] [Google Scholar]
- 44. Su H. L., Liao C. L., Lin Y. L. 2002. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76:4162–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sully G., et al. 2004. Structural and functional dissection of a conserved destabilizing element of cyclo-oxygenase-2 mRNA: evidence against the involvement of AUF-1 [AU-rich element/poly(U)-binding/degradation factor-1], AUF-2, tristetraprolin, HuR (Hu antigen R) or FBP1 (far-upstream-sequence-element-binding protein 1). Biochem. J. 377:629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sumiyoshi H., et al. 1987. Complete nucleotide sequence of the Japanese encephalitis virus genome RNA. Virology 161:497–510 [DOI] [PubMed] [Google Scholar]
- 47. Ta M., Vrati S. 2000. Mov34 protein from mouse brain interacts with the 3′ noncoding region of Japanese encephalitis virus. J. Virol. 74:5108–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tilgner M., Deas T. S., Shi P. Y. 2005. The flavivirus-conserved penta-nucleotide in the 3′ stem-loop of the West Nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology 331:375–386 [DOI] [PubMed] [Google Scholar]
- 49. Tourrière H., et al. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50. Varnavski A. N., Khromykh A. A. 1999. Noncytopathic flavivirus replicon RNA-based system for expression and delivery of heterologous genes. Virology 255:366–375 [DOI] [PubMed] [Google Scholar]
- 51. Vashist S., Anantpadma M., Sharma H., Vrati S. 2009. La protein binds the predicted loop structures in the 3′ non-coding region of Japanese encephalitis virus genome: role in virus replication. J. Gen. Virol. 90:1343–1352 [DOI] [PubMed] [Google Scholar]
- 52. Villordo S. M., Gamarnik A. V. 2009. Genome cyclization as strategy for flavivirus RNA replication. Virus Res. 139:230–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vindigni A., Ochem A., Triolo G., Falaschi A. 2001. Identification of human DNA helicase V with the far upstream element-binding protein. Nucleic Acids Res. 29:1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Westaway E. G., Khromykh A. A., Mackenzie J. M. 1999. Nascent flavivirus RNA colocalized in situ with double-stranded RNA in stable replication complexes. Virology 258:108–117 [DOI] [PubMed] [Google Scholar]
- 55. Westaway E. G., Mackenzie J. M., Khromykh A. A. 2003. Kunjin RNA replication and applications of Kunjin replicons. Adv. Virus Res. 59:99–140 [DOI] [PubMed] [Google Scholar]
- 56. Yang S. H., Liu M. L., Tien C. F., Chou S. J., Chang R. Y. 2009. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) interaction with 3′ ends of Japanese encephalitis virus RNA and colocalization with the viral NS5 protein. J. Biomed. Sci. 16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang W. H., Bloch D. B. 2007. Probing the mRNA processing body using protein macroarrays and “autoantigenomics.” RNA 13:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. You S., Falgout B., Markoff L., Padmanabhan R. 2001. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′- and 3′-terminal regions that influence RNA structure. J. Biol. Chem. 276:15581–15591 [DOI] [PubMed] [Google Scholar]
- 59. Yu L., Markoff L. 2005. The topology of bulges in the long stem of the flavivirus 3′ stem-loop is a major determinant of RNA replication competence. J. Virol. 79:2309–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Z., Harris D., Pandey V. N. 2008. The FUSE binding protein is a cellular factor required for efficient replication of hepatitis C virus. J. Virol. 82:5761–5773 [DOI] [PMC free article] [PubMed] [Google Scholar]