

Abstract
We have previously shown that only endotheliotropic strains of human cytomegalovirus (HCMV), such as TB40E, infect monocytes and impair their chemokine-driven migration. The proteins encoded by the UL128-131A region (UL128, UL130, and UL131A) of the HCMV genome, which assemble into a pentameric gH-gL-UL128-UL130-UL131A envelope complex, have been recognized as determinants for HCMV endothelial cell tropism. The genes for these proteins are typically inactivated by mutations in all fibroblast-adapted strains that have lost the diversified tropism of clinical isolates. By using mutant HCMV reconstituted from TB40E-derived bacterial artificial chromosomes (BAC) encoding a wild-type (wt) or mutated form of UL128, we show here that UL128-131A products are essential determinants of infection in monocytes and that pUL128, in particular, can block chemokine-driven motility. The virus BAC4, encoding wt UL128, established infection in monocytes, induced the intracellular retention of several chemokine receptors, and rendered monocytes unresponsive to different chemokines. In contrast, the virus BAC1, encoding a mutated UL128, failed to infect monocytes and to downregulate chemokine receptors. BAC1-exposed monocytes did not express immediate-early (IE) products, retained virions in cytoplasmic vesicles, and exhibited normal chemokine responsiveness. A potential role of second-site mutations in the observed phenotype was excluded by using the revertant viruses BAC1rep and BAC4mut. By incubating noninfected monocytes with soluble recombinant pUL128, we observed both the block of migration and the chemokine receptor internalization. We propose that among the gH-gL-UL128-UL130-UL131A complex subunits, the UL128 protein is the one that triggers monocyte paralysis.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus found in 50 to 85% of the human population (7). HCMV infects only humans, and primary infection results in the establishment of a lifelong persistent infection. During persistence, either reactivations may occur from established latency or constant low-level HCMV replication may take place in particular cell types (14). With its large genome of approximately 240 kbp, encoding over 200 gene products, HCMV displays the most genetic complexity among the eight human herpesviruses (9). The large proportion of genes dedicated to interaction with the host may justify the uncommon ability of HCMV to infect a broad spectrum of cell types in vivo, including epithelial and endothelial cells, fibroblasts, monocytes, macrophages, dendritic cells, hepatocytes, myocytes, neurons, and glial cells.
In particular, monocytes represent a key cell type for HCMV infection, and they have been described as a potential site of latency and as efficient vehicles for viral dissemination (5, 26, 28, 29, 33, 34). In previous work, we provided evidence that a large percentage of human monocytes are susceptible to infection by endotheliotropic strains of HCMV and that as a result of mere contact with the virus, monocytes show a severe impairment of chemokine-driven migration (11). Since monocyte migration is fundamental for biological processes and is critical for the pathogenesis of several diseases, it is important to understand which mechanisms are used by HCMV to interfere with monocyte motility. Responsiveness to chemokines depends upon the expression of chemokine receptors on the cell surface (4, 21, 22). We observed that endotheliotropic strains of HCMV, such as TB40E and clinical isolates, rapidly and specifically downregulated CCR1, CCR2, CCR5, and CXCR4 on the surface of monocytes. Neither viral gene expression nor soluble factors produced during HCMV infection were responsible for these alterations, which were therefore attributed to a structural component of the virus. Interestingly, the nonendotheliotropic laboratory strain AD169 was unable to induce chemokine receptor downregulation, and monocytes maintained responsiveness to inflammatory (CCL5/RANTES, CCL3/MIP-1a, CCL4/MIP-1b, and CCL2/MCP-1) and homeostatic (CXCL12/SDF-1) chemokines (11).
In recent years, several independent studies have shown that for infection of epithelial and endothelial cells, polymorphonuclear leukocytes, and dendritic cells, HCMV depends on the products of the three genes within the viral locus UL128-131A (UL128, UL130, and UL131A). Disruption of any of the three genes abolishes infection of endothelial cells (13, 27, 38, 39), and conversely, repairing or complementing the mutation regenerates the original tropism (13, 38). The rapid restriction of tropism that accompanies HCMV fibroblast adaptation is explained by the emergence of disabling mutations in at least one of the UL128-131A genes. In the aforementioned AD169 strain, for example, a frameshift mutation has been mapped within the UL131A coding sequence (cds) (2).
The products of the UL128-131A genes have been characterized as structural components of the viral envelope (1, 17, 24, 39). In the model proposed by Ryckman et al. (24), the viral glycoprotein gH, a type I transmembrane protein and a major HCMV envelope fusion component, binds to glycoprotein L (gL), pUL131A, and pUL130 through its ectodomain; pUL130 in turn binds pUL128 through noncovalent interactions. Thus, the envelope of endotheliotropic strains contains the pentameric glycoprotein complex gH-gL-UL128-UL130-UL131A, which is absent or incomplete in nonendotheliotropic strains whose envelope includes either only gH-gL or the alternative trimeric gH-gL-glycoprotein O (gO) complex.
In light of these data, the UL128-131A genes could be suspected of being responsible for the chemokine receptor downregulation and migration block induced by HCMV in monocytes. Here we directly addressed this issue and specifically tested the hypothesis that pUL128 possesses monocyte-modulating activity.
The phenotypes of viruses reconstituted from four bacterial artificial chromosomes (BACs) harboring either an intact or mutated UL128 cds within the backbone of one of two distinct TB40E-derived clones were compared. In detail, BAC-derived viruses TB40-BAC4 and TB40-BAC1, renamed BAC4 and BAC1 for brevity (25), differ by the presence of an adenine inserted at position 332 of UL128 in TB40-BAC1 that results in a predicted truncation of pUL128. BAC-derived viruses TB40-BAC4-UL128insA332 (shortened to BAC4mut) and TB40-BAC1-UL128repaired (shortened to BAC1rep) (27), were used as controls.
In addition, to check whether pUL128 could induce chemokine receptor internalization and blocking of chemotaxis, monocytes were treated with a soluble recombinant pUL128 protein (rpUL128) (16). Altogether, our results support a model wherein contacts between pUL128 exposed on the HCMV envelope and monocyte surface receptors are the triggers of multiple chemokine receptor downregulation and migration block, in a process that is separable from virus entry.
MATERIALS AND METHODS
Cells.
Primary human monocytes were isolated from peripheral blood by a negative immunoselection procedure (monocyte isolation kit II; Miltenyi Biotec, Bergisch Gladbach, Germany) and then cultured nonadherently in endotoxin-free RPMI 1640 medium supplemented with 10% human AB serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin in polypropylene tubes (Falcon; BD Biosciences, Le Pont de Claix, France) as previously described (11). Human foreskin fibroblasts (HFF) were cultivated in minimal essential medium (MEM) with 10% fetal calf serum (FCS; Gibco BRL, Grand Island, NY), 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. Cell viability was determined by trypan blue (Biochrom, Berlin, Germany) exclusion.
Preparation of viral stocks and HCMV infection.
Different HCMV variants were used for the infection of monocytes (Table 1). The endotheliotropic strain TB40E and the TB40E-derived BAC viruses TB40-BAC4, TB40-BAC1, TB40-BAC4-UL128insA332, and TB40-BAC1-UL128repaired (called BAC4, BAC1, BAC4mut, and BAC1rep, respectively, for the sake of brevity) were created, extensively characterized, and kindly provided by C. Sinzger (27). Cell-free viral stocks were prepared from supernatants of infected HFF and frozen at −80°C, and then titers were determined by plaque assay as previously described (41). Viral stocks were negative for contamination with Mycoplasma, as determined by MycoAlert (Cambrex, Rockland, MD). UV-inactivated virus (subjected to 200 kJ two times) (11) was used in the same manner as “live” virus. Monocytes were infected at a multiplicity of infection (MOI) of 5 PFU per cell in complete medium overnight.
Table 1.
Summary of viral propertiesa
| Virus | Abbreviation | Tropism for human umbilical vein endothelial cellsb | Description of UL128 |
|---|---|---|---|
| TB40E | + | wt | |
| TB40E-BAC4 | BAC4 | + | wt |
| TB40E-BAC4-UL 128insA332 | BAC4mut | − | In vitro A insertion at nt 332 |
| TB40E-BAC1 | BAC1 | − | Natural A insertion at nt 332 |
| TB40E-BAC1-UL 128repaired | BAC1rep | + | In vitro A deletion at nt 332 |
The wt UL130 and UL131A sequences were present in all viruses.
All viruses had tropism for HFF.
UL128-131A sequence analysis.
The UL128-UL131A regions of all viruses used in this study were analyzed by sequencing of PCR products. Viral DNA was purified from viral stocks with a High Pure viral nucleic acid kit (Roche Applied Science, Mannheim, Germany) following the manufacturer's instructions. One microgram of viral DNA was then used as a template for PCR amplification using the following primers: UL128for (5′-TTGGATCACAGCCGCGTGC-3′), UL128rev (5′-CCACGATCCGGGTTATCTTGTCG-3′), UL130for (5′-GCTAACGGCGAACCAGAATCC-3′), UL130rev or UL131Arev (5′-GGCTGTGATCCAATAACAGCCAC-3′), and UL131Afor (5′-CCCATCACCTCGCCTATACTATGTG-3′). The PCR products were monitored by agarose gel electrophoresis, and bands at the expected sizes were purified using a HiYield PCR cleanup/gel extraction kit (SLG, Gauting, Germany). Sequencing was performed by 4baseLab, Reutlingen, Germany. Finally, sequences were analyzed and aligned using VectorNTI (Invitrogen, Eugene, OR).
Indirect immunofluorescence.
To analyze the intracellular localization of viral proteins as well as the distribution of cytoskeletal components, the following monoclonal antibodies (MAbs) were used: anti-immediate-early 1-2 (anti-IE 1-2), anti-pp65 (Argene-Biosoft, Varilhes, France), and anti-vimentin (Oncogene Research Products, Boston, MA). Actin was detected by using BODIPY FL phallacidin (Molecular Probes). At 24 h postinfection, monocytes were washed, resuspended in phosphate-buffered saline (PBS), and allowed to adhere to glass slides for 30 min at 37°C prior to fixation/permeabilization with ice-cold methanol-acetone (1:1) or with 4% paraformaldehyde (PFA)–PBS–0.2% Triton for investigation of the cytoskeletal proteins. Monocytes were incubated first with MAbs for 60 min at 37°C and then with Alexa Fluor488-conjugated goat anti-mouse immunoglobulins (Molecular Probes). Nuclear and cytoplasmic counterstaining was performed with 4′,6-diamidino-2-phenylindole (DAPI) and Evans blue (Sigma-Aldrich, Munich, Germany), respectively. Microphotographs were generated with a Zeiss Observer.Z1 microscope (Zeiss, Oberkochen, Germany).
Electron microscopy.
Monocytes were infected at an MOI of 50 for 1 h or 24 h prior to high-pressure freezing in cellulose capillary tubes (inner diameter, 200 μm) as previously described (37). Briefly, the cellulose tubes were filled with the monocyte suspension by capillary forces, cut to a length of 2 mm, and loaded in standard aluminum cups. The cavities between cellulose tubes and aluminum cups were filled with hexadecane. High-pressure freezing was performed with an HPF 01 freezing apparatus (Engineering Office M. Wohlwend GmbH, Switzerland). Samples were freeze substituted in acetone containing 0.1% (wt/vol) uranyl acetate, 0.2% (wt/vol) osmium tetroxide, and 5% (vol/vol) water and embedded in Epon (8, 37). After being thin sectioned, samples were imaged with a Zeiss EM10 transmission electron microscope at an acceleration voltage of 80 kV.
Western blot analysis.
Monocytes were lysed for 30 min on ice with lysis solution (50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40) containing protease inhibitors and dithiothreitol (DTT). Viral stocks were centrifuged for 1 h at 14,000 rpm, and pellets were lysed for 30 min on ice with RIPA buffer containing protease inhibitors. Lysis was followed by freezing the samples at −80°C for 1 h and thawing them by sonication. Protein concentrations were determined with a precision Red Advance protein assay (Cytoskeleton, Denver, CO). Equal amounts of cell or viral extracts were separated in 12% SDS-polyacrylamide gels and blotted onto nitrocellulose membranes (Bio-Rad Laboratories, Munich, Germany). Membranes were blocked with PBS containing 0.3% Tween 20 and 5% dry milk powder and incubated with the indicated monoclonal or polyclonal antibodies followed by peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgGs (Pierce Biotechnology, Rockford, IL). A MAb specific for pUL128 (MAb 17) was a gift from Giuseppe Gerna (Fondazione IRCCS Policlinico San Matteo, Pavia, Italy). Rabbit polyclonal antisera directed against gH, gL, pUL130, and pUL131A were gifts from Brent Ryckman (Oregon Health and Science University, Portland, OR). For detection of cellular components of the cytoskeleton, anti-actin (Sigma-Aldrich, St. Louis, MO), anti-α-tubulin (Molecular Probes, Eugene, OR), anti-vimentin (Oncogene Research Products, Boston, MA), and anti-WASP (Santa Cruz Biotechnology, Santa Cruz, CA) were used. A chemiluminescence detection kit (SuperSignal West Dura; Pierce Biotechnology) was used according to the manufacturer's instructions.
Flow cytometry.
Analysis of chemokine receptor expression was performed as previously described (11). Briefly, monocytes were incubated for 1 h in blocking buffer (10% human immunoglobulin [Flebogamma; Grifols Deutschland GmbH, Langen, Germany], 3% FCS, and 0.01% sodium azide in PBS) containing specific antibodies directed against CD14, CCR1, CCR2, or CCR5 (R&D Systems, Minneapolis, MN), followed by a 45-min incubation with phycoerythrin (PE)-conjugated rabbit anti-mouse immunoglobulins (DAKOCytomation, Hamburg, Germany). Isotype-matched immunoglobulins (R&D Systems) were used as controls. For determination of the total (cell surface and intracellular) chemokine receptor proteins, monocytes were fixed and permeabilized with a Cytofix/Cytoperm kit (BD Biosciences, San Diego, CA) prior to incubation with primary MAbs or isotype-matched controls. Data were analyzed using CELLQuest software (BD Biosciences), and for each antigen, the expression level was measured as the percentage of positive cells as well as the mean fluorescence intensity (MFI) for the respective antibody compared to the isotype-matched control.
Chemotaxis assay.
Human recombinant chemokines CCL2/MCP1 and CCL5/Rantes (R&D Systems) were used at a final concentration of 100 ng/ml in RPMI 1640–1% FCS. N-Formyl-Met-Leu-Phe (fMLP; Sigma-Aldrich) at a final concentration of 10−8 M served as a positive control. Cell migration was evaluated with a 48-well Boyden chamber (Neuroprobe, Pleasanton, CA) with 5-μm-pore-size polycarbonate filters. Stimuli were assayed in triplicate, and the number of cells that migrated in five visual fields (original magnification, ×100) was counted for each well as previously described (11, 31). For each experiment, the results are expressed as means for three replicates ± standard deviations (SD).
Statistical analysis.
Statistical analysis of the results was performed using a paired, two-tailed Student t test, setting the level of statistical significance to P values of ≤0.05.
RESULTS
UL128-131A are essential for infection of primary human monocytes.
In our previous work, we have shown that peripheral blood monocytes are susceptible to in vitro infection by endotheliotropic HCMV strains, such as TB40E and clinical isolates, but resistant to fibroblast-adapted strains (11). Since it has become clear that the viral genes UL128-131A define endothelial and epithelial cell tropism (12, 13), we decided to investigate whether the gene products are also determinants of monocyte susceptibility to HCMV infection and whether they are involved in chemokine receptor downregulation and blocking of migration.
For this purpose, we took advantage of 4 different TB40E BAC derivatives that were recently characterized for the ability to infect endothelial cells (25, 27). BAC-derived viruses TB40-BAC4 and TB40-BAC1 (BAC4 and BAC1) differ by two point mutations within UL128: an adenine-to-cytosine change at nucleotide position 282 and an adenine insertion at nucleotide position 332 of the UL128 cds in BAC1 (27). The 332A insertion is located within the second exon and causes a frameshift resulting in a truncated pUL128 protein (27). BAC-derived revertant viruses TB40-BAC4-UL128insA332 (BAC4mut) and TB40-BAC1-UL128repaired (BAC1rep) (27), engineered to reproduce the UL128 sequences found in BAC1 and BAC4 within the otherwise unchanged BAC4 and BAC1 backbones, respectively, were used as controls. As summarized in Table 1, BAC4 and BAC1rep, carrying the wild-type (wt) UL128 sequence and an overall functional UL128-131A locus, exhibited full tropism for endothelial cells, whereas BAC1 and BAC4mut, harboring the disabling mutation within UL128, did not infect endothelial cells.
Monocytes were inoculated at an MOI of 5 with the four viruses, and at 24 h postinfection (p.i.), the initiation of the viral cycle was determined by detection of IE 1-2 proteins in the monocyte nuclei. As shown in Fig. 1, the endotheliotropic viruses BAC4 and BAC1rep were able to express IE gene products in up to 60% of the monocytes, whereas no IE 1-2-positive nuclei were detected in monocytes inoculated with the nonendotheliotropic viruses BAC1 and BAC4mut.
Fig. 1.

UL128 to UL131A are determinants of HCMV tropism in monocytes. (A) Human primary monocytes were inoculated at an MOI of 5 with BAC4, BAC4mut, BAC1, and BAC1rep. At 24 h postinfection, the viral IE 1-2 and early-late (pp65) antigens were detected by indirect immunofluorescence staining (green). Mock-infected monocytes served as controls. All pictures (original magnification, ×60) are representative of 5 donors. Cell nuclei were counterstained with DAPI, and the cytoplasm was counterstained with Evans blue (blue and red signals, respectively). (B) In each experiment, the percentages of both IE- and pp65-positive nuclei were calculated by counting and correlating blue (DAPI) and green (IE or pp65) nuclei in five randomly selected microscopic fields. (C) The percentage of pp65-positive cells was calculated by counting and correlating blue (DAPI) nuclei and green (pp65) nuclei and cytoplasm in five randomly selected microscopic fields. Statistical analysis was performed for 5 independent experiments.
The infectivity of the four viruses correlated with different efficiencies of nuclear translocation of pp65, an HCMV tegument protein delivered rapidly into the nucleus after virus entry (19). At 24 h p.i., pp65 was detected in the nuclei of monocytes infected with BAC4 and BAC1rep but remained in the cytoplasm of monocytes inoculated with the BAC1 and BAC4mut viruses (Fig. 1).
To investigate the fate of the viral particles, ultrastructural analysis of infected monocytes was performed by electron microscopy. All four viruses were internalized into monocytes, and shortly after infection, numerous virions and dense bodies accumulated in large vacuoles in the monocyte cytoplasm, irrespective of the virus used. At 1 h p.i., monocytes infected by BAC4 were undistinguishable from those inoculated with BAC1 (Fig. 2). At 24 h p.i., however, BAC1 viral particles were still present in large vesicles, while BAC4 viral particles were not visible anymore, consistent with the virions carrying the pentameric gH-gL-UL128-UL130-UL131A complex being released from the vesicles and uncoated.
Fig. 2.
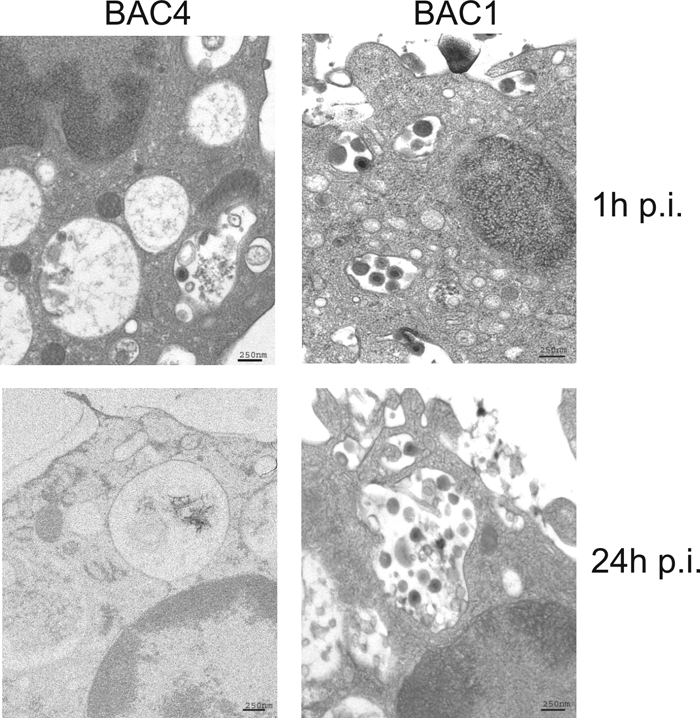
UL128-131A are necessary for release of viral particles from endocytic vesicles. Monocytes were infected at an MOI of 50 with BAC4 or BAC1. Ultrastructural analysis was performed at 1 h and 24 h p.i. by electron microscopy after high-pressure freezing and freeze substitution as described in Materials and Methods. Samples were imaged with a Zeiss EM10 transmission electron microscope at an acceleration voltage of 80 kV.
Altogether, these data demonstrate for the first time that an intact UL128-131A region is necessary to establish HCMV infection of monocytes. Since monocytes are not productively infected by HCMV, as they are not permissive for the entire replicative cycle of the virus (20), it was not possible to measure viral growth/release in these cells. Here and throughout this work, we therefore define HCMV-infected cells as those cells that show expression of IE 1-2 products and nuclear translocation of pp65.
UL128 mutation suppresses pentameric gH-gL-UL128-UL130-UL131A complex incorporation into HCMV virion envelope.
Since pUL128 is part of the pentameric gH-gL-UL128-UL130-UL131A complex, it was important to define the impact of UL128 mutations on the actual complex composition found in HCMV virions. For this purpose, purified viral particles of the four BAC viruses were analyzed in Western blots for their content of gH, gL, pUL128, pUL130, and pUL131A. As shown in Fig. 3, while BAC4 and BAC1rep virions showed the same complex composition as TB40E, with all five subunits present, BAC4mut and BAC1 virions did not contain pUL128, pUL130, and pUL131. A parallel analysis of UL128-131A product synthesis by the HCMV-infected fibroblast cells used to produce virions was performed (data not shown). BAC4- and BAC1rep-infected fibroblasts contained all three proteins (pUL128 to pUL131A), as expected. BAC4mut- and BAC1-infected fibroblasts, on the other hand, accumulated levels of pUL130 and pUL131A similar to those found in BAC4 and BAC1rep infections, while a truncated pUL128 form was not found.
Fig. 3.
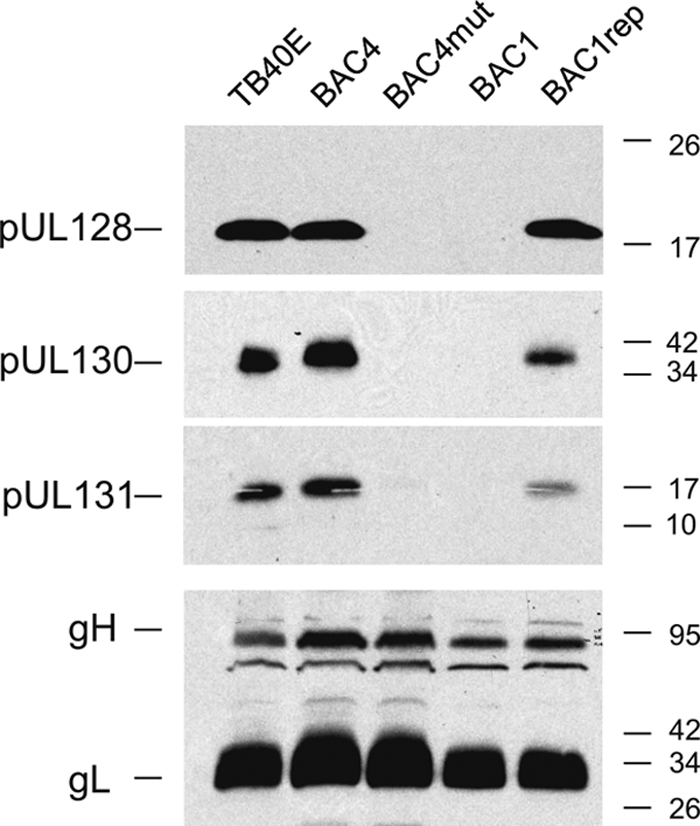
Immunoblot of HCMV virions. Expression of gH, gL, pUL128, pUL130, and pUL131A was investigated in the parental TB40E virus and the four different TB40E-derived virus mutants encoding wild-type or mutant UL128. Viral stocks produced in HFF were lysed in reducing buffer and resolved by SDS-PAGE, and proteins were analyzed with MAb 17, directed against pUL128, or with rabbit antisera directed against gH, gL, pUL130, and pUL131A.
Overall, this analysis suggests that the truncated pUL128 protein encoded by BAC4mut and BAC1 viruses is unstable and therefore undetectable by immunoblotting. The absence of pUL128 more generally inhibits the incorporation of pUL130 and pUL131A into the viral envelope.
pUL128 is necessary to impair the chemokine-driven migration of monocytes.
We previously observed that the mechanism used by TB40E to inhibit the chemokine-driven migration of monocytes was independent of viral gene expression but was dependent on exposure to some structural component of the viral particles (11). To test the possible involvement of the UL128-131A proteins, monocytes were inoculated for 24 h at an MOI of 5 with all four viruses prior to the measurement of monocyte chemotaxis. As shown in Fig. 4A, only BAC4 and BAC1rep inhibited CCL2- and CCL5-driven chemotaxis, whereas BAC4mut and BAC1 did not. The inhibitory effect was specific for chemokine-driven migration, as none of the viruses affected basal migration or the responsiveness to fMLP (Fig. 4A and B). Exactly identical patterns were obtained by using UV-inactivated, replication-incompetent viruses (data not shown), thus confirming our previous results that de novo viral gene expression is not required for the block of migration.
Fig. 4.

pUL128 is necessary to block chemokine-driven migration of monocytes. The chemotactic migration of monocytes toward the inflammatory chemokines CCL5 and CCL2 (100 ng/ml) was evaluated using a Boyden chamber as described in Materials and Methods. As controls, migration toward fMLP (10−8 M) and basal/spontaneous migration (toward medium alone) were evaluated. The numbers of cells that migrated were obtained from at least 5 independent experiments using cells from different donors and are means ± SD. (A) Only viruses carrying the wild-type UL128-131A locus are able to impair monocyte chemotaxis. Monocytes were mock infected or incubated overnight with BAC4, BAC4mut, BAC1, or BAC1rep at an MOI of 5 prior to measurement of chemotaxis. *, P ≤ 0.05 compared to mock-infected cells. (B) Soluble recombinant pUL128 impairs chemotaxis toward CCL2 and CCL5. Monocytes were incubated overnight with increasing concentrations of rpUL128 prior to assessment of cell migration. Untreated monocytes (n.t.) and monocytes infected with BAC4 (MOI of 5) were used as controls. (C) The inhibitory effect of rpUL128 is specific. Monocytes were infected with BAC4 or BAC1 (MOI of 5) or treated overnight with 1 μg/ml of rpUL128 or the irrelevant protein rProtX, and migration toward CCL2 and CCL5 was evaluated.
A conservative interpretation of these results is that the structural component responsible for monocyte paralysis belongs to the UL128-131A region. To address the issue of a specific role for pUL128 in migration inhibition, monocytes were treated with increasing doses of soluble recombinant pUL128 (rpUL128). Figure 4B shows that starting at a concentration of 1 ng/ml, rpUL128 blocked the CCL2- and CCL5-driven migration of monocytes. Monocytes treated with rpUL128 exhibited normal basal migration as well as normal fMLP responsiveness. The inhibitory effect exhibited by rpUL128 was specific (Fig. 4C), as monocytes treated with an unrelated recombinant protein (rProtX, a hepatitis B virus N-terminally His-tagged Pres1-Pres2 protein), produced and purified in the same way as rpUL128 (16), migrated like mock-infected untreated monocytes. Altogether, these data indicate that pUL128 is specifically involved in inhibiting the chemokine-driven migration of monocytes and that it does so even as a soluble recombinant molecule without HCMV infection.
The block of chemokine-driven migration is not dependent on modification of the cellular cytoskeleton.
Since an intact and functional cytoskeleton is a prerequisite for monocyte migratory behavior, we investigated whether a defect in the cytoskeletal architecture could account for the block of migration. Monocytes infected with BAC4 and BAC1rep were compared to monocytes inoculated with BAC1 and BAC4mut regarding the total amount and architecture of the three main components of the cytoskeleton, namely, actin, vimentin, and tubulin. As shown in Fig. 5A, irrespective of the virus used, the distributions of actin and vimentin appeared identical to those in mock-infected cells, presenting as intense peripheral accumulations of actin and vimentin with protruding spikes (short plasma membrane extensions). Additionally, by Western blot analysis, we observed that the total intracellular amounts of the structural components actin, vimentin, and tubulin, as well as the regulatory protein WASP (42), remained unaltered in monocytes infected by all four viruses (Fig. 5B). Since all monocytes exhibited the same cytoskeletal features, we can conclude that HCMV infection of monocytes, in contrast to HCMV infection of monocyte-derived macrophages (10), does not induce dramatic alterations of the cytoskeletal architecture.
Fig. 5.

Inhibition of monocyte migration is not due to cytoskeletal modification. Monocytes were mock infected or incubated overnight with BAC4, BAC4mut, BAC1, or BAC1rep (MOI of 5). (A) Morphological analysis of actin and intermediate (vimentin) filaments was performed by indirect immunofluorescence at 24 h p.i. Monocyte nuclei are blue (DAPI staining), the cytoplasm is red (Evans blue staining), and actin and vimentin are green. Original magnification, ×60. The images are for one donor representative of three. (B) Western blot analysis of major components of the monocyte cytoskeleton. Data for one donor representative of five are shown. Equal protein amounts of cell lysates were separated by SDS-PAGE, blotted onto nitrocellulose membranes, and probed with antibodies raised against actin, vimentin, tubulin, and WASP.
pUL128 is necessary to downregulate the surface expression of chemokine receptors.
We finally compared, by cytofluorimetric analysis, the cell surface expression of different chemokine receptors on monocytes inoculated with the four viruses or treated with 1 μg/ml of rpUL128 or rProtX. As shown in Fig. 6A, the surface expression of CCR1, CCR2, and CCR5, the cognate receptors for the CCL2 and CCL5 chemokines used previously in migration assays, was strongly reduced in monocytes infected with BAC4 and BAC1rep as well as in monocytes treated with 1 μg/ml of rpUL128. However, monocytes inoculated with BAC4mut or BAC1 or treated with 1 μg/ml of rProtX exhibited expression levels of CCR1, CCR2, and CCR5 identical to those in mock-infected untreated monocytes. Under all conditions, monocytes showed high expression levels of CD14, indicating that the downregulation of chemokine receptors was specific and also reflecting the purity of the monocyte population.
Fig. 6.

pUL128 is necessary to downregulate the surface expression of chemokine receptors. Monocytes were mock infected or incubated overnight with BAC4, BAC4mut, BAC1, or BAC1rep (all used at an MOI of 5) or with rpUL128 or rProtX (both used at 1 μg/ml). The expression of the indicated chemokine receptors as well as of CD14 was evaluated by fluorescence-activated cell sorter analysis of viable or fixed-permeabilized cells for quantification of surface (A) and total (B) protein amounts. The percentages of cells expressing the indicated molecules were evaluated in independent experiments and analyzed statistically. Values are means ± SD for 5 independent experiments. *, P ≤ 0.05 compared to mock-infected cells.
To investigate whether the reduced cell surface expression was dependent on receptor degradation or internalization, we compared the total amounts of chemokine receptor proteins in BAC4-infected and rpUL128-treated monocytes. As controls, we considered mock-infected and rProtX-treated monocytes. All cells—uninfected, infected, and treated with rpUL128 or rProtX—were shown to possess the same amounts of chemokine receptors after fixation and permeabilization (Fig. 6B), thus indicating that the downregulation on the cell surface was not due to protein degradation but to a redistribution of receptor molecules to intracellular compartments.
DISCUSSION
HCMV uses monocytes as an important target of infection, subverting their roles in both innate and adaptive immune responses. In fact, monocytes are regarded as a site of viral latency in vivo (26, 30) and as vehicles for viral dissemination (28, 32, 35). At present, little is known about the mechanisms of HCMV entry and establishment of infection in these cells or about the influence of HCMV on their functions. Monocytes originate from CD34+ progenitors in the bone marrow, circulate in the bloodstream for 1 to 3 days, and then move into peripheral tissues in order to replenish the resident populations of tissue macrophages and dendritic cells. Monocytes can encounter HCMV during all of these stages: in the bone marrow, in the bloodstream, and in the peripheral tissues during or after transendothelial migration from the bloodstream. Monocyte movements are tightly controlled by members of the superfamily of chemoattractant cytokines called chemokines and by their receptors (22). The deregulation of the chemokine-chemokine receptor system can favor HCMV infection, spread, and persistence in the host. Accordingly, more than 30 distinct virally encoded proteins have been identified that are able to corrupt the chemokine system (15); in particular, HCMV encodes two CXC chemokine homologues (18) and four chemokine receptor homologues (36).
In previous work, we demonstrated that structural components present in the viral particles of clinical isolates and endotheliotropic HCMV strains account for the impaired expression and function of the cellular chemokine receptors CCR1, CCR2, CCR5, and CXCR4 in monocytes (11). At that time, we could not identify the specific viral factors involved. Recently, the viral genes UL128, UL130, and UL131A have been shown to be mutated consistently in all fibroblast-adapted strains and to be essential for infection of endothelial, epithelial, and dendritic cells (1, 12, 13, 23, 38, 39). Importantly, the three encoded proteins have also been shown to be structural components of the viral envelope. Wang and Shenk described complexes containing gH/gL and the UL128 and UL130 proteins in infected cells as well as in virions (39). Adler et al. extended these studies by showing that UL131A is associated with gH in virions (1). Finally, Ryckman et al. showed that gH/gL and UL128-131A assemble to form one complex consisting of all five proteins, modeled in the following way. gH binds both gL and pUL130, thus forming a pocket for pUL131A. pUL128 is not covalently bound to the other members of the complex but is held in an exposed position by pUL130 (24). We therefore investigated whether these three proteins could be important for monocyte infection, on the one hand, and whether they could play a role in impairment of chemotaxis on the other. Since the UL128 gene encodes a protein with a CC chemokine domain (13) and, moreover, soluble recombinant pUL128 shows virus-inhibitory and cell surface-binding properties (16), we hypothesized that pUL128 could have a particular role within the complex and could be involved directly in chemokine receptor downregulation.
The TB40E-derived BAC4 and BAC1 viruses exhibit opposite endothelial cell tropism properties, carry identical UL130 and UL131A sequences, and differ by the presence of a critical adenine insertion within the UL128 cds of BAC1 (27). Removal of the additional adenine in the genomic background of BAC1, in the virus called BAC1rep, restored endothelial cell tropism. Conversely, insertion of the additional adenine in the genomic background of BAC4, in the virus called BAC4mut, abolished endothelial cell tropism.
While BAC4 and BAC1rep were able to initiate the viral cycle in primary human monocytes, the BAC1 and BAC4mut viruses were not. Thus, the genetically pure and well-defined BAC4 and BAC1 viruses and their derivatives nicely reproduced the phenotypic differences previously observed between TB40E, its nonendotheliotropic counterpart AD169 (11), and TB40F (our unpublished data), formally demonstrating that the UL128-131A locus defines HCMV tropism for monocytes.
However, the inability to infect endothelial cells and monocytes exhibited by BAC1 and BAC4mut cannot be referred exclusively to the lack of UL128 product, because our analysis of virion composition showed that in the absence of wild-type UL128, the entire complex was altered. Mutation in UL128 leads to the joint lack of all three UL128-131A products from the viral envelope. Our results are similar and consistent with those of Ryckman et al. (24) showing that pUL128, pUL130, and pUL131A must all bind simultaneously to gH/gL for the production of complexes that can function in entry into epithelial and endothelial cells. When any single component of a multiprotein complex is missing or misfolded, the entire complex is frequently retained in the endoplasmic reticulum (ER) or is degraded by ER-associated degradation. Whereas HFF infected by BAC1 and BAC4mut expressed pUL130 and pUL131A, we did not detect full-length or shorter forms of pUL128. It is probable that truncated pUL128 has a short half-life because it is identified as misfolded and disposed of by the ER quality control of the infected HFF.
An important point to be verified was whether the BAC1 and BAC4mut viruses were unable to enter monocytes or suffered from a postentry block instead (i.e., an arrest at any stage along the postfusion pathway of uncoating, virus particle translocation to the nuclear pores, and release of the viral genomes into the nucleus). Earlier studies with endothelial and epithelial cells established that pUL128-131A are essential for HCMV fusion and entry. The specific site of fusion in these cells seems to be influenced by the cell of origin of the virions. For example, in Arpe-19 epithelial cells, endotheliotropic HCMV entered by direct fusion at the plasma membrane if the virions were produced in the same cell line but was endocytosed and fused at endosomal membranes if virions were produced in fibroblasts (40). In our experiments using viral stocks produced in HFF, we observed both BAC4 and BAC1 virions accumulating inside endosome-like structures in the cytoplasm of monocytes by electron microscopy at early times p.i. These vesicular structures retained BAC4 virions for only a limited period, and at 24 h p.i., the cytoplasmic vesicles were filled by an amorphous material that might represent the remains of the uncoating process, but none contained intact virions. In contrast, at the same time point, BAC1 virion particles were retained in large cisternae that nicely reflected the pattern given by pp65 immunofluorescence staining. Therefore, monocytes seem to mirror the features of the working model for endothelial and epithelial cells in which fibroblast-produced HCMV enters the cell by endocytosis and fuses with endosomal membranes.
The data shown here lead to the hypothesis that the products of the UL128-131A locus are necessary to allow the fusion of the endosomal membrane with the viral envelope. However, an additional or alternative role of the UL128-131A products in docking the virions to a specific receptor, thus mediating a “nondegrading” type of endocytosis, cannot be excluded.
The mechanism linking the UL128-131A proteins to migration impairment and chemokine receptor downregulation in HCMV-infected monocytes was analyzed in turn by testing the expression and functionality of the chemokine receptors CCR1, CCR2, and CCR5 in monocytes exposed to the four clonal viruses. Both BAC4 and BAC1rep inhibited monocyte migration toward CCL2 and CCL5 and induced surface downregulation of the cognate receptors CCR1, CCR2, and CCR5 through cytoplasmic accumulation. Importantly, the same happened when UV-inactivated BAC4 and BAC1rep virions were used. In contrast, BAC1 and BAC4mut did not alter migration or chemokine receptor expression on the cell surface. Since we compared viruses expressing wt and mutated versions of pUL128 in two different genomic backgrounds, we can rule out the possibility that adventitious mutations outside UL128 are involved in this phenomenon. Also, the observation that BAC4- and BAC1rep-infected monocytes retained their basal migration and fMLP responsiveness, together with a cytoskeletal structure comparable to that of uninfected cells, indicated that the inhibition of migration was a selective effect. The persistence of an intact cytoskeleton in infected monocytes was surprising because HCMV infection of macrophages, which are derived from monocytes, induces a dramatic alteration of the cellular architecture (10).
These data indicated that a product(s) of the UL128-131A locus is required for the chemotaxis inhibition and receptor downregulation effects. However, these data did not allow us to define whether any of the UL128-131A products are directly responsible for chemotaxis inhibition or if they are required to enable entry and the subsequent release of a critical component (e.g., a tegument protein) into monocytes. A hint of a direct role for pUL128 was obtained by treating uninfected monocytes with a recombinant pUL128 protein (16) prior to measurement of their chemotaxis and assessment of chemokine receptor expression. The soluble rpUL128 protein potently inhibited chemokine-driven migration as well as the surface expression of the chemokine receptors CCR1, CCR2, and CCR5.
This result suggests a couple of conclusions. First, pUL128 may be the only HCMV protein required to induce effects on monocyte chemotaxis and chemokine receptors, and in our experimental setting, its action does not require presentation on the surface of the virion envelope. Second, its mechanism of action is most probably dependent on physical interaction with a cellular receptor(s). The hypothesis that pUL128 can bind one or more chemokine receptors is particularly tempting because chemokine receptors have been known to undergo internalization by endocytosis (desensitization) after ligand engagement. Alternatively, pUL128 could induce ligand-independent internalization of other receptors of the family under conditions of sustained signaling (heterologous desensitization) (6). However, the observation that rpUL128 did not induce calcium fluxes in monocytes (data not shown), together with the broad spectrum of chemokine receptor downregulation, supports the hypothesis that pUL128 activates a general negative signaling pathway such as the protein kinase A (PKA) or PKC pathway (3).
ACKNOWLEDGMENTS
This study was supported by Baustein 3.2 (G. Frascaroli), by BMBF FKZ01KI0772 project C, and by DFG SPP1175.
We thank Anke Lüske for excellent technical assistance and Ingrid Bennett for editing the English text. We are very grateful to Christian Sinzger (University of Tübingen, Germany) for the generous gift of the four BAC clones. We thank David C. Johnson and Giuseppe Gerna for supplying important antibodies.
We declare that we have no conflicting financial interests.
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Adler B., et al. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 87:2451–2460 [DOI] [PubMed] [Google Scholar]
- 2. Akter P., et al. 2003. Two novel spliced genes in human cytomegalovirus. J. Gen. Virol. 84:1117–1122 [DOI] [PubMed] [Google Scholar]
- 3. Ali H., Richardson R. M., Haribabu B., Snyderman R. 1999. Chemoattractant receptor cross-desensitization. J. Biol. Chem. 274:6027–6030 [DOI] [PubMed] [Google Scholar]
- 4. Ancuta P., et al. 2003. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J. Exp. Med. 197:1701–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Booss J., Dann P. R., Griffith B. P., Kim J. H. 1989. Host defense response to cytomegalovirus in the central nervous system. Predominance of the monocyte. Am. J. Pathol. 134:71–78 [PMC free article] [PubMed] [Google Scholar]
- 6. Borroni E. M., Mantovani A., Locati M., Bonecchi R. 2010. Chemokine receptors intracellular trafficking. Pharmacol. Ther. 127:1–8 [DOI] [PubMed] [Google Scholar]
- 7. Britt W. J., Alford C. A. 1996. Cytomegalovirus, p. 2493–2523 In Fields B. N., Knipe D. M. (ed.), Fields virology, vol. 2 Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 8. Buser C., Walther P. 2008. Freeze-substitution: the addition of water to polar solvents enhances the retention of structure and acts at temperatures around −60 degrees C. J. Microsc. 230:268–277 [DOI] [PubMed] [Google Scholar]
- 9. Chee M. S., et al. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:126–169 [DOI] [PubMed] [Google Scholar]
- 10. Frascaroli G., et al. 2009. Human cytomegalovirus paralyzes macrophage motility through down-regulation of chemokine receptors, reorganization of the cytoskeleton, and release of macrophage migration inhibitory factor. J. Immunol. 182:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frascaroli G., et al. 2006. Human cytomegalovirus subverts the functions of monocytes, impairing chemokine-mediated migration and leukocyte recruitment. J. Virol. 80:7578–7589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gerna G., et al. 2005. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 86:275–284 [DOI] [PubMed] [Google Scholar]
- 13. Hahn G., et al. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78:10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mocarski E. S. 1996. Cytomegaloviruses and their replication, p. 2447–2492 In Fields B. N., Knipe D. M., Howley P. M. (ed.), Fields virology fs, vol. 1 Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 15. Murphy P. M. 2001. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat. Immunol. 2:116–122 [DOI] [PubMed] [Google Scholar]
- 16. Patrone M., Secchi M., Bonaparte E., Milanesi G., Gallina A. 2007. Cytomegalovirus UL131-128 products promote gB conformational transition and gB-gH interaction during entry into endothelial cells. J. Virol. 81:11479–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patrone M., et al. 2005. Human cytomegalovirus UL130 protein promotes endothelial cell infection through a producer cell modification of the virion. J. Virol. 79:8361–8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Penfold M. E., et al. 1999. Cytomegalovirus encodes a potent alpha chemokine. Proc. Natl. Acad. Sci. U. S. A. 96:9839–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Revello M. G., Percivalle E., Di Matteo A., Morini F., Gerna G. 1992. Nuclear expression of the lower matrix protein of human cytomegalovirus in peripheral blood leukocytes of immunocompromised viraemic patients. J. Gen. Virol. 73:437–442 [DOI] [PubMed] [Google Scholar]
- 20. Rice G. P., Schrier R. D., Oldstone M. B. 1984. Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc. Natl. Acad. Sci. U. S. A. 81:6134–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rollins B. J. 1997. Chemokines. Blood 90:909–928 [PubMed] [Google Scholar]
- 22. Rossi D., Zlotnik A. 2000. The biology of chemokines and their receptors. Annu. Rev. Immunol. 18:217–242 [DOI] [PubMed] [Google Scholar]
- 23. Ryckman B. J., Jarvis M. A., Drummond D. D., Nelson J. A., Johnson D. C. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryckman B. J., et al. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuessler A., Sampaio K. L., Sinzger C. 2008. Charge cluster-to-alanine scanning of UL128 for fine tuning of the endothelial cell tropism of human cytomegalovirus. J. Virol. 82:11239–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sinclair J., Sisson P. 1996. Latent and persistent infections of monocytes and macrophages. Intervirology 39:239–301 [DOI] [PubMed] [Google Scholar]
- 27. Sinzger C., et al. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 28. Smith M. S., Bentz G. L., Alexander J. S., Yurochko A. D. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 78:4444–4453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith M. S., Bentz G. L., Smith P. M., Bivins E. R., Yurochko A. D. 2004. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J. Leukoc. Biol. 76:65–76 [DOI] [PubMed] [Google Scholar]
- 30. Soderberg-Naucler C., et al. 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol. 75:7543–7554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sozzani S., et al. 1997. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 159:1993–2000 [PubMed] [Google Scholar]
- 32. Stoddart C. A., et al. 1994. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol. 68:6243–6253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taylor-Wiedeman J., Hayhurst G. P., Sissons J. G., Sinclair J. H. 1993. Polymorphonuclear cells are not sites of persistence of human cytomegalovirus in healthy individuals. J. Gen. Virol. 74:265–268 [DOI] [PubMed] [Google Scholar]
- 34. Taylor-Wiedeman J., Sissons J. G., Borysiewicz L. K., Sinclair J. H. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059–2064 [DOI] [PubMed] [Google Scholar]
- 35. van der Strate B. W., et al. 2003. Dissemination of rat cytomegalovirus through infected granulocytes and monocytes in vitro and in vivo. J. Virol. 77:11274–11278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vischer H. F., Leurs R., Smit M. J. 2006. HCMV-encoded G-protein-coupled receptors as constitutively active modulators of cellular signaling networks. Trends Pharmacol. Sci. 27:56–63 [DOI] [PubMed] [Google Scholar]
- 37. Walther P., Ziegler A. 2002. Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J. Microsc. 208:3–10 [DOI] [PubMed] [Google Scholar]
- 38. Wang D., Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 79:10330–10338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang D., Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang D., Yu Q. C., Schroer J., Murphy E., Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 104:20037–20042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wentworth B. B., French L. 1970. Plaque assay of cytomegalovirus strains of human origin. Proc. Soc. Exp. Biol. Med. 135:253–258 [DOI] [PubMed] [Google Scholar]
- 42. Zicha D., et al. 1998. Chemotaxis of macrophages is abolished in the Wiskott-Aldrich syndrome. Br. J. Haematol. 101:659–665 [DOI] [PubMed] [Google Scholar]