

Abstract
The parvovirus minute virus of mice (MVM) packages a single copy of its linear single-stranded DNA genome into preformed capsids, in a process that is probably driven by a virus-encoded helicase. Parvoviruses have a roughly cylindrically shaped pore that surrounds each of the 12 5-fold vertices. The pore, which penetrates the virion shell, is created by the juxtaposition of 10 antiparallel β-strands, two from each of the 5-fold-related capsid proteins. There is a bottleneck in the channel formed by the symmetry-related side chains of the leucines at position 172. We report here the X-ray crystal structure of the particles produced by a leucine-to-tryptophan mutation at position 172 and the analysis of its biochemical properties. The mutant capsid had its 5-fold channel blocked, and the particles were unable to package DNA, strongly suggesting that the 5-fold pore is the packaging portal for genome entry.
INTRODUCTION
Members of the family Parvoviridae package their ∼5-kb, single-stranded, linear DNA genome into a preformed, nonenveloped protein capsid (25, 33, 36, 43). Viruses in the genus Dependovirus package DNA strands of both senses with equal frequency, but members of the genus Parvovirus, including minute virus of mice (MVM), package predominantly negative-sense strands (37). MVM DNA replication initiates with complementary strand synthesis and proceeds through a series of duplex concatameric intermediates via a unidirectional strand displacement mechanism called rolling hairpin replication (12). The polarity of strands selected for encapsidation is determined by the relative efficiency of the two telomeric origins (14).
Parvoviruses encode a large nonstructural replication initiator protein, called NS1 in MVM and Rep68/78 in adeno-associated virus 2 (AAV2), which comprises two distinct enzymatic domains. The amino-terminal half of the molecule contains a site-specific single-stranded nuclease (21, 27, 32, 34). The carboxy-terminal half contains a 3′-to-5′ helicase domain, related to the AAA+ family of cellular ATPases (8, 19, 22, 23). The insertion of single-stranded DNA into particles of parvoviruses, including MVM, starts from the 3′ end (13, 25). This correlates with the 3′-to-5′ processivity of the helicase and has led to the suggestion that the viral helicase functions as the molecular motor that translocates progeny DNA molecules into the capsid (13, 25, 42).
The icosahedral parvovirus capsid of MVM has a maximum external diameter of 280 Å and is constructed from 60 polypeptide subunits, of which VP2 is the major species, while VP1, an N-terminal extension of VP2, is present in about 10 copies per particle (38). The crystal structure of the MVM virion showed that there are 547 ordered amino acids in each subunit (1, 29). The capsid protein has a β-sandwich fold formed by β-strands B through I that make two β-sheets consisting of strands BIDG and CHEF. No structural information is currently available for the N-terminal extensions of the capsid proteins, which consist of 38 residues for VP2 and a further 142 residues for VP1. These N-terminal peptides appear to serve as major mediators of cellular exit and reentry pathways, becoming sequentially externalized from the MVM particle (15).
The portal through which these N termini are externalized is the pore that surrounds each of the 12 vertices in parvovirus particles. This pore is formed by the juxtaposition of 10 antiparallel β-strands from the 5-fold-related capsid proteins (7, 40). Genetic evidence supports the idea that the pore may also serve as the portal for packaging the viral genome and, subsequently, its extrusion in the next infectious cycle (4, 10, 11, 17, 18, 28, 30, 31). The tightest constriction in the 5-fold pore is near its base and is formed by leucine 172 in VP2 (1). Previous analysis of MVM mutants carrying a full range of substitutions at this position showed that, with the exception of two viable mutants (L172I and L172V), all of the mutants were defective in establishing infection (18). While some of these modifications rendered capsids temperature sensitive or unconditionally defective for assembly and/or subsequent cell entry, one mutant (L172W) produced only empty particles, indicating that this substitution might interfere with packaging. Here we further characterize this mutant and provide structural evidence that its phenotype results from a blockage of the pore, indicating that the channel along one of the 5-fold axes is the packaging portal for the parvovirus genome.
MATERIALS AND METHODS
Generation and purification of L172W virus-like particles (VLPs).
A full-length copy of the immunosuppressive MVM (MVMi) VP2 gene containing the L172W mutation was cloned into pFastBac and recovered as a recombinant baculovirus by using the Bac-to-Bac strategy, following the manufacturer's protocols (Invitrogen, Carlsbad, CA). Primary stocks were amplified to tertiary stocks, which were then used to infect 10-cm plates containing 1.2 × 107 SF9 Spodoptera frugiperda cells in SF900 II serum-free medium. Cell extracts in batches of 10 plates per gradient were purified on iodixanol step gradients as previously described (17).
Crystallization and data collection.
Crystals of L172W mutant VLPs were obtained using the hanging drop technique, with a well solution containing 10 mM Tris-HCl (pH 7.5), 8 mM CaCl2, and 0.75% (wt/vol) polyethylene glycol 8000 (PEG 8000). The drops were prepared by mixing 5 μl of the well solution with an equal volume of a 10-mg/ml virus solution in 10 mM Tris-HCl (pH 7.5). Crystals formed in 4 to 8 weeks. For data collection, crystals were soaked for 30 to 90 s in mother liquor containing 16% PEG 8000 and 20% glycerol and immediately frozen in liquid nitrogen. Data were collected from a single crystal at 100 K on the MarCCD 165 detector at beamline BioCARS of the APS synchrotron. An oscillation range of 0.2° was used during data collection. Three data sets were collected, the best of which contained data to 4.2-Å resolution. All three data sets (Table 1) were processed and scaled using the HKL2000 package (35), but only the 4.2-Å-resolution data set was used for structure determination once the data sets had been independently phased.
Table 1.
Scaling and refinement statistics
| Characteristic | Valuea for data set no.: |
||
|---|---|---|---|
| 1 | 2 | 3 | |
| Space group | C2 | C2 | C2 |
| Unit cell dimensions (Å) | |||
| a | 442.9 | 435.6 | 438.5 |
| b | 411.1 | 407.0 | 407.2 |
| c | 301.8 | 299.2 | 297.5 |
| β (°) | 95.88 | 95.73 | 95.27 |
| Resolution limit (Å range) | 35.0–4.2 (4.39–4.20) | 35.0–4.3 (4.50–4.30) | 35.0–4.4 (4.60–4.40) |
| Completeness (%) | 55.3 (29.4) | 45.4 (44.5) | 43.4 (40.8) |
| Rmergeb | 10.0 (26.0) | 10.3 (25.8) | 14.4 (39.6) |
| Avg redundancy | 1.5 (1.2) | 2.2 (2.1) | 2.1 (1.5) |
| <I>/<σI> | 5.52 (1.86) | 7.50 (2.02) | 4.94 (1.92) |
| Reciprocal space correlation coefficient of Fobs and Fcalc after convergence of map | 0.86 (0.63) | 0.89 (0.73) | 0.85 (0.69) |
| R-factor | 0.303 (0.432) | ||
| Avg B-factor | 113.4 | ||
| Ramachandran plot outliers (%)c | 3.7 | ||
| Ramachandran plot of most-favored regions (%)c | 82.8 | ||
| Rotamer outliersc | 2.6 | ||
| RMSD, bonds (Å) | 0.011 | ||
| RMSD, angles (°) | 1.25 | ||
| No. of unique reflections | 214,730 | 159,098 | 147,418 |
Values in parentheses are for the high-resolution bin.
Rmerge = ∑h∑j |Ihj − <Ih>|/∑∑|Ihj|.
Determined according to the criterion of Molprobity (16).
X-ray structure determination.
The parameters of the MVMi L172W crystal in space group C2 obtained from postrefinement were a = 443.1 Å, b = 411.3 Å, c = 301.8 Å, β = 95.93°. The packing considerations indicated that there were two particles per unit cell, with one half of a virus particle occupying a crystallographic asymmetric unit. A rotation function for κ of 180° calculated with the program GLRF (39) established the orientation of the icosahedral symmetry axes relative to the crystal axes. The position of the particle center on a crystallographic 2-fold axis was arbitrarily chosen to be at y = 0. A correctly placed MVMi particle (pdb code 1z1c) was used to calculate initial phases for reflections with a resolution lower than 10 Å by using the program CNS (5). The phases were refined with 15 cycles, using 30-fold noncrystallographic symmetry averaging with the program AVE (26). The mask defining the volume of electron density to be averaged was derived from the wild-type MVMi atomic model by including all grid points within 5 Å of each atom. Grid points outside the capsid were set to the average value of the density outside the capsid. Phase information for reflections immediately outside the current resolution limit was obtained by extending the resolution by (1/a)Å−1 followed by four cycles of averaging. This procedure was repeated until 4.2-Å resolution was reached. Particle orientation and unit cell parameters were refined by searching for their best value as judged by the correlation coefficient between observed and calculated structure amplitudes (Table 1). During the final 10 averaging cycles, the volume on the inside of the capsid was included in the averaging procedure as opposed to being flattened.
The structure was built using the program O (24) starting from the MVMi virion structure (pdb code 1z1c) with residue 172 mutated to tryptophan. The structure was built manually and subjected to coordinate and B-factor refinement using the program CNS. The atoms from each residue were constrained to have the same B-value, because of the limited resolution of the diffraction data. The structure refinement utilized noncrystallographic symmetry constraints. Other calculations used the CCP4 suite of programs (9). No water molecules were added because of the low resolution of the data. Had it been calculated, Rfree would have been very close to the Rworking value, due to the high noncrystallographic redundancy (2).
Analysis of virus particles produced by transfection.
Confluent dishes of iD5 cells were transfected with 5 μg each of MVMi wild-type or L172W genomic DNA by using Superfect (Qiagen) according to the manufacturer's instructions. After 3 h of incubation, the cells were washed and fed with fresh medium containing 0.04 U/ml neuraminidase to prevent expansion of the viable wild-type virus.
For analysis of viral DNA replication, transfected cells were harvested at 48 h posttransfection, and total DNA was extracted. Purified DNA was digested with DpnI, to distinguish replicating viral DNA from input plasmid, separated on a 1.4% neutral agarose gel, and subjected to Southern blotting using a randomly labeled probe derived from an internal 3,961-bp fragment of the viral genome.
For analysis of viral protein expression, cultures were harvested at 48 h posttransfection in the presence of protease inhibitors and separated on a 10% acrylamide gel containing SDS, as described previously (17). NS1 was detected using an antibody raised against the N terminus and the viral polypeptides with an antipeptide antibody against residues 311 to 327 of VP2. A rabbit polyclonal antibody to β-actin served as a loading control.
For analysis of virus particle assembly and packaging, transfected cells were harvested at 72 hours postinfection and concentrated by centrifugation, and the pellets were extracted by freezing and thawing three times in 500 μl of TE buffer (10 mM Tris [pH 8.7], 1 mM EDTA). The cell extracts and culture medium were combined, clarified, and sedimented to equilibrium in iodixanol step gradients as described previously (17). Individual fractions were analyzed by Southern or Western blotting as described previously (17), except that samples for Western blot analysis were further concentrated by ethanol precipitation.
Protein Data Bank accession number.
The MVMi L172W coordinates together with the observed structure amplitudes have been deposited in the Protein Data Bank with accession number 2xgk.
RESULTS AND DISCUSSION
The L172W mutation induces a reorganization of the N-terminal part of VP2.
The crystal structure of the MVMi L172W mutant has been determined to 4.2-Å resolution (Table 1). Crystals of the L172W mutant were nearly isomorphous with that of the wild-type virus. The root mean square deviation between equivalent Cα atoms of wild-type MVMi capsid protein (1z1c) and the L172W MVMi mutant was 0.3 Å for 542 residues. In contrast to the wild type, the mutant structure did not show any evidence for the presence of DNA in the capsid.
The side chain of leucine 172 interacts with the side chain of valine 40 in the wild-type structure (Fig. 1A). This interaction is modified in the L172W mutant, resulting in a different conformation of the N-terminal region than in the wild-type structure (Fig. 1B). The wild-type VP2 is ordered from residue 39 onwards (1), whereas the N-terminal region of the MVMi L172W mutant is well-ordered only from residue 46 onward. Residues 46 and 47 in the mutant structure were shifted toward the particle interior relative to their position in the wild-type capsid (Fig. 1B). In the L172W mutant capsid, there was an additional density resembling a β-strand of about 5 residues located next to residues 263 to 267 from β-strand G (Fig. 1C), extending the BIDG sheet. This density has ∼1/2 the height of the main chain density of the capsid protein. There is an even-lower density that extends the additional β-strand toward the inner opening of the 5-fold channel (Fig. 1C). The extra β-strand could be formed by the missing residues 39 to 45, although there was no connecting density between this peptide and the first modeled residue 46 in VP2. In the wild-type structure, residues 39 to 47 form a part of β-strand A and a loop that interacts with the ordered N-terminal regions of symmetry-related subunits around the 5-fold axis. The loss of interaction between the hydrophobic side chains of L172 and V40 probably resulted in the displacement of the VP2 N-terminal region. The volume where the β-strand density was located in the mutant is not occupied by density belonging to icosahedrally ordered DNA in the wild-type virion structure (1), indicating that the dislocation of this strand from its wild-type position is not due to the absence of DNA from the VLP.
Fig. 1.

The channels along the 5-fold symmetry axes of the L172W mutant capsid are blocked. (A) Detail of the hydrophobic interaction between the side chains of Val 40 and Leu 172 (green). The position of the Trp 172 side chain in the L172W mutant is shown in orange. (B) Comparison of VP2 N termini of wild-type MVMi (green) and of mutant L172W MVMi (orange). The map of L172W MVMi contoured at 2σ is shown in blue. The protein subunits are shown in cartoon representation, and side chains of Leu 172, Trp 172, and Val 172 are shown as sticks. The position of the icosahedral 5-fold axis is indicated by a black line. (C) Electron density of the putative β-strand positioned next to residues 263 to 267 of VP2. The map covering VP2 was contoured at 2σ (blue). The putative β-strand and the density on the 5-fold axis are contoured at 1σ (green) and 0.1σ (red). The position of the icosahedral 5-fold axis is indicated by a black line. (D) Comparison of Leu 172 in wild-type MVMi (green) and Trp 172 in the mutant L172W MVMi (orange) at the 5-fold pore. The proteins are shown in cartoon representation, and the side chains of Leu and Trp are shown as sticks. The map of the mutant L172W MVMi, contoured at 1σ, is shown as a blue mesh.
The crystal structures of parvoviruses (40), including MVMi (1), show that the N terminus of one of the five VP2 subunits runs through the channel at most of the 5-fold vertices. The cleavage of the VP2 N-terminal regions, when they become exposed on the particle surface, appears to be a prerequisite for infection (17). As the VP2 N-terminal regions are displaced from the 5-fold pore opening in the L172W mutant capsid, other substitutions that disrupt the L172-V40 interaction, without abrogating packaging, may also suffer a similar disruption. This is consistent with the observation that only those mutant viruses that have isoleucine or valine at position 172 are viable (18), whereas other substitutions with residues whose side chains are less likely to interact with V40 have severely reduced infectivity relative to wild-type virus. Although it might be supposed that disruption of the L172-V40 interaction would impair externalization of VP2 N termini, this appears not to be the case. While L172F, L172G, and L172T mutant virions, generated in a single burst from transfection, had sequestered VP1 N termini, their VP2 N termini were exposed and could be cleaved with trypsin to the VP3 form, as seen for the wild-type virus (18). However, following such cleavage, the VP1 N termini of mutant virions became exposed and vulnerable to proteolysis (18), suggesting that failure to establish the normal L172-V40 interaction may have serious consequences for the subsequent stability of the cylinder. This phenotype was further explored for the L172T mutant in the prototype strain of MVM (MVMp), because unlike MVMi, MVMp can be grown productively at 32°C, and the L172T mutant is temperature sensitive (17). The MVMp L172T mutant is viable at 32°C, but not at 37°C, because it has a conditional cell entry defect that correlates inversely with the integrity of its VP2 N termini (17). At the permissive temperature, mutant viruses that had substantial numbers of intact VP2 termini at the time they were added to cells were infectious, whereas viruses with fully cleaved VP2 N termini were unable to initiate infection at either temperature. Moreover, in vitro, VP2 cleavage of the L172T mutant at 37°C and neutral pH induced exposure of both the VP1 N termini and the viral genome, suggesting that L172-V40 interactions help to provide a level of cylinder stability that is critical for the controlled virion dynamics required to mediate sequential steps in the cell entry process.
The 5-fold channel is the MVMi packaging portal.
The crystal structure of the L172W capsid shows that the diameter of the 5-fold channel is reduced from 8 Å in the wild-type to 6 Å in the mutant structure (Fig. 1D). Furthermore, a spherical density at a height comparable to that of the VP2 backbone was observed on the icosahedral 5-fold axis in the vicinity of the tryptophan 172 side chains (Fig. 1D). The electron density map of the MVMi L172W mutant was independently determined from three different data sets (Table 1). Each of these maps contained a similar density on the 5-fold axis. Thus, this density is unlikely to be noise that could not be eliminated because of its location on the 5-fold axis. The nature of the moiety could not be identified based on the shape of the density, because the diffraction data used to calculate the electron density map represented the 5-fold average of the molecule. However, the density could represent a 5-fold average of a small hydrophobic molecule that interacted with tryptophan side chains, or it could belong to a residue from the extra β-strand, discussed above, that interacts with the mutant's tryptophan 172 side chains. The presence of the low density connecting the extra β-strand with the density on the 5-fold axis favors the second option. The connecting density should be weak, because a residue belonging to only one of the five β-strands can occupy this position on a 5-fold axis. The L172W mutant therefore has two defects, the first being the misplaced N-terminal region and the second being the blockage of the 5-fold channel, perhaps by a residue from the dislocated A strand.
L172W is the only mutation at this position that generates capsids devoid of progeny DNA (18). Cells transfected with L172W and wild-type MVMi genomes synthesized equivalent amounts of monomer (5-kb) and dimer (10-kb) double-stranded replicative form DNA (Fig. 2A) as well as the viral polypeptides NS1, VP1, and VP2 (Fig. 2B).
Fig. 2.
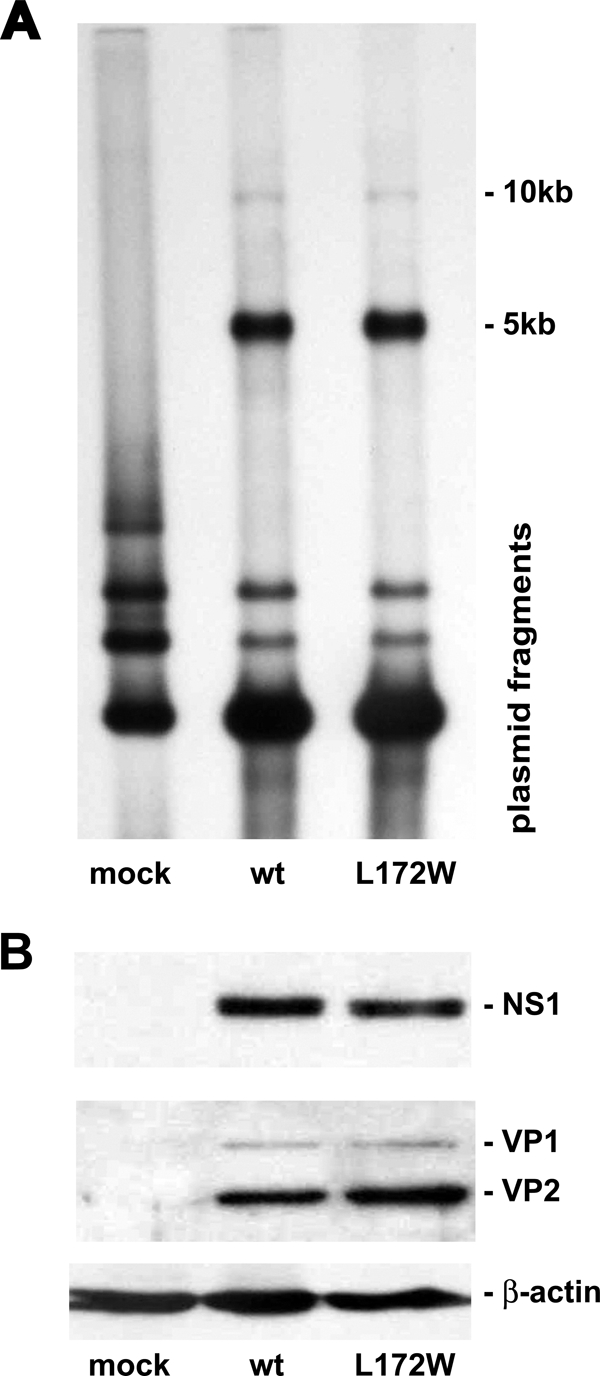
Viral DNA replication and gene expression is unaltered for the L172W MVMi mutant. (A) Southern blot of total DNA isolated from A9 cells after transfection with L172W and wild-type MVMi genomes, digestion with DpnI, and probing for MVM sequences. (B) Western blot analysis of protein isolated from A9 cells following transfection with L172W and wild-type MVMi genomes and probing with antibodies specific for the viral polypeptides NS1 or VP1 and VP2.
Following transfection, the L172W mutant produced empty particles with a composition and density similar to those of the wild type (Fig. 3A, fractions 7 to 9). This region of the gradient contains about 2-fold-fewer empty particles for the mutant than for the wild type, although the steady-state level of capsid protein expression is the same in wild-type- and mutant-transfected cells, as shown in Fig. 2B, middle panel. However, the gradient position normally occupied by full virions (Fig. 3A, fractions 3 to 5) contains minimal amounts of viral protein for the mutant, so that the disparity between viral protein for the mutant versus the wild type is substantially greater for full particles than for empty capsids. Fractions 3 to 5 (Fig. 3B, lower panel) are also completely devoid of 5-kb viral DNA for the L172W mutant, being lower than the wild type (Fig. 3B, upper panel) by more than 70-fold at a 5-fold-longer exposure, indicating that L172W empty capsids fail to be converted to full virions. Further experiments will be required to determine whether the lower L172W empty particle yield is due to an assembly defect or a stability problem. However, the difference in empty capsid production is insufficient to explain the failure to observe full virions in the mutant virus gradient by more than 2 orders of magnitude. The lack of L172W full particles could also result from mutant virions uncoating concomitantly with, or immediately following, packaging of the viral genome. However, the biochemical evidence, taken together with the channel blockage seen in the crystal structure, favors the conclusion that DNA packaging is impaired in the mutant and identifies the 5-fold channel as the packaging portal for the parvovirus genome.
Fig. 3.
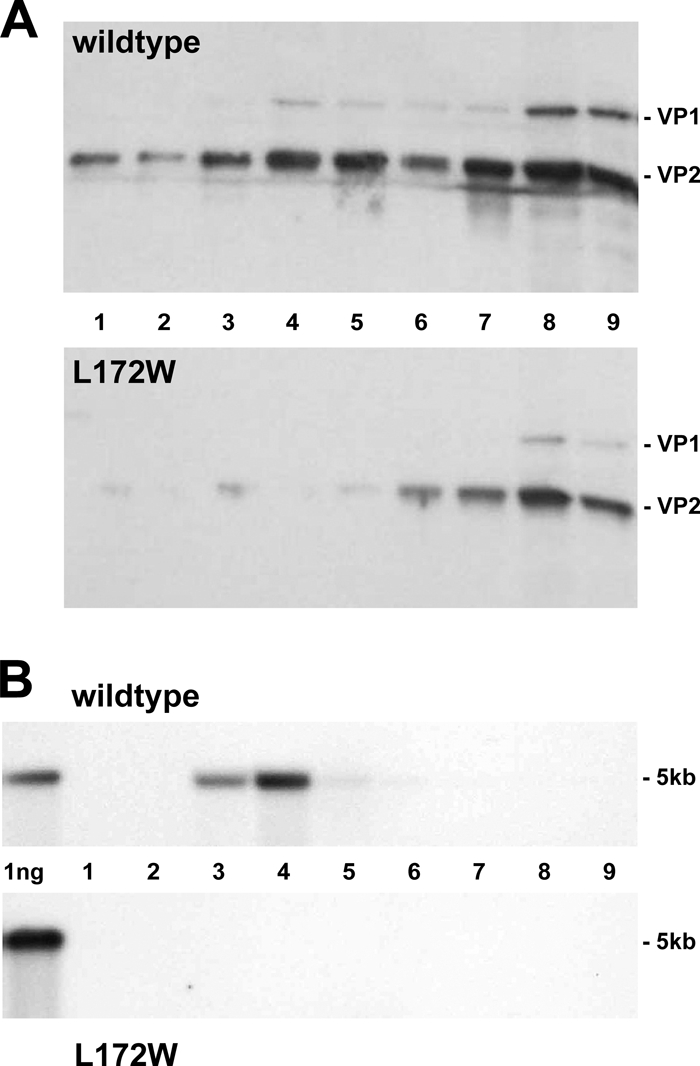
The L172W MVMi mutant produces only empty particles. (A) Iodixanol gradient of wild-type (upper panel) and L172W (lower panel) cell extracts analyzed for viral proteins by Western blotting. (B) Iodixanol gradient of wild-type (upper panel) and L172W (lower panel) cell extracts analyzed for viral genomes by alkaline agarose gel electrophoresis and probed for MVM DNA sequences.
The orthologous mutation in the related Dependovirus AAV2, VP1 L336W, has also been shown to result in defective packaging (3), although this is not as profound a defect as seen here for the MVM mutant, perhaps reflecting the slightly different architecture of the base of the cylinder and wider pore in AAV2 compared to MVM (41). The AAV VP1 L336W mutant was also defective in binding the viral Rep protein, shown to be essential for packaging, suggesting that the mutation might disrupt association of the packaging motor with the capsid (3). However, no structural rearrangements of the MVM capsid surface that might prevent NS1 from binding were evident in our study, and the least complicated explanation of this mutant's nonpackaging phenotype appears to be the significant density blocking the pore.
The structural verification that MVMi has a unique vertex for genome packaging is probably also valid for other parvoviruses. By analogy to bacteriophages, the special vertex could serve not only for genome entry but also for genome egress. However, we have recently shown that uncoating occurs in the same 3′-to-5′ direction as packaging and is not simply a reversal of the packaging process (11). Thus, there may be two distinctive vertices in the parvovirus particle, one in the empty capsid for packaging and another in the full virion for uncoating. The presence of a special vertex may also influence the asymmetric binding of the cellular receptor to canine parvovirus (20). Furthermore, such a vertex has also been noted for poliovirus (6), suggesting that the existence of a special vertex might be a common feature for viruses with linear genomes that lack a membrane envelope.
ACKNOWLEDGMENTS
We thank Sheryl Kelly for help with preparation of the manuscript. We thank the staff of BioCARS sector 14 at the Advanced Photon Source (APS) of the Argonne National Laboratory for their help with data collection. The APS facility is supported by the U.S. Department of Energy and the National Institutes of Health.
This work was supported by Public Health Service grants AI11219 to M.G.R., AI26109 and CA29303 to P.T., and AI79271 to S.H. from the National Institutes of Health, as well as a Purdue University reinvestment grant awarded to the Purdue Structural Biology Group.
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Agbandje-McKenna M., Llamas-Saiz A. L., Wang F., Tattersall P., Rossmann M. G. 1998. Functional implications of the structure of the murine parvovirus, minute virus of mice. Structure 6:1369–1381 [DOI] [PubMed] [Google Scholar]
- 2. Arnold E., Rossmann M. G. 1990. Analysis of the structure of a common cold virus, human rhinovirus 14, refined at a resolution of 3.0 Å. J. Mol. Biol. 211:763–801 [DOI] [PubMed] [Google Scholar]
- 3. Bleker S., Pawlita M., Kleinschmidt J. A. 2006. Impact of capsid conformation and Rep.-capsid interactions on adeno-associated virus type 2 genome packaging. J. Virol. 80:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bleker S., Sonntag F., Kleinschmidt J. A. 2005. Mutational analysis of narrow pores at the fivefold symmetry axes of adeno-associated virus type 2 capsids reveals a dual role in genome packaging and activation of phospholipase A2 activity. J. Virol. 79:2528–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brünger A. T., et al. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921 [DOI] [PubMed] [Google Scholar]
- 6. Bubeck D., Filman D. J., Hogle J. M. 2005. Cryo-electron microscopy reconstruction of a poliovirus-receptor-membrane complex. Nat. Struct. Mol. Biol. 12:615–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chapman M. S., Rossmann M. G. 1993. Structure, sequence, and function correlations among parvoviruses. Virology 194:491–508 [DOI] [PubMed] [Google Scholar]
- 8. Christensen J., Tattersall P. 2002. Parvovirus initiator protein NS1 and RPA coordinate replication fork progression in a reconstituted DNA replication system. J. Virol. 76:6518–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 10. Cotmore S. F., D'Abramo A. M., Jr., Ticknor C. M., Tattersall P. 1999. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 254:169–181 [DOI] [PubMed] [Google Scholar]
- 11. Cotmore S. F., Hafenstein S., Tattersall P. 2010. Depletion of virion-associated divalent cations induces parvovirus minute virus of mice (MVM) to eject its genome in a 3′-to-5′ direction from otherwise intact viral particles. J. Virol. 84:1945–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cotmore S. F., Tattersall P. 2006. Parvoviruses, p. 593–608 In DePamphilis M. (ed.), DNA replication and human disease. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 13. Cotmore S. F., Tattersall P. 2005. Encapsidation of minute virus of mice DNA: aspects of the translocation mechanism revealed by the structure of partially-packaged genomes. Virology 336:100–112 [DOI] [PubMed] [Google Scholar]
- 14. Cotmore S. F., Tattersall P. 2005. Packaging sense is controlled by the efficiency of the nick site in the right-end replication origin of parvoviruses MSM and LuIII. J. Virol. 79:2287–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cotmore S. F., Tattersall P. 2007. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 70:183–232 [DOI] [PubMed] [Google Scholar]
- 16. Davis I. W., et al. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farr G. A., Cotmore S. F., Tattersall P. 2006. VP2 cleavage and the leucine ring at the base of the fivefold cylinder control pH-dependent externalization of both the VP1 N terminus and the genome of minute virus of mice. J. Virol. 80:161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farr G. A., Tattersall P. 2004. A conserved leucine that constricts the pore through the capsid fivefold cylinder plays a central role in parvoviral infection. Virology 323:243–256 [DOI] [PubMed] [Google Scholar]
- 19. Gorbalenya A. E., Koonin E. V., Wolf Y. I. 1990. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 262:145–148 [DOI] [PubMed] [Google Scholar]
- 20. Hafenstein S., et al. 2007. Asymmetric binding of transferrin receptor to parvovirus capsids. Proc. Natl. Acad. Sci. U. S. A. 104:6585–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hickman A. B., Ronning D. R., Perez Z. N., Kotin R. M., Dyda F. 2004. The nuclease domain of adeno-associated virus rep coordinates replication initiation using two distince DNA recognition interfaces. Mol. Cell 13:13. [DOI] [PubMed] [Google Scholar]
- 22. Iyer L. M., Leipe D. D., Koonin E. V., Aravind L. 2004. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146:11–31 [DOI] [PubMed] [Google Scholar]
- 23. James J. A., et al. 2003. Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure 11:1025–1035 [DOI] [PubMed] [Google Scholar]
- 24. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110–119 [DOI] [PubMed] [Google Scholar]
- 25. King J. A., Dubielzig R., Grimm S. W., Kleinschmidt J. A. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 20:3282–3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kleywegt G. J., Read R. J. 1997. Not your average density. Structure 5:1557–1569 [DOI] [PubMed] [Google Scholar]
- 27. Koonin E. V., Ilyina T. V. 1993. Computer-assisted dissection of rolling circle DNA replication. Biosystems 30:241–268 [DOI] [PubMed] [Google Scholar]
- 28. Kronenberg S., Bottcher B., der Lieth C. W., Bleker S., Kleinschmidt J. A. 2005. A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J. Virol. 79:5296–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Llamas-Saiz A. L., et al. 1997. Structure determination of minute virus of mice. Acta Crystallogr. D Biol. Crystallogr. 53:93–102 [DOI] [PubMed] [Google Scholar]
- 30. Lombardo E., Ramírez J. C., Garcia J., Almendral J. M. 2002. Complementary roles of multiple nuclear targeting signals in the capsid proteins of the parvovirus minute virus of mice during assembly and onset of infection. J. Virol. 76:7049–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maroto B., Valle N., Saffrich R., Almendral J. M. 2004. Nuclear export of the nonenveloped parvovirus virion is directed by an unordered protein signal exposed on the capsid surface. J. Virol. 78:10685–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mouw M., Pintel D. J. 1998. Amino acids 16-275 of minute virus of mice NS1 include a domain that specifically binds (ACCA)2-3-containing DNA. Virology 251:123–131 [DOI] [PubMed] [Google Scholar]
- 33. Myers M. W., Carter B. J. 1980. Assembly of adeno-associated virus. Virology 102:71–82 [DOI] [PubMed] [Google Scholar]
- 34. Nuesch J. P., Cotmore S. F., Tattersall P. 1995. Sequence motifs in the replicator protein of parvovirus MVM essential for nicking and covalent attachment to the viral origin: identification of the linking tyrosine. Virology 209:122–135 [DOI] [PubMed] [Google Scholar]
- 35. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 36. Richards R., Linser P., Armentrout R. W. 1977. Kinetics of assembly of a parvovirus, minute virus of mice, in synchronized rat brain cells. J. Virol. 22:778–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tattersall P., et al. 2005. Parvoviridae, p. 593–608 In Fauquet C. M., Mayo M. A., Maniloff J., Desselberger U., Ball L. A. (ed.), Virus taxonomy. Eightht report of the International Committee on Taxonomy of Viruses Elsevier Academic Press, London, England [Google Scholar]
- 38. Tattersall P., Cawte P. J., Shatkin A. J., Ward D. C. 1976. Three structural polypeptides coded for by minute virus of mice, a parvovirus. J. Virol. 20:273–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tong L., Rossmann M. G. 1997. Rotation function calculations with GLRF program. Methods Enzymol. 276:594–611 [PubMed] [Google Scholar]
- 40. Tsao J., et al. 1991. The three-dimensional structure of canine parvovirus and its functional implications. Science 251:1456–1464 [DOI] [PubMed] [Google Scholar]
- 41. Xie Q., et al. 2002. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 99:10405–10410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoon-Robarts M., et al. 2004. Residues within the B′ motif are critical for DNA binding by the SF3 helicase Rep40 of adeno-associated virus type 2. J. Biol. Chem. 279:50472–50481 [DOI] [PubMed] [Google Scholar]
- 43. Yuan W., Parrish C. R. 2001. Canine parvovirus capsid assembly and differences in mammalian and insect cells. Virology 279:546–557 [DOI] [PubMed] [Google Scholar]