

Abstract
The human immunodeficiency virus (HIV) capsid (CA) protein assembles into a hexameric lattice that forms the mature virus core. Contacts between the CA N-terminal domain (NTD) of one monomer and the C-terminal domain (CTD) of the adjacent monomer are important for the assembly of this core. In this study, we have examined the effects of mutations in the NTD region associated with this interaction. We have found that such mutations yielded modest reductions of virus release but major effects on viral infectivity. Cell culture and in vitro assays indicate that the infectivity defects relate to abnormalities in the viral cores. We have selected second-site compensatory mutations that partially restored HIV infectivity. These mutations map to the CA CTD and to spacer peptide 1 (SP1), the portion of the precursor Gag protein immediately C terminal to the CTD. The compensatory mutations do not locate to the molecularly modeled intermolecular NTD-CTD interface. Rather, the compensatory mutations appear to act indirectly, possibly by realignment of the C-terminal helix of the CA CTD, which participates in the NTD-CTD interface and has been shown to serve an important role in the assembly of infectious virus.
INTRODUCTION
Retroviruses are composed of a glycoprotein-studded lipid membrane enveloping a ribonucleoprotein core. The major structural protein of retroviruses is the PrGag polypeptide, which is composed of several subdomains. At the N terminus, the matrix (MA) domain interacts with the membrane via a myristoyl moiety and N-terminal basic amino acids (3, 6, 18, 19, 22, 25, 50, 56, 57, 61, 64, 73, 74). The capsid (CA) domain, which includes an N-terminal domain (NTD) and a C-terminal domain (CTD) connected by a flexible linker, is the major component responsible for Gag-Gag protein interactions (24, 29, 31, 32, 34, 39, 67, 70). Following capsid is spacer peptide 1 (SP1), which joins to nucleocapsid (NC). Nucleocapsid binds the viral RNA; through this interaction, NC both initiates PrGag oligomerization and assembly and ensures that the viral genome is packaged in budding virions (4, 12, 14, 20, 23). Nucleocapsid is connected to spacer peptide two (SP2), which is followed by the p6, or late, domain, at the C terminus of the Gag polyprotein. The p6 domain is involved in protein interactions with cellular components that ensure PrGag budding at the plasma membrane, the site of productive assembly (28, 33, 35, 36, 44, 53, 61, 65, 71).
PrGag undergoes proteolytic cleavage during the maturation of the virus, which is critical for infectivity. This process may occur during assembly at the plasma membrane or shortly after budding from the producer cell. Prior to cleavage by the HIV protease, immature virions are spherical in nature, with as many as 5,000 densely packed PrGag proteins assembled into a hexameric lattice (10, 11, 54). After cleavage, the spherical shell of PrGag reassembles to form a cone or tube; this viral core is composed of ∼1,500 CA proteins (32). The CA proteins in the core are also assembled as hexamers, but with 12 pentamers at the ends to enable closure of the cone (11, 32, 43, 52). In electron micrographs of HIV produced from cells, mature HIV cores can be seen in both tube and cone forms, with the cone form predominating (16, 17, 42, 70). Within viral cores, NC proteins are bound to two copies of viral genomic RNA, which associate with viral reverse transcriptase (RT) and integrase (IN) proteins as well as other viral and cellular factors that contribute to infectivity.
The structural organization of CA accommodates both the immature and the mature forms of hexameric assembly. The seven alpha helices of the NTD (CA helices 1 to 7) are connected via a linker to the four alpha helices (CA helices 8 to 11) of the CTD (Fig. 1A). At the N-terminal end of the NTD, 13 residues fold after proteolytic cleavage to form a β-hairpin, which is required for infectivity (1, 37, 41, 49, 51, 67). The first three helices of the NTD interact with the first three helices of other CA NTDs, forming an 18-helix bundle in a hexameric lattice (30, 52, 58). The CA CTD dimerizes to connect the hexameric subunits to each other (13, 30, 58). Intermolecular interactions between NTD helices 3 and 4 and helix 8 of the adjacent CTD stabilize the hexamer (30, 58). Further stabilization is provided from contacts between helix 7 of the NTD and helix 11 of the CTD (30, 58). A further interaction is between the NTD-CTD interface and helix 9 of the CTD: this connection is proposed to have a role in cooperativity of assembly (30). Evidence of the importance of the intermolecular NTD-CTD interface in vivo comes from a study of the Rous sarcoma virus capsid: mutations in the CTD major homology region (MHR) elicited second-site revertants in the NTD (7). Further support for the importance of NTD-CTD interactions derives from studies of the assembly inhibitor CAP-1. CAP-1 binds to a hydrophobic groove formed by the N-terminal end of helix 4 and the C-terminal end of helix 7; this same groove is normally occupied by the CTD. Disruption of this interaction in cell culture by incubation with CAP-1 leads to inhibition of HIV-1 infectivity and abnormal core morphologies (46, 58, 66).
Fig. 1.

Effects of mutations on virus particle assembly and release. (A) Shown is a pair of CA proteins (Protein Data Bank [PDB] accession no. 3GV2) in HIV-1 CA hexamers viewed roughly from the center of the hexamer outward. NTDs (blue) and CTDs (green) are marked, as are the β-hairpin (β) and cyclophilin A binding (CypA) loops and helices 1 to 11 on one of the capsid proteins. H62 residues are indicated in red. (B) HEK 293T cells were transfected with the indicated HIVLuc constructs. At 72 h posttransfection, virus samples were collected, and Gag proteins were detected after gel electrophoresis by immunoblotting using a primary anti-CA antibody. Full-length PrGag and CA bands are as indicated. (C) For quantification of viral particle release at 72 h posttransfection, virus and cell samples were collected and subjected to electrophoresis and immunoblotting using an anti-CA antibody. Cell and virus Gag levels were quantitated densitometrically; raw virus release levels (Virus Gag/Cell PrGag) were calculated and are shown, normalized to WT HIV release levels. Results derive from two (H62K and H62W), three (H62C and H62F), four (H62Y), five (H62A), or eight (WT) separate experiments. Standard deviations are as shown.
To further understand how the NTD-CTD interface contributes to HIV-1 assembly, we mutated a key residue, histidine 62 (H62), within the loop between NTD helices 3 and 4 (Fig. 1A). This residue is of particular interest for several reasons. It is highly conserved (27, 62), and it has been shown to move upon CAP-1 binding. Furthermore, histidine is unique in that the pKa of its imidazole side chain is 6.04, within physiologic levels. Protonation of histidine, occurring at a pH that is thought to trigger the “mature” form of CA in vitro, could be responsible for rearrangements of the capsid hexamer lattice involved in maturation (27). Indeed, structural evidence suggests that the imidazole group of H62 forms ring-stacking interactions with F32 and Y145 of the NTD, which is buttressed by the guanidium group of the CTD at R162 (58). In our study, mutations of H62 to alanine, cysteine, tryptophan, lysine, tyrosine, or phenylalanine had modest effects on assembly and release but pronounced effects on infectivity. Remarkably, we discovered three independent second-site compensatory mutations of H62 mutants. These second-site mutations bracketed helix 11 of the CTD: one was in SP1, and remarkably, two mapped to the same G208 residue in the CTD. The compensatory mutants showed enhanced viral replication and assembly in vitro over the respective H62 mutants but were not better than wild-type (WT) HIV-1. The locations of the compensatory mutations, in SP1 and in the loop between helices 10 and 11, are surprising, as they are not spatially located near the H62 residue or directly involved in any CA NTD-CTD interactions. Our data suggest that compensatory changes for H62 mutations act indirectly by repositioning CTD elements to offset NTD structural alterations.
MATERIALS AND METHODS
Recombinant DNA constructs.
A vesicular stomatitis virus (VSV) glycoprotein (G) expression construct, pVSV-G, was a generous gift from Randy Taplitz. The parental HIVLuc construct (pNL-LucE-R+) (21) was kindly provided by Nathaniel Landau. The parental NL4-3 construct (2) is available from the AIDS reagent database. The bacterial expression vector used in the in vitro assembly assay, pWISP-WTCAH6, has been described previously (5).
To create the H62 mutants in the HIVLuc and pNL4-3 backgrounds, the BssHII (nucleotide [nt] 711)-HindIII (nt 1712) fragments were amplified using overlap PCR with primers mutagenic at the nt 1369-to-1371 (H62) site. All nucleotide numbers refer to NL4-3 as a reference sequence. Mutant fragments were cloned into the pGEMTeasy vector (Promega), and mutations were confirmed by sequencing. The BssHII-SpeI fragments then were subcloned into HIVLuc and pNL4-3. The nucleotides covering CA residues 60 to 65 (NL4-3 nt 1363 to 1377) are as follows, with mutations underlined for each variant: for H62A, 5′GGGGGAGCTCAAGCA3′; H62C, 5′GGGGGATGCCAAGCA3′; H62F, 5′GGGGGATTTCAAGCA3′; H62K, 5′GGGGGAAAGCAAGCA3′; H62W, 5′GGGGGATGGCAAGCA3′; and H62Y, 5′GGGGGATATCAAGCA3′.
Compensatory mutants cloned from longitudinal passages were isolated by extraction of DNA from infected cells. Cells were lysed in 250 μl IPB (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.02% sodium azide, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]). An equal volume of water was added, and cells were then digested with 50 μg of proteinase K for 2 h. DNA was isolated by phenol-chloroform extraction and ethanol precipitation. The sequence between nt 671 and nt 2096 was amplified using Accuprime Pfx polymerase (Invitrogen) and then cloned into the EcoRV site of the pBluescript-SK plasmid (Stratagene) for sequencing. Potential revertants were subcloned into pNL4-3 and HIVLuc via the BssHII-ApaI (nt 711 to 2006) sites. For bacterial expression, the H62 variants were subcloned into pWISP-WTCAH6 as Nsi-SpeI fragment (nt 1247 to 1507) sites from HIVLuc.
Cell culture and transfections.
HEK 293T (26) and HiJ (45) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 10 mM HEPES (pH 7.4), penicillin, and streptomycin at 37°C/5% CO2. CEM-SS and MT-4 T cell lines were maintained in RPMI medium supplemented with 10% fetal bovine serum, 10 mM HEPES (pH 7.4), penicillin, and streptomycin at 37°C/5% CO2.
Quantification of viral particle assembly and release was performed using calcium phosphate transfections of HEK 293T cells by 24 μg of HIVLuc plasmid constructs as described previously (68, 69). Virus samples were collected at 72 h by filtering them through a 0.45-μm filter to remove cell debris and then concentrated by centrifugation (68, 69). Viruses were then resuspended in phosphate-buffered saline (PBS) and stored at −80°C. Cell samples were lysed in IPB, spun for 15 min in a microcentrifuge at 13,000 rpm to pellet the insoluble membrane material, and then stored at −80°C. Virus and cell samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (62, 63) and subsequently immunoblotted using a primary anti-CA antibody (183-H12-5C; available from the AIDS Reagent and Reference program) (15), followed by secondary alkaline phosphatase-conjugated anti-mouse antibodies (Promega). Cell and virus Gag levels were quantified densitometrically using NIH ImageJ. Mean gray values for each CA band were measured for the same-sized areas on blots, and virus release levels were calculated as virus Gag/cell Gag ratios and normalized to WT HIVLuc levels.
To perform luciferase-based infectivity assays, HEK 293T cells were transfected as described above, but with 18 μg of HIVLuc plasmids and 6 μg of pVSV-G. At 72 h posttransfection, viruses were collected, filtered to remove cell debris, and then stored at −80°C until use. Aliquots of transfected cell samples also were collected in luciferase assay buffer (100 mM sodium phosphate, pH 8.0, 4 mM adenosine triphosphate, 1 mM sodium pyrophosphate, 6 mM magnesium chloride, 0.2% Triton X-100) to be run as transfection controls. For infections, HiJ cells were plated from a 10-cm dish the day before. One-fortieth of cells from a 10-cm dish were plated in each well of a 6-well dish and 1 day later were exposed to 1 ml virus plus 1 ml complete DMEM in duplicate and then incubated at 37°C/5% CO2 for 72 h. After infections, wells were washed with PBS and lysed in 250 μl luciferase assay buffer. Samples from both transfected and infected cells were diluted 1:10 in luciferase assay buffer without Triton X-100 and run on an Auto Lumat LB 953 luminometer (Berthold) with injections set for 100 μl of a 1 mM luciferin solution (BD Monolight). After collection of raw luminometer counts, results were calculated as (average of infection duplicate counts)/(average of transfection counts) and normalized to the level for WT samples run in parallel.
For the longitudinal infections, 24 μg of pNL4-3 WT or H62 variants was transfected via calcium phosphate into HEK 293T cells. At 3 days posttransfection, viral supernatants were collected and filtered to remove cell debris. Viral supernatants were subjected to SDS-PAGE and immunoblotting, as described above, and CA bands were quantified with NIH ImageJ. Starting inocula were normalized based on CA levels for infection of either MT4 or CEM-SS cells. After infections were initiated, aliquots of cells (20%) were collected every 3 to 4 days, centrifuged to remove virus, and lysed in IPB. Infections were tracked by immunoblot detection of infected cell Gag levels. Potential compensatory mutant viruses from late-stage collections were used to reinfect fresh stocks of cells after normalization against the WT level. These passages were monitored by CA immunoblot as described above, and mutations were cloned as described above.
Analysis of capsid assembly products.
HIVLuc WT and H62 variant virus-like particles were produced by calcium phosphate transfection, concentrated by ultracentrifugation, and suspended in PBS. Concentrated virus particle samples were lifted for 2 min onto 400-mesh carbon-Formvar-coated, UV-treated electron microscopy (EM) grids (Ted Pella 01822-F), rinsed for 15 s in water, wicked, stained for 1 min in filtered 1.3% uranyl acetate, wicked, and dried. EM images were collected at 100 kV on a Philips CM120/Biotwin transmission electron microscope (TEM). Images were collected as 1,024- by 1,024-pixel, 14-bit grayscale Gatan digital micrograph 3 (DM3) files on a Gatan 794 multiscan charge-coupled-device multiscan camera and converted into 8-bit grayscale TIFF images using the program Digital Micrograph 3.4.0. Samples were viewed at ×37,000.
To examine in vitro assembly of CA proteins, bacterial expression plasmids were transformed into the Escherichia coli strain BL21(DE3)/pLysS (Novagen) for protein expression and purification, which were done in accordance with previous procedures (5). Purified fractions were subjected to three rounds of buffer exchange by dialysis in buffer (20 mM Tris, pH 8.0, 5 mM β-mercaptoethanol [βME]) for 6 h at 8°C, and protein aliquots were flash frozen on dry ice and stored at −80°C. Protein identities were assessed by immunoblotting, and protein purities of >90% were verified by Coomassie blue staining of samples fractionated by SDS-PAGE. Protein concentrations were determined via 280-nm absorbance readings assuming a molar extinction coefficient of 33,580 and by densitometric comparison of stained SDS-PAGE protein bands versus known standards.
Imaging of assembly reactions by electron microscopy (EM) was performed as follows. Typically, 10-μl assembly reaction mixtures included 100 μM protein and 1× assembly buffer (50 mM Tris, pH 7.0, 1 M NaCl, and 5 mM βME) and were incubated for 48 h at 4°C. After the incubations, samples were lifted onto 400-mesh carbon-Formvar grids. Samples were lifted onto grids for 3 min, washed for 1 min in water, wicked on filter paper, stained for 45 s in filtered 1.33% uranyl acetate, wicked, and air dried for 5 min. Samples were viewed at 100 kV on a Philips CM120 TEM, and images were collected as 1,024- by 1,024-pixel, 14-bit grayscale Gatan Digital Micrograph 3 (DM3) files on a Gatan 794 CCD multiscan camera and converted into 8-bit grayscale TIFF images using the program Digital Micrograph 3.4.0.
For quantitation of EM results, at least six 4.136-μm by 4.136-μm grayscale TIFF images for each variant were analyzed. To do so, image features were highlighted using the Feature J/FJ Laplacian command, with a smoothing step of 10 and the zero crossings parameter unselected. Laplacian images were thresholded to low values of 0.10 and maximum high values. For tube coverage tabulation, the Analyze/Analyze Particles command was used, with the Analyze/Set Measurements area and Feret's diameter options highlighted. The Analyze Particles command was called with size and circularity arguments as for FM analysis, and the results (including particle identification numbers, particle areas, and Feret diameters) were saved in table format. Tube candidates were then excluded if they did not meet the criteria of having a Feret's diameter of at least 50 and a (Feret)2/(area) value of at least 4. After these qualification steps were performed, total areas covered by tubes were summed and divided by total areas to obtain area fractions covered by tubes. The results are depicted as areas covered by tubes, normalized to the results obtained with WT incubations.
RESULTS
Analysis of capsid mutants.
There are five histidines in HIV-1 capsid, three of which are conserved (H12, H62, and H84) (27). Of these three, our previous work delineated a role in assembly for H84, located on an external surface of the NTD hexamer (62). The residue H62 is also in the NTD (Fig. 1A) and locates to the assembly inhibitor CAP-1 binding region. Additionally, H62 forms ring-stacking interactions with F32 and Y145, which are thought to be critical for the stability of the NTD-CTD intermolecular interface (46, 58). We interrogated the role of H62 in this interaction by substituting alanine, cysteine, lysine, tyrosine, phenylalanine, and tryptophan for histidine and assessed these mutants in vitro and in cell culture.
To measure the effects of mutations on the release of virus-like particles (VLP), we transfected 293T cells with each mutant construct in the context of an HIV Luc background. Levels of virus release were monitored by anti-CA immunoblotting of SDS-PAGE-fractionated VLP samples taken from the supernatants of transfected cells. As shown in Fig. 1B, the VLP levels of the H62 mutants were slightly reduced compared to those of the WT, and there appeared to be minor variations in CA-to-PrGag ratios. To quantify the viral release, parallel cell and VLP samples were electrophoresed and immunoblotted for PrGag and CA detection. Gag band intensities were determined via densitometry, and virus-to-cell Gag ratios were calculated, after which the mutant construct ratios were normalized to those of the WT (Fig. 1C). Whereas H62 mutants exhibited slight decreases in virus release, the defects appeared moderate in that all of the mutant viruses were released at least half as efficiently as the WT. Thus, none of the H62 mutants appear to be significantly impaired for VLP release.
As a second step of analysis, we assessed the impact of H62 mutations on virus infectivity. To do so, HIVLuc luciferase reporter (21) versions of WT and mutant constructs were cotransfected into 293T cells with a VSV envelope glycoprotein expression construct to generate pseudotyped virus. Viruses were used to infect CD4+ HiJ cells (45), and after 72 h, infected and transfected cells were assayed for luminescence. While these assays did not correct for the minor release effects observed in Fig. 1C, Fig. 2A demonstrates that several H62 mutants exhibited substantial defects in their abilities to support infection. Consistent with the notion that H62 forms important aromatic interactions with F32 and Y145, aromatic substitutions for H62 (H62F, H62W, and H62Y) yielded viruses that were at least 5% as infectious as the WT. In contrast, substitutions lacking an aromatic side chain (H62A, H62C, and H62K) were at most 1% as infectious as the WT.
Fig. 2.

Analysis of virus infectivity. Luciferase gene-transducing HIV viruses were produced by cotransfection of HEK 293T cells with a VSV-G expression vector plus WT and H62 mutant HIVLuc constructs (A) or the indicated HIV variants that derived from selection of H62A or H62F second-site revertants (B). At 3 days posttransfection, virus-containing medium supernatants were collected, filtered through 0.45-μm sterile filters, and used to infect HiJ cells. At 3 days postinfection, HiJ cells were collected and processed for luciferase assays. Results are depicted as normalized luciferase activity signals in infected cells, relative to signals from cells infected with WT HIVLuc virus in parallel: they are not corrected for variations observed in virus particle release. Results are derived from 46 (WT), 16 (H62A and H62F), 2 (H62C and H62F/G208A), 3 (H62K), 4 (H62W and H62Y), 5 (G208A), 8 (G208R and H62F/SP1P10L), and nine (H62A/G208R) separate experiments. Standard deviations are as shown.
Examination of core structures.
As mentioned previously, H62 locates to a region involved in intermolecular NTD-CTD interfaces, which are crucial for the stabilization of the hexameric CA lattices (7, 30, 31, 47, 48). Therefore, we examined the core structures of released VLP for possible anomalies. Consequently, VLP from cells transfected with HIVLuc variants were collected, filtered to remove cell debris, pelleted by ultracentrifugation, resuspended, adhered to carbon-coated grids, and negatively stained with uranyl acetate for visualization by EM. We chose to use negative staining of isolated viral particles as opposed to EM of cells, as in our hands the quantitative effects of various treatments have proven difficult to assess in EM of sectioned cell or virus material (62, 63). Using this approach, WT viruses exhibited tubular or conical structures associated with mature virions (Fig. 3A). For WT samples, such cores were observed in over half of the VLP (Table 1). Typical HIV-1 cores also were evident with the H62Y variant (Fig. 3G), albeit at a slightly lower frequency (47%) (Table 1). With the exception of H62Y, which was the most infectious of all the H62 variants (Fig. 2A), the other H62 mutant viruses exhibited aberrant core morphologies (Fig. 3B to F). Indeed, tube or core forms were observed in only 17 to 21% of the H62A, H62C, H62K, H62F, and H62W VLP. Thus, core morphology abnormalities roughly correlated with defects in infectivity.
Fig. 3.
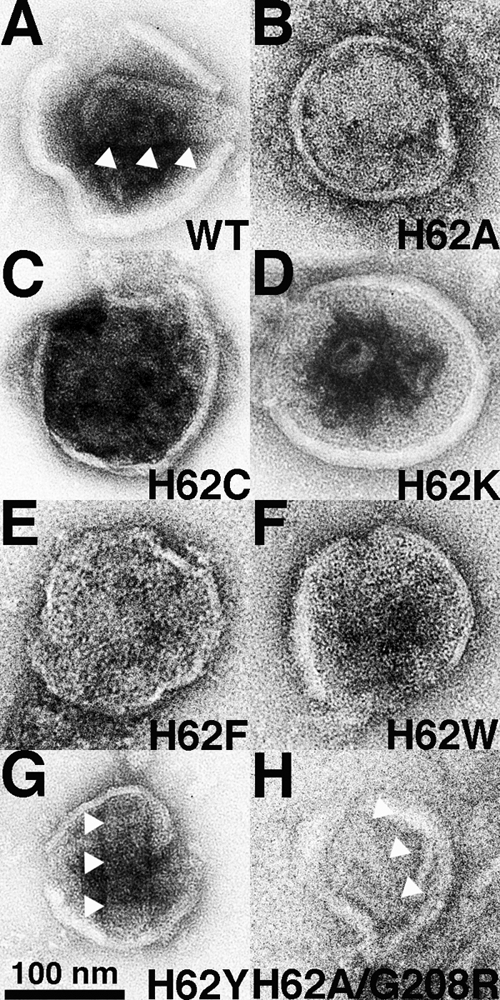
Effects of H62 CA mutations on virus-like-particle morphologies. The indicated HIVLuc viruses from transfected cells were sedimented through 20% sucrose cushions, resuspended, adhered to carbon-coated EM grids, and visualized by EM. Conical and tubular cores in panels A, G, and H are indicated by three white arrowheads each. The size bar for all panels is provided at the bottom of panel G, and observed core morphologies are tabulated in Table 1.
Table 1.
Morphologies of HIV particlesa
| Capsid variant | % conical or cylindrical | No. viewed |
|---|---|---|
| WT | 55 | 64 |
| H62A | 19 | 52 |
| H62C | 19 | 73 |
| H62K | 17 | 41 |
| H62F | 17 | 24 |
| H62W | 21 | 34 |
| H62Y | 47 | 15 |
| H62A/G208R | 47 | 30 |
Morphologies of virus-like particles. Virus-like particles from cells transfected with the indicated HIVLuc viruses were sedimented through 20% sucrose cushions, resuspended, and processed for EM. The percentages of particles with discernibly roughly conical or cylindrical cores were determined from the indicated numbers of separate virus particle images.
As a further examination of the role H62 plays in assembly of virions, we utilized an in vitro assembly assay with purified CA proteins. We chose to compare the WT CA versus the H62A mutant, as H62A exhibited a large defect in infectivity (Fig. 2A), but the protein was as readily purified from bacteria as the WT protein. For analysis, purified proteins (100 μM) were incubated in assembly reactions (5), after which products were lifted onto EM grids for EM imaging. WT CA assembled into long tubes (Fig. 4A to C), representative of a mature assembly phenotype (11, 31, 32, 37, 38, 54). In contrast, H62A CA completely failed to assemble tubes (Fig. 4D to F). These results are consistent with the appearance of aberrant cores assembled from cells expressing H62A proteins and support the notion that H62 mutants are noninfectious because their cores are defective.
Fig. 4.

In vitro assembly of HIV capsid proteins. WT or variant CA proteins were expressed in bacteria, purified, and employed in in vitro assembly reactions. Following assembly incubations, samples were processed and imaged by EM at low magnification (A, B, D, E, G, H, J, and K; size bar in panel J) or high magnification (C, F, I, and L; size bar in panel L). For each capsid variant, assembly efficiency was assessed by the appearance of capsid tubes, and tube assembly was quantified by tube coverage in EM images as described in Materials and Methods. Average tube coverage percentages, normalized to the WT level, were calculated from 12 pictures each and are as follows: WT, 100%; H62A, 0%; H62A/G208R, 11%; G208R, 68%.
Characterization of compensatory mutations.
Although our luciferase-based infectivity assays (Fig. 2A) were informative, they reflect only a single round of infection. To examine the role of H62 in the context of a more physiologically relevant infection model, we cloned H62 mutations into the HIV-1 NL4-3 provirus. We then transfected the proviral mutants into 293T cells, collected viral supernatants, and normalized the viral samples by immunoblotting for CA. We initially infected CEM-SS T cells with equalized amounts of the WT, H62A, H62C, H62K, and H62Y viruses. As anticipated from luciferase infectivity assays, the WT virus replicated well in cell culture, peaking at 11 days, and the H62Y mutant virus replicated with slightly slower kinetics, peaking at 14 days (Fig. 5A). In contrast, the H62A, H62C, and H62K viruses failed to replicate in CEM T cells (Fig. 5A). We subsequently examined the WT, H62F, and H62W viruses in the more permissive MT-4 T cells (40). Along with these viruses, we also tested H62Y in MT-4 cells as a representative of a slightly impaired virus as well as H62A as a representative of a severely impaired virus. As shown in Fig. 5B, the H62Y virus replicated almost as well as the WT, peaking on day 4 of culture, albeit with lower total Gag levels. The H62W and H62F viruses both peaked around 11 days, reflecting the greater defects observed in single-round infectivity assays (Fig. 2A). Finally, the H62A mutation again failed to replicate. A small amount of CA was detected after 27 days of virus passage, but this appeared to represent a compensatory mutation, as discussed in further detail below.
Fig. 5.

Replication kinetics of HIV mutants. Stocks of WT and CA variant viruses on an NL4-3 HIV background were generated by transfection of constructs into HEK 293T cells and were normalized with respect to Gag levels. CEM-SS (A) or MT4 (B to D) cells were infected in parallel with normalized virus stocks of WT or variant viruses and passaged at 3- to 4-day intervals. Virus spread was monitored by measuring Gag levels in aliquots of infected cells by immunoblotting and densitometry. Replication is plotted as relative Gag level in infected cells versus number of days postinfection.
The H62A, H62F, and H62W mutants were chosen for extended serial passages to examine the capability of the virus to evolve compensatory mutations. We chose these mutants because their rates of replication in MT4 cells were distinguishable from those for the WT and because the replication delays that we observed (Fig. 5B) could have resulted from the acquisition of second-site mutations. For these studies, MT4 cells were infected with equalized stocks of virus and split every 3 to 4 days for several weeks. Late-passage infections were pelleted to remove cell debris, and the supernatants were added to fresh MT4 cells. This was repeated twice, for a total of three sequential series of infections. With the H62W virus, serial passage resulted in the derivation of a virus with enhanced replication relative to that of the mutant. To identify mutations that might account for this phenotypic change, the gag gene of the virus was cloned and sequenced. Our analysis indicated that the virus had reverted to the original H62 sequence and contained no other mutations (data not shown).
During our analysis of H62F replication, two of the H62F replicates were much more infectious than the H62F mutant. The first replicate was sequenced as before, and we confirmed the maintenance of the H62F mutation. Interestingly, we discovered a P10L mutation in the SP1 spacer peptide; this mutation also has arisen in response to an HIV mutant lacking stem-loop 1 of viral genomic RNA, a region involved in dimerization and packaging into virions (59). The second replicate also maintained the H62F mutation but encoded a G208A mutation in the C-terminal domain of capsid and an E21K mutation in NC. It is noteworthy that residue 208 is also an alanine in other HIV strains. Further sequencing confirmed that this was not a viral contaminant but indeed a compensatory mutation (see below). Remarkably, another compensatory mutation arose at the same codon from the H62A mutant during the third round of serial passages. After cloning and sequencing, the stock was found to have a G208R mutation in addition to the original H62A mutation.
To determine the roles of the above-described mutations in counteracting the H62F and H62A defects, we first cloned them into the HIVLuc background. In doing so, we separated the NC(E21K) and CA(G208A) mutations in the H62F background, as it was possible that only one was responsible for correcting the H62F defect. Indeed, the H62F/E21K mutant was still defective in single-round and longitudinal infectivity assays (data not shown). In contrast, the H62F/G208A, H62F/SP1P10L, and H62A/G208R double mutations improved single-round infectivities more than 6-fold over the respective H62F and H62A viruses (Fig. 2B). Because the G208R and G208A variants identified the same residue as being important for overcoming the effects of H62 mutations, we focused on these in the studies described below. To ensure that the G208A and G208R mutations were compensating for the H62 defects and not independently enhancing HIV-1 infectivity, we assessed the single-round infectivities of G208R and G208A in the context of the WT H62 sequence. As seen in Fig. 2B, the G208A and G208R single mutations were not superinfectious; if anything, the mutants exhibited slight defects relative to WT infections (Fig. 2B).
To compare compensatory mutants with the original mutants in T cell infections, we cloned the single and double mutants into the HIV-1 strain NL4-3 background for passages in MT4 cells. The results for these investigations are shown in Fig. 6. With the H62F set of viruses (Fig. 5C), the original H62F virus showed delayed kinetics, as expected, but the double H62F/G208A virus replicated nearly as fast as the WT (day 7 versus day 4 peaks), and the single G208A mutant virus was similar to the WT. These results indicate that G208A compensates for the H62F defect. A similar pattern was observed for the H62A set of viruses (Fig. 5D). Specifically, the original H62A virus was noninfectious, the G208R virus mimicked the WT, and the H62A/G208R double mutant showed intermediate replication kinetics. The compensatory mutations thus converted replication-defective viruses into replication-competent viruses.
Fig. 6.
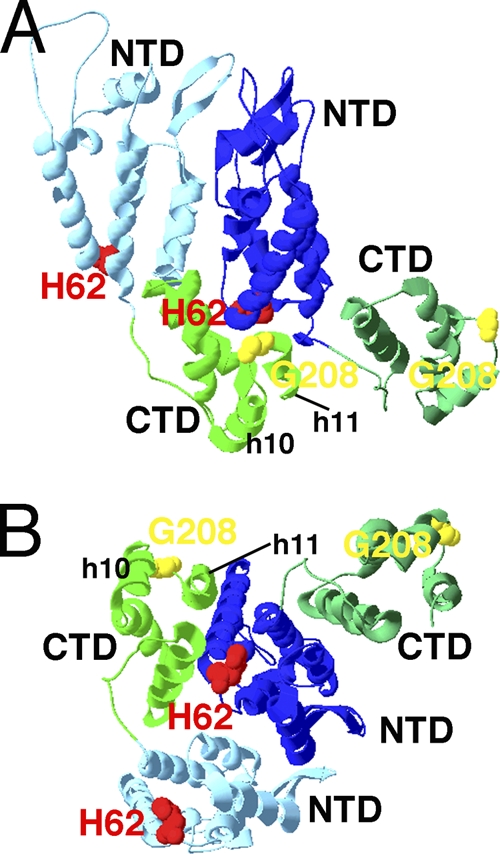
Proximity of mutant and revertant residues on models of HIV CA. The locations of CTD helices 10 and 11 and residues H62 (red) and G208 (yellow) on pairs of CA proteins in HIV CA hexamers (PDB accession no. 3GV2) are depicted. Panel A is viewed roughly parallel to the plane of capsid coats, with the predicted outer capsid surface up and the inner surface down. Panel B is viewed roughly perpendicular to the capsid surface from the outside. Light and dark blue N-terminal domains and light and dark green C-terminal domains are as labeled.
To examine how at least one of our compensatory mutants might affect CA protein interactions, we examined the cores of H62A/G208R mutant viruses by EM. Relative to the H62A mutant, which showed predominantly immature-type cores, the double mutant yielded an increased number of cone-like or tube-like cores (47% versus 19%) (Table 1 and Fig. 3). To further examine the ability of proteins with the G208R mutation to compensate for the assembly defect of H62A CA proteins, we utilized our in vitro assembly assay. Importantly, while H62 CA proteins assembled no tubes in vitro (Fig. 4D to F), the H62A/G208R double mutant proteins assembled tubes (Fig. 4G to I), albeit at 10.8% of WT levels. Because the single G208R mutant CA proteins did not assemble any more efficiently than the WT (Fig. 4J to L) (68% of WT levels), our data indicate that mutation at CA CTD residue 208 specifically rescues the assembly defect of mutations at CA NTD residue H62.
DISCUSSION
Intermolecular interactions between HIV-1 capsid NTDs and CTDs regulate HIV assembly (7, 30, 31, 58). NTD residue H62 locates to this interface, which involves NTD helices 3 and 4 and CTD helices 8 and 11. Although H62 is highly conserved and has the potential to act as a pH switch, mutation of histidine did not have major effects on release of virus-like particles from transfected cells (Fig. 1A and B). However, replacement of H62 in single-round and longitudinal infectivity studies produced a more dramatic phenotype, with significant defects in both assays (Fig. 2 and 5). In in vitro assembly assays, the H62A mutant failed to assemble capsid tubes compared to the WT (Fig. 4), and infectivity defects correlated with anomalies in core morphologies visualized by electron microscopy (Fig. 3 and Table 1). In each assay, substitutions with aromatic side chains (phenylalanine, tryptophan, and tyrosine) were less affected than those without (alanine, cysteine, and lysine). This is not surprising, as the aromatic rings of H62 are implicated in orthogonal ring-stacking interactions at the NTD-CTD interface.
Could HIV-1 evolve second-site gag mutations to compensate for the structural defects caused by changing H62? Longitudinal cell cultures in a permissive cell line yielded four viral clones that were able to replicate at an increased rate compared to that of the original H62 mutants. When sequenced, these included an H62W clone that reverted to the WT. We also discovered an H62F clone that maintained the H62F mutation but changed the P10 residue of SP1 to a leucine. Curiously, a previous study identified this residue as a compensatory mutation for a deletion in SL1 of the viral genomic RNA (59). This study showed that SP1 P10L did not enhance viral replication over the level for the WT but that it enabled Gag to maintain specificity in packaging genomic viral RNA. How this relates to the defect in H62 core assembly is unclear. However, SP1 has been modeled to form a hexameric basket of PrGag contacts (8, 9, 72), and the SP1 P10L change may compensate for compromised NTD-CTD hexamer contacts resulting from the H62F mutation. In particular, we envision a possibility wherein SP1 changes may reposition CTD helix 11 so that it offsets the original NTD mutation. By this scenario, SP1-mediated helix 11 placement in PrGag would be maintained at least to some degree in mature CA.
Two other clones with increased replicative capacity compared to that of the original mutants were isolated from H62F and H62A longitudinal cultures. Remarkably, the compensatory mutations both mapped to residue 208 of the CA CTD. Our data show that G208A (which compensates for H62F) and G208R (which compensates for H62A) are not found to be superinfectious when examined in the WT background (Fig. 2B and 5). However, when expressed in the context of H62 mutations, the compensatory mutations improved infectivity, and it is plausible that continued passage of these variants might evolve even more-fit viruses. As shown in Fig. 6A, residue 208 lies between helix 10 and helix 11 in the CTD and locates just below the CA NTDs. However, atomic models indicate that CTD residue 208 is not in a position to interact directly with NTD residue 62 (Fig. 6B). How then do these second-site mutations compensate for the structural defects caused by a change of H62? One possibility is that they alter a newly described 3-fold axis of symmetry defined by CTD-CTD dimers (13). Alternately, residue 208 mutations may reposition helix 11 so that it accommodates H62 NTD mutation. In effect, this compensatory mutant may act in the same manner as the one we have proposed above for the P10L mutation and may help explain the pronounced effects that helix 11 mutations have been shown to have on virus infectivity (24, 55, 60). In summary, our work here supports the idea that NTD-CTD intermolecular interactions of the CA core are crucial to the survival of HIV: the virus will evolve mutations to compensate for structural defects caused by losing this interaction.
ACKNOWLEDGMENTS
The HIVLuc construct was kindly provided by Nathaniel Landau, and the VSV-G protein expression construct was a generous gift from Randy Taplitz. We appreciate the help and support of Ayna Alfadhli, Robin Barklis, Seyram Tsagli, and Mike Webb.
This work was supported by the following grants from the National Institutes of Health to E.B.: GM060170 and AI071798.
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Abdurahman S., Youssefi M., Hoglund S., Vahlne A. 2007. Characterization of the invariable residue 51 mutations of human immunodeficiency virus type 1 capsid protein on in vitro CA assembly and infectivity. Retrovirology 4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adachi A., et al. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alfadhli A., Barklis R. L., Barklis E. 2009. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alfadhli A., Dhenub T. C., Still A., Barklis E. 2005. Analysis of human immunodeficiency virus type 1 Gag dimerization-induced assembly. J. Virol. 79:14498–14506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barklis E., et al. 2009. Characterization of the in vitro HIV-1 capsid assembly pathway. J. Mol. Biol. 387:376–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bouamr F., Scarlata S., Carter C. 2003. Role of myristylation in HIV-1 Gag assembly. Biochemistry 42:6408–6417 [DOI] [PubMed] [Google Scholar]
- 7. Bowzard J. B., Wills J. W., Craven R. C. 2001. Second-site suppressors of Rous sarcoma virus Ca mutations: evidence for interdomain interactions. J. Virol. 75:6850–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Briggs J. A., Johnson M. C., Simon M. N., Fuller S. D., Vogt V. M. 2006. Cryo-electron microscopy reveals conserved and divergent features of gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J. Mol. Biol. 355:157–168 [DOI] [PubMed] [Google Scholar]
- 9. Briggs J. A., et al. 2009. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U. S. A. 106:11090–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Briggs J. A., et al. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672–675 [DOI] [PubMed] [Google Scholar]
- 11. Briggs J. A., Wilk T., Welker R., Krausslich H. G., Fuller S. D. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22:1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burniston M. T., Cimarelli A., Colgan J., Curtis S. P., Luban J. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol. 73:8527–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Byeon I. J., et al. 2009. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Campbell S., Vogt V. M. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chesebro B., Wehrly K., Nishio J., Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J. Virol. 66:6547–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chrystie I. L., Almeida J. D. 1988. Further studies of HIV morphology by negative staining. AIDS 2:459–464 [DOI] [PubMed] [Google Scholar]
- 17. Chrystie I. L., Almeida J. D. 1988. The morphology of human immunodeficiency virus (HIV) by negative staining. J. Med. Virol. 25:281–288 [DOI] [PubMed] [Google Scholar]
- 18. Chukkapalli V., Hogue I. B., Boyko V., Hu W. S., Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 82:2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chukkapalli V., Oh S. J., Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. U. S. A. 107:1600–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cimarelli A., Sandin S., Hoglund S., Luban J. 2000. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol. 74:3046–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Connor R. I., Chen B. K., Choe S., Landau N. R. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944 [DOI] [PubMed] [Google Scholar]
- 22. Davis M. R., Jiang J., Zhou J., Freed E. O., Aiken C. 2006. A mutation in the human immunodeficiency virus type 1 Gag protein destabilizes the interaction of the envelope protein subunits gp120 and gp41. J. Virol. 80:2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dawson L., Yu X. F. 1998. The role of nucleocapsid of HIV-1 in virus assembly. Virology 251:141–157 [DOI] [PubMed] [Google Scholar]
- 24. Dorfman T., Bukovsky A., Ohagen A., Hoglund S., Gottlinger H. G. 1994. Functional domains of the capsid protein of human immunodeficiency virus type 1. J. Virol. 68:8180–8187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorfman T., Mammano F., Haseltine W. A., Gottlinger H. G. 1994. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 68:1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DuBridge R. B., et al. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ehrlich L. S., Liu T., Scarlata S., Chu B., Carter C. A. 2001. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: structure switching by pH-induced conformational changes. Biophys. J. 81:586–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finzi A., Orthwein A., Mercier J., Cohen E. A. 2007. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J. Virol. 81:7476–7490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gamble T. R., et al. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853 [DOI] [PubMed] [Google Scholar]
- 30. Ganser-Pornillos B. K., Cheng A., Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79 [DOI] [PubMed] [Google Scholar]
- 31. Ganser-Pornillos B. K., von Schwedler U. K., Stray K. M., Aiken C., Sundquist W. I. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ganser B. K., Li S., Klishko V. Y., Finch J. T., Sundquist W. I. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83 [DOI] [PubMed] [Google Scholar]
- 33. Garrus J. E., et al. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65 [DOI] [PubMed] [Google Scholar]
- 34. Gitti R. K., et al. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273:231–235 [DOI] [PubMed] [Google Scholar]
- 35. Goff A., Ehrlich L. S., Cohen S. N., Carter C. A. 2003. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J. Virol. 77:9173–9182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gottlinger H. G., Dorfman T., Sodroski J. G., Haseltine W. A. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U. S. A. 88:3195–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gross I., Hohenberg H., Huckhagel C., Krausslich H. G. 1998. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72:4798–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gross I., et al. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 19:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hansen M. S., Barklis E. 1995. Structural interactions between retroviral Gag proteins examined by cysteine cross-linking. J. Virol. 69:1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harada S., Koyanagi Y., Yamamoto N. 1985. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 229:563–566 [DOI] [PubMed] [Google Scholar]
- 41. Hockley D. J., Nermut M. V., Grief C., Jowett J. B., Jones I. M. 1994. Comparative morphology of Gag protein structures produced by mutants of the gag gene of human immunodeficiency virus type 1. J. Gen. Virol. 75(11):2985–2997 [DOI] [PubMed] [Google Scholar]
- 42. Hockley D. J., Wood R. D., Jacobs J. P., Garrett A. J. 1988. Electron microscopy of human immunodeficiency virus. J. Gen. Virol. 69(10):2455–2469 [DOI] [PubMed] [Google Scholar]
- 43. Jin Z., Jin L., Peterson D. L., Lawson C. L. 1999. Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol. 286:83–93 [DOI] [PubMed] [Google Scholar]
- 44. Jouvenet N., et al. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabat D., Kozak S. L., Wehrly K., Chesebro B. 1994. Differences in CD4 dependence for infectivity of laboratory-adapted and primary patient isolates of human immunodeficiency virus type 1. J. Virol. 68:2570–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kelly B. N., et al. 2007. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 373:355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lanman J., et al. 2003. Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 325:759–772 [DOI] [PubMed] [Google Scholar]
- 48. Lanman J., et al. 2004. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11:676–677 [DOI] [PubMed] [Google Scholar]
- 49. Lee E., Yeo A., Kraemer B., Wickens M., Linial M. L. 1999. The gag domains required for avian retroviral RNA encapsidation determined by using two independent assays. J. Virol. 73:6282–6292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee Y. M., Tang X. B., Cimakasky L. M., Hildreth J. E., Yu X. F. 1997. Mutations in the matrix protein of human immunodeficiency virus type 1 inhibit surface expression and virion incorporation of viral envelope glycoproteins in CD4+ T lymphocytes. J. Virol. 71:1443–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leschonsky B., Ludwig C., Bieler K., Wagner R. 2007. Capsid stability and replication of human immunodeficiency virus type 1 are influenced critically by charge and size of Gag residue 183. J. Gen. Virol. 88:207–216 [DOI] [PubMed] [Google Scholar]
- 52. Li S., Hill C. P., Sundquist W. I., Finch J. T. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409–413 [DOI] [PubMed] [Google Scholar]
- 53. Martin-Serrano J., Zang T., Bieniasz P. D. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794–4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mayo K., et al. 2003. Retrovirus capsid protein assembly arrangements. J. Mol. Biol. 325:225–237 [DOI] [PubMed] [Google Scholar]
- 55. McDermott J., Farrell L., Ross R., Barklis E. 1996. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions, using cysteine-specific reagents. J. Virol. 70:5106–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morikawa Y., Hockley D. J., Nermut M. V., Jones I. M. 2000. Roles of matrix, p2, and N-terminal myristoylation in human immunodeficiency virus type 1 Gag assembly. J. Virol. 74:16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ono A., Orenstein J. M., Freed E. O. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 74:2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pornillos O., et al. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ristic N., Chin M. P. 2010. Mutations in matrix and SP1 repair the packaging specificity of a human immunodeficiency virus type 1 mutant by reducing the association of Gag with spliced viral RNA. Retrovirology 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Roldan A., et al. 2004. In vitro identification and characterization of an early complex linking HIV-1 genomic RNA recognition and Pr55Gag multimerization. J. Biol. Chem. 279:39886–39894 [DOI] [PubMed] [Google Scholar]
- 61. Saad J. S., et al. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 103:11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scholz I., Arvidson B., Huseby D., Barklis E. 2005. Virus particle core defects caused by mutations in the human immunodeficiency virus capsid N-terminal domain. J. Virol. 79:1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scholz I., et al. 2008. Analysis of human immunodeficiency virus matrix domain replacements. Virology 371:322–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spearman P., Wang J. J., Vander Heyden N., Ratner L. 1994. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 68:3232–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Strack B., Calistri A., Gottlinger H. G. 2002. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 76:5472–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tang C., et al. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013–1020 [DOI] [PubMed] [Google Scholar]
- 67. von Schwedler U. K., et al. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17:1555–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang C. T., Barklis E. 1993. Assembly, processing, and infectivity of human immunodeficiency virus type 1 gag mutants. J. Virol. 67:4264–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang C. T., Zhang Y., McDermott J., Barklis E. 1993. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J. Virol. 67:7067–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Welker R., Hohenberg H., Tessmer U., Huckhagel C., Krausslich H. G. 2000. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 74:1168–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Welsch S., et al. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wright E. R., et al. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26:2218–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yu X., Yuan X., Matsuda Z., Lee T. H., Essex M. 1992. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J. Virol. 66:4966–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou W., Parent L. J., Wills J. W., Resh M. D. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]