

Abstract
The fusogenic reoviruses induce syncytium formation using the fusion-associated small transmembrane (FAST) proteins. A recent study indicated the p14 FAST protein transmembrane domain (TMD) can be functionally replaced by the TMDs of the other FAST proteins but not by heterologous TMDs, suggesting that the FAST protein TMDs are modular fusion units. We now show that the p15 FAST protein is also a modular fusogen, as indicated by the functional replacement of the p15 ectodomain with the corresponding domain from the p14 FAST protein. Paradoxically, the p15 TMD is not interchangeable with the TMDs of the other FAST proteins, implying that unique attributes of the p15 TMD are required when this fusion module is functioning in the context of the p15 ecto- and/or endodomain. A series of point substitutions, truncations, and reextensions were created in the p15 TMD to define features that are specific to the functioning of the p15 TMD. Removal of only one or two residues from the N terminus or four residues from the C terminus of the p15 TMD eliminated membrane fusion activity, and there was a direct correlation between the fusion-promoting function of the p15 TMD and the presence of N-terminal, hydrophobic β-branched residues. Substitution of the glycine residues and triserine motif present in the p15 TMD also impaired or eliminated the fusion-promoting activity of the p15 TMD. The ability of the p15 TMD to function in an ecto- and endodomain-specific context is therefore influenced by stringent sequence requirements that reflect the importance of TMD polar residues and helix-destabilizing residues.
INTRODUCTION
The current membrane fusion paradigm, based on enveloped virus fusion proteins, predicts that bilayer merger is intimately linked with triggered structural changes occurring in the large, complex ectodomains of these viral fusogens, which function as autonomous, metastable fusion machines (29). Among the known viral fusogens, the fusion-association small transmembrane (FAST) proteins challenge this mechanistic paradigm (17). The FAST proteins, encoded by the nonenveloped fusogenic orthoreoviruses, are the smallest known viral fusogens. As nonstructural viral proteins, the FAST proteins do not mediate virus-cell fusion. The FAST proteins are instead expressed and trafficked to the cell surface of virus-infected or transfected cells, where their sole function is the induction of cell-cell fusion and multinucleated syncytium formation. FAST protein-induced syncytiogenesis enhances pathogenicity through a two-step dissemination process, initially serving to promote localized cell-cell viral transmission followed by an apoptosis-induced burst of progeny virus release and systemic spread of infection caused by extensive late-stage fusion events (7, 40). When reconstituted into liposomes, the purified p14 FAST protein is both necessary and sufficient to induce membrane fusion (53). However, exhibiting no inherent receptor-binding activities, the FAST proteins rely on surrogate cellular adhesins to mediate early membrane attachment events during cell-cell fusion (39). The FAST proteins also use their cytoplasmic endodomains to recruit cellular pathways involved in the expansion of stable fusion pores to the lumen-sized openings required for syncytiogenesis (52). Therefore, although the FAST proteins are bona fide viral fusogens, they recruit or rely on cellular cofactors to promote the pre- and postfusion stages of syncytium formation. How these rudimentary viral fusogens function to induce the actual merger of closely apposed membranes remains unclear.
The FAST protein family consists of four members, each named according to its predicted molecular mass: p15 of baboon reovirus (BRV), p14 of reptilian reovirus (RRV), the p10 proteins encoded by avian reovirus (ARV) and Nelson Bay reovirus (NBV), and the newly discovered p22 protein of Atlantic salmon reovirus (AtSRV) (12, 16, 38, 44). In the absence of a cleavable signal peptide, a single-pass transmembrane domain (TMD), flanked on the cytoplasmic side by a cluster of basic residues, functions as both an insertion signal and a membrane anchor to direct the FAST proteins into an Nexoplasmic/Ccytoplasmic (Nexo/Ccyt) membrane topology (12, 15, 44). Beyond this common topology, the FAST proteins lack significant sequence similarity and show an exceptional diversity in the arrangement of shared structural motifs. Each protein is modified by fatty acylation: an N-terminal myristate moiety in the cases of p14 and p15 (and possibly p22) or a membrane-proximal cytoplasmic palmitoylated dicysteine motif in p10 (14, 46). In addition, both p14 and p15 have a polyproline region in their endo- or ectodomain, respectively, that is absent from the p22 and p10 proteins. Finally, each FAST protein carries a stretch of moderately hydrophobic amino acids, termed the hydrophobic patch, found in the ectodomains of p14 and p10 but in the endodomains of p15 and p22 (12, 15, 38, 44). Mutagenic and peptide analyses suggest that the hydrophobic patches of p10 and p14 may function analogously to the fusion peptides or fusion loops of enveloped viral fusion proteins (3, 14, 45). The 19-residue p15 ectodomain lacks any sequence that resembles a traditional fusion peptide.
The asymmetric membrane topology of the FAST proteins is unusual for a viral fusion protein, with the bulk of each protein allocated to the transmembrane and endodomains (12, 15, 38, 44). With ectodomains of only ∼20 to 40 amino acids, it is highly unlikely that the FAST proteins utilize mechanical energy released by extensive ectodomain conformational changes to drive close membrane apposition and fusion, as occurs with the enveloped viral fusion proteins (20, 57). Instead, other regions of the FAST proteins, such as their TMDs, may play a more dominant role in the fusion reaction. Beyond their role as simple membrane anchors, TMDs in many proteins are also implicated in protein-protein interactions, in membrane topology, and in some cases as having a direct role in the fusion reaction itself (6, 42, 56). Previous studies indicate that the TMDs of enveloped virus fusion proteins must fully span the membrane to allow the formation and expansion of a stable fusion pore (1, 26). There is little consensus, however, as to the specific primary sequence requirements of the TMD needed to mediate membrane fusion (25). While TMD structural flexibility conferred by glycine residues may be important for membrane fusion with some viral fusogens (11), not all fusion systems support this concept (1, 26, 32). It has also been posited that the helix-breaking tendencies of β-branched amino acids may be required for TMD conformational plasticity during membrane fusion. β-branched residues, which are over represented in the TMDs of some fusion proteins, have been implicated in the fusion reaction through studies involving artificial peptides of SNARE and vesicular stomatitis virus (VSV) G protein TMDs (23, 24, 37). However, the role of TMD β-branched residues within the context of the fusion protein has not yet been explored.
A recent study of the RRV p14 FAST protein revealed that the TMD is directly involved in mediating membrane fusion at a step prior to the formation of a stable fusion pore (10). The results indicated that the p14 TMD could be functionally replaced by the TMDs of the BRV p15 or ARV p10 FAST proteins but not by TMDs from heterologous viral or cellular membrane proteins (10). The FAST protein TMDs can therefore function in a modular fashion, at least in a p14 backbone, suggesting that a family-specific structural motif required for fusion may exist within their TMDs. It was proposed that a funnel-shaped TMD, with bulky aromatic residues clustering toward the C-terminal half of the TMD, may help the FAST proteins induce positive curvature of the inner leaflet to promote pore formation (10). Further support for this model derives from recent results indicating that chlorpromazine (CPZ), which induces the transition from hemifusion to pore formation by promoting positive curvature of the inner leaflet, has no effect on p14-mediated membrane fusion (9). These results are consistent with the possibility that the role of CPZ in promoting curvature of the inner leaflet may instead be fulfilled by the funnel-shaped p14 transmembrane domain. However, there is some evidence to suggest that features beyond a funnel-shaped geometry may influence the function of the FAST protein TMDs. For example, glycine residues in the p14 TMD are not required for fusion activity, while a triglycine motif in the p10 TMD is essential for syncytium formation (10, 46).
The only investigations of the BRV p15 FAST protein established p15 as a viral fusogen and determined its Nexo/Ccyt topology (15, 16). To further explore the role of the FAST protein TMDs in the fusion reaction, we undertook a comprehensive investigation of the primary sequence requirements of the p15 TMD. Similar to the TMD sequences of p14 and p10, the p15 TMD also contains glycine residues, but in place of the p10 triglycine motif, the p15 protein has a triserine motif in the TMD. The p15 TMD is also four amino acids longer than that of either p14 or p10 (Fig. 1 A). With these distinctive properties in mind, we carried out a mutagenic and chimeric analysis of the p15 TMD. The results indicate that N-terminal β-branched residues, glycine residues, and the triserine motif in the p15 TMD are all essential for p15-mediated fusion. Additionally, the p15 ectodomain, but not the TMD or endodomain, can be functionally replaced by the corresponding domain of other FAST proteins. Unique characteristics of the p15 FAST protein therefore influence the modularity of the FAST protein TMDs, necessitating stringent TMD sequence requirements to generate a fusion-competent FAST protein.
Fig. 1.

The FAST protein TMDs do not function modularly within the context of p15. (A) Linear representation of the TMD regions of p15, ARV p10, and p14. Sequences in bold delineate the predicted TMDs and the sequences that were exchanged to create chimeric p15 proteins whose TMDs were replaced by those of p14 (TM14) or p10 (TM10). (B) QM5 cells were transfected with pcDNA3 (a), authentic p15 (b), p15TM10 (c), or p15TM14 (d) and immunostained using polyclonal anti-p15 antiserum at 9 (a and b) or 24 (c and d) h posttransfection. Arrows indicate antigen-positive syncytia (b) or single-cell foci (c and d). (C) Dual-color pore formation assay. QM5 cells were cotransfected with pEGFP and pcDNA3, authentic p15, p15TM10, or p15TM14 and overlaid with calcein red-labeled Vero cells. Cells were resuspended and fixed at 9 h posttransfection, and EGFP-positive cells were gated. The dot plots are representatives of duplicate samples from a single experiment, and the average percentage of EGFP-expressing cells that acquired calcein red due to pore formation is shown above the horizontal gating line, relative to forward scatter (FSC-H). The indicated percentage values varied by <1% between the duplicate samples and between two separate experiments.
MATERIALS AND METHODS
Cells and antibodies.
The QM5 and Vero cells were maintained at 37°C in a 5% CO2 atmosphere and were grown in medium 199 with Earle's salts containing 100 U of penicillin and streptomycin per ml and 10% or 5% heat-inactivated fetal bovine serum. Production of polyclonal rabbit antisera against the p15 endodomain, residues 90 to 140 (anti-p15 C-terminal), or the full-length p15 protein (anti-p15) was described previously (15, 16). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was from KPL. Alkaline phosphatase-conjugated goat anti-rabbit secondary antibody was from Jackson ImmunoResearch. Monoclonal mouse antihemagglutinin (anti-HA) antiserum was produced by concentrating the cell supernatant from 12CA5 mouse hybridoma cells by precipitation with 35% ammonium sulfate (approximate concentration of IgG, 1.6 μg/μl).
Cloning and plasmids.
The procedure for cloning pcDNA3-p15 from BRV was described previously (16). This clone was used as a template for point substitutions and deletions in the TMD using the QuikChange (Stratagene) method according to the manufacturer's specifications. The ExCite (Stratagene) mutagenesis method was used, according to the manufacturer's specifications, to create chimeric p15 mutants containing the TMD of either the ARV p10 or RRV p14 FAST protein (p15TM10 and p15TM14, respectively). The myristoylation-minus p15G2A mutant was previously described (15). All mutants were also subcloned into the pEGFP-N1 expression vector (Clontech) for C-terminal green fluorescent protein (GFP) tagging. The p15HAC clone was described previously (15).
Transfections, cell staining, and syncytial indexing.
Cluster plates containing subconfluent monolayers of QM5 cells were transfected with expression plasmids using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Monolayers were methanol fixed and stained using Wright-Giemsa stain at various times posttransfection, and a syncytial index was determined by counting the number of syncytial nuclei in random microscopic fields, as previously described (12). Alternatively, cells were methanol fixed and immunostained using 1:800 anti-p15 antibody and 1:1,000 goat anti-rabbit alkaline phosphatase-conjugated secondary antibody, as described previously (15). For immunofluorescent detection of intracellular p15 and p15 mutants, QM5 cells were transfected in culture plates containing glass coverslips. At 24 h posttransfection, cells were fixed for 20 min in 3.7% formaldehyde and then permeabilized in 0.1% Triton X-100 for 20 min. The cells were then stained using 1:800 anti-p15 C-terminal primary antibody and 1:800 Alexa-Fluor 488-conjugated secondary antibody, as described previously (12). For cell surface localization studies of p15 mutants, QM5 cells were transfected with C-terminally GFP-tagged p15G2A or TMD mutants in culture plates containing glass coverslips, as described above. The intracellular pool of p15 was depleted by treatment with 200 μg/ml cycloheximide (Sigma; C7698-1G) at 20 h posttransfection in serum-free medium for 3 h at 37°C. Cells were then fixed in 3.7% formaldehyde, as described above, and then visualized and photographed using a Zeiss LSM510 scanning argon laser confocal microscope and the ×100 objective. Images shown represent two or three merged Z sections.
Membrane fractionation and Western blotting.
The membrane and soluble fractions from transfected QM5 cells were obtained by vesiculating cells by passage through a 29-gauge syringe 10 times at 10 h posttransfection. Cell debris was removed by low-speed centrifugation at 700 × g, and the resulting cell lysates were separated by ultracentrifugation at 100,000 × g for 45 min, as described previously (44). Fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 15% acrylamide gels. Proteins were visualized using 1:10,000 anti-p15 C-terminal primary antibody and 1:10,000 goat anti-rabbit immunoglobulin G(H+L) peroxidase-labeled antibody. Western blots were imaged on a Typhoon 9410 variable-mode imager (Amersham Biosciences).
Pore formation assay.
Low-density QM5 cells (<25% confluent) were cotransfected with pEGFP and pcDNA3, authentic p15, or various p15 mutants at a 1:4 ratio. Vero cells were stained with CellTrace calcein AM red-orange (Molecular Probes) and added to QM5 cells at 4 or 8 h posttransfection, as described previously (10). Briefly, the cells were cocultured for 5 h (until small syncytia were visible in cells expressing authentic p15) or for up to 15 h (for p15 syncytiogenic-defective p15 constructs). Single-cell suspensions were generated by trypsin treatment and then fixed in 3.7% formaldehyde. Cofluorescence was analyzed by flow cytometry (FACSCalibur [Becton Dickinson]) with gating for enhanced GFP (EGFP)-expressing cells. Ten thousand EGFP-positive cells were counted and analyzed using FCS Express 2.0 (DeNovo Software).
Coimmunoprecipitation assay for p15 clustering.
QM5 cells were cotransfected in 10-cm2 culture dishes at a 1:1 ratio with pcDNA3 and untagged versions of authentic p15 or the indicated p15 chimeras or point substitutions. Cells were also cotransfected with pcDNA3 and C-terminally HA-tagged p15 (p152HAC) or with untagged p15 and p152HAC. Cells were lysed at 8 to 10 h posttransfection using 500 μl of a stringent ionic detergent-containing (radioimmunoprecipitation assay [RIPA]) buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] Igepal CA-630 [octylphenoxypolyethoxyethanol; Sigma-Aldrich], 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] sodium dodecyl sulfate) or a less stringent Triton X-100 buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] Triton X-100), both containing protease inhibitors (200 nM aprotinin, 1 μM leupeptin, and 1 μM pepstatin). A fraction of each lysate (100 μl) was retained for SDS-PAGE analysis, and the remainder was immunoprecipitated overnight (12 to 16 h) at 4°C using a 1:100 dilution of mouse monoclonal anti-HA antiserum. Antibody-antigen complexes were recovered by incubation with 40 μl IgGSORB (The Enzyme Center), prepared according to the manufacturer's specifications, with gentle shaking at 4°C for 30 min and then pelleting at 16,000 × g for 1 min. Pellets were washed three times with lysis buffer containing protease inhibitors, and antibody-antigen complexes were released by boiling in protein sample buffer for 5 min. Cell lysates and immune precipitates were analyzed by SDS-PAGE and detected by Western blotting with 1:10,000 anti-p15 C-terminal antisera and 1:10,000 goat anti-rabbit horseradish peroxidase-conjugated secondary antibody.
RESULTS
Modularity of the FAST protein TMDs depends on the endo- and ectodomain contexts.
The observation that the RRV p14 TMD can be functionally replaced by that of the other FAST proteins, but not by TMDs from outside the FAST protein family, led to the proposal that the FAST protein TMDs may function as modular fusion units (10). However, the p15 FAST protein has some unusual features, most notably an exceptionally small (19-residue), proline-rich ectodomain and a hydrophobic patch that resides in the endodomain, not in the ectodomain as in the p10 and p14 FAST proteins. It was therefore unclear whether the modular nature of the FAST protein TMDs would apply in the context of a p15 backbone. To investigate this question, the TMD of p15 was replaced by either the ARV p10 or RRV p14 TMD to create the chimeric constructs p15TM10 and p15TM14, respectively (Fig. 1A). The TMD boundaries were defined as described previously (10) and predicted by one or more TMD prediction algorithms. The N-terminal boundaries were defined by the first amino acid following a negatively charged residue in the case of p14 and p15 or a proline in the case of p10. The C-terminal boundaries were set at the last residue preceding a lysine (p14 and p15) or palmitoylated dicysteine motif (p10). The p15TM14 and p15TM10 constructs were used to transfect QM5 monolayers, and the progression of syncytiogenesis was followed by microscopic observation. In stark contrast to p14 TMD chimeras, heterologous FAST protein TMDs did not support p15-mediated syncytiogenesis (Fig. 1B). Monolayers transfected with p15TM10 and p15TM14 were devoid of syncytia, and immunostaining with anti-p15 antiserum revealed only single antigen-positive cells even up to 24 h posttransfection (Fig. 1B).
Numerous studies have implicated the TMDs of enveloped viral proteins in the formation or expansion of stable fusion pores (25, 27, 31, 42). Syncytial indexing does not permit the detection of mutants for which fusion is initiated but arrested prematurely at an earlier step, such as hemifusion or pore formation. To determine if the p15 TMD chimeras were capable of inducing the formation of stable fusion pores, a previously described fluorescence-activated cell sorting (FACS)-based pore formation assay was used (52). QM5 cells were cotransfected with the pEGFP expression plasmid and a plasmid encoding either authentic p15 or a p15 TMD substitution construct and were briefly incubated to allow initial transgene expression. The transfected donor cells were then overlaid with target Vero cells labeled with the aqueous fluor calcein red. Donor and target cells were cocultured to allow fusion events to occur and then trypsin treated to generate single-cell suspensions or small syncytia that were analyzed by flow cytometry. Quantifying the percentage of GFP-expressing donor cells that acquired the small, 800-Da calcein red fluor from target cells is indicative of stable pore formation. An increase in red fluorescence in cells coexpressing p15 and GFP clearly indicated that p15 mediated the formation of stable fusion pores (Fig. 1C). However, when donor cells were cotransfected with the p15 TMD chimeras (p15TM14 or p15TM10), no dye transfer above background levels was observed (Fig. 1C). While the dot plots in Fig. 1 were obtained at 9 h posttransfection (to avoid large syncytia that are incompatible with FACS analysis), no dye transfer above background levels was observed for the nonsyncytiogenic chimeric constructs even when donor cells were incubated with target cells for up to 24 h (data not shown). Therefore, unlike the case for p14, heterologous FAST protein TMDs do not support pore formation when present in a p15 backbone.
The chimeric TMD results suggested that undefined features of the ecto-and/or endodomain of the p15 FAST protein work in conjunction with unique attributes of the p15 TMD to generate a functional membrane fusion protein. To determine whether the p15 ecto- and/or endodomain works cooperatively with the p15 TMD, two additional p15 chimeric constructs were created, which contained the p14 ectodomain with the p15 endodomain and TMD (p15ect14) or the p14 endodomain with the p15 TMD and ectodomain (p15end14) (Fig. 2 A). The syncytiogenic activities of these constructs in transfected QM5 cells were determined by microscopic examination of Giemsa-stained monolayers. The rate and extent of syncytium formation induced by the p15ect14 construct were visually indistinguishable from the syncytiogenic activity of authentic p15. Both p15 and p15ect14 generated multinucleated syncytia by 4 to 6 h posttransfection, which rapidly progressed to encompass the majority of the monolayer by 10 to 12 h posttransfection (Fig. 2B). In contrast, the p15end14 construct failed to generate any syncytia even by 24 h posttransfection (Fig. 2B). The p15 FAST protein is therefore also a modular fusogen, but undefined features of the ecto-, endo-, and transmembrane domains influence the modularity of these p15 fusion domains. For the remainder of this study, we focused our attention on defining the unique attributes of the p15 TMD that allow this fusion module to function in its natural context.
Fig. 2.

The p15 FAST protein is a modular fusogen. (A) Linear representation of the p15 and p14 FAST proteins, indicating the locations of the TMD and hydrophobic patch (HP) in each protein. Numbers refer to amino acid positions demarcating the boundaries of the indicated motifs. The p15 domains (shaded rectangles) and p14 domains (white rectangles) present in the chimeric constructs (p15ect14 and p15end14) are indicated. (B) QM5 cells were transfected with authentic p15 or the indicated chimeric construct and then methanol fixed and Giemsa-stained at 6 h (a to c), 10 h (d and e), or 24 h (f) posttransfection to detect syncytium formation. The arrows in panels a and b indicate small syncytia.
Truncation of the p15 TMD eliminates membrane fusion activity.
The p14 and p10 TMDs are both predicted to be 19 residues in length, while the p15 TMD is predicted to be 23 residues. To determine whether the terminal residues of the extended p15 TMD influence p15-induced membrane fusion, a series of deletions were created in which the p15 TMD was truncated by either one or two residues from the N-terminal end (p15Δ21 and p15Δ21/22, respectively) or by four residues from the C-terminal end (p15Δ40-43) (Fig. 3 A). Giemsa stains of transfected QM5 monolayers revealed that each deletion of the authentic p15 TMD resulted in the complete loss of syncytiogenesis (Fig. 3B). Generally, when membrane fusion is lost due to insufficient transmembrane length, fusion of these mutants can be recovered by restoring the length of the TMD (1, 26, 58). Replacement of the N-terminal valine and/or isoleucine residues that were deleted from the p15Δ21 and p15Δ21/22 constructs with alanine did not restore membrane fusion activity, with transfected monolayers containing only individual antigen-positive cells as late as 24 h posttransfection (Fig. 3C, panels a and b). The cell-cell fusion activity of the p15 FAST protein is therefore highly sensitive to either truncation or replacement of the terminal residues of the TMD.
Fig. 3.
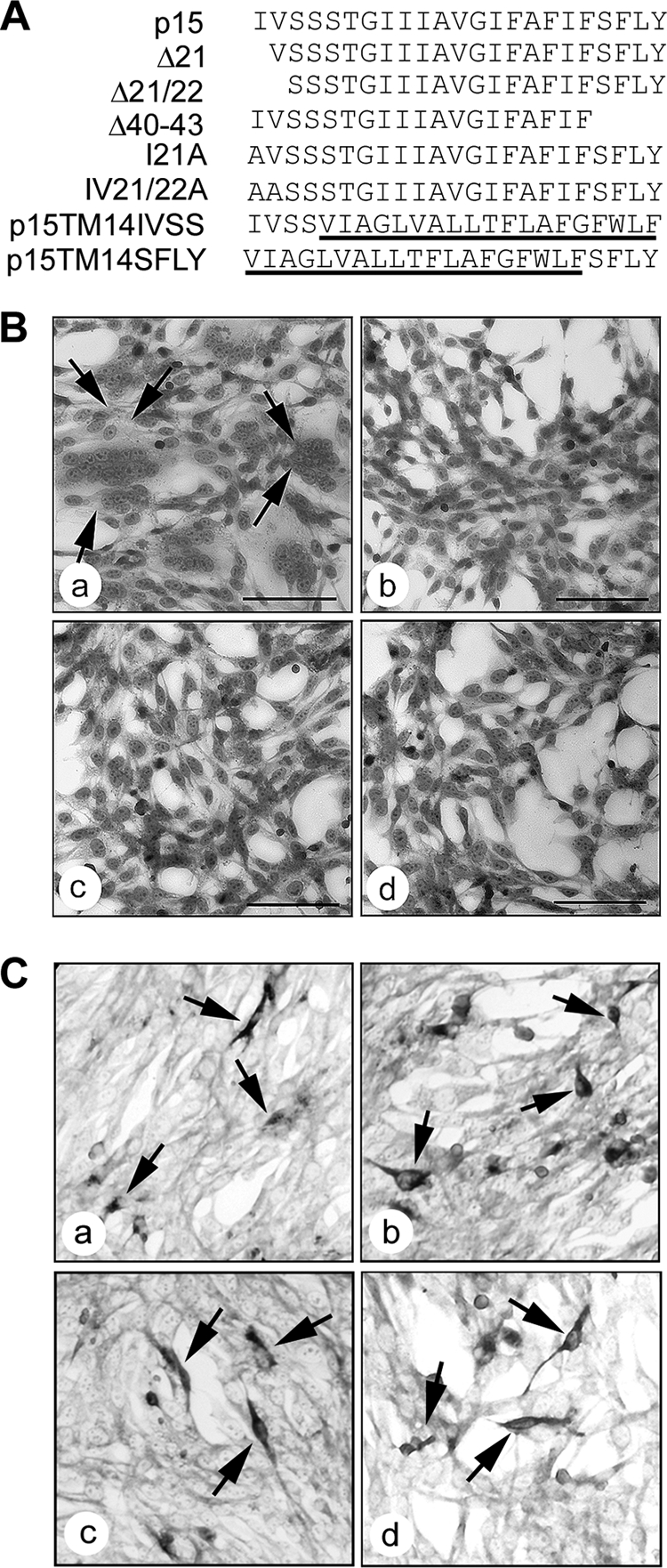
The terminal residues of the p15 TMD are required for membrane fusion. (A) Linear representation of the TMDs of authentic p15 and p15 deletion constructs (Δ21, Δ21/22, and Δ40–43) or N-terminal substitution constructs (I21A and I21/22A). The p15TM14 construct (Fig. 1) containing the 19-residue p14 TMD was also reextended to the 23-residue length of the p15 TMD by addition of the four N-terminal (p15TM14IVSS) or C-terminal (p15TM14SFLY) residues of the p15 TMD (underlined sequences are those from the p14 TMD). (B) QM5 cells were transfected with authentic p15 (a), p15Δ21 (b), p15Δ21/22 (c), or p15Δ40–43 (d) and then methanol fixed and Giemsa stained at 10 h posttransfection to detect syncytium formation (indicated by arrows in panel a). Scale bars = 100 μm. (C) QM5 cells were transfected with p15I21A (a), p15I21/22A (b), p15TM14IVSS (c), or p15TM14SFLY (d) and immunostained at 24 h posttransfection using polyclonal anti-p15. Arrows indicate antigen-positive single-cell foci.
To determine if disparities in TMD length were responsible for the loss of fusion activity of the chimeric TMD p15 constructs, p15TM14 was extended from 19 to 23 residues. Either the N- or C-terminal four residues of the authentic p15 TMD were added to the p15TM14 TMD to create the p15TM14IVSS and p15TM14SFLY constructs, respectively (Fig. 3A). The p15TM14 chimera was chosen as a backbone for these extensions since the p14 TMD can be functionally replaced by that of p15, suggesting that the TMDs share some common features (10). Both p15TM14IVSS and p15TM14SFLY failed to induce syncytium formation in transfected cells, generating instead single antigen-positive cells detected by immunostaining using anti-p15 antiserum (Fig. 3C, panels c and d). These results suggested that length alone is not a defining feature of the p15 TMD, implying that other TMD features restrict the ability of heterologous FAST protein TMDs to function in the context of the p15 ecto- and endodomains.
The FAST protein TMDs function as reverse signal-anchors to direct cotranslational membrane insertion. To determine whether the truncated p15 TMDs could still function as signal-anchors, transfected cell lysates were separated into the cytoplasmic and membrane fractions by high-speed ultracentrifugation. Cells were transfected with a plasmid expressing EGFP as a soluble, cytoplasmic control for the membrane fractionation. SDS-PAGE analysis and Western blotting using anti-p15 C-terminal antiserum indicated that each deletion construct was found exclusively in the pelleted membrane fraction (Fig. 4 A), confirming that the truncated TMDs were still capable of directing membrane insertion of p15 as an integral membrane protein. Although we cannot exclude the possibility that TMD truncations might alter the membrane topologies of these constructs, this is highly unlikely in view of the “positive-inside” rule of membrane protein topology (4, 55) and the preponderance of basic residues present in the p15 polybasic motif and throughout the cytoplasmic tail of p15.
Fig. 4.

Truncated p15 TMDs still function as signal-anchors and support localization to the plasma membrane. (A) Transfected QM5 cell lysates (L) were fractionated into soluble (S) and membrane (M) fractions by ultracentrifugation. The presence of p15, p15Δ21, p15Δ21/22, p15Δ40–43, or GFP (as a soluble protein control) in each fraction was detected by SDS-PAGE and immunoblotting using polyclonal anti-p15 C-terminal antiserum or anti-GFP. (B) QM5 cells were transfected with C-terminally GFP-tagged p15G2A, p15Δ21, p15Δ21/22, or p15Δ40–43 or with GFP as a non-membrane-associated protein, chased to the cell surface with a 3-h cycloheximide treatment at 20 h posttransfection, and then fixed with formaldehyde. The images shown represent two or three merged fluorescent Z sections (left panels) overlaid on the differential interference contrast (DIC) image (right panels). Arrows indicate plasma membrane-localized p15 mutant constructs. Scale bars = 20 μm.
Some viral fusion protein TMD truncation constructs exhibit trafficking defects (1, 26). Fluorescence microscopy was therefore used to determine whether truncation of the p15 TMD disrupted trafficking to the plasma membrane. In the absence of an antiserum that specifically recognizes the p15 ectodomain, the fusion-dead p15Δ21, p15Δ21/22, and p15Δ40-43 constructs were C-terminally tagged with GFP. A GFP-tagged, fusion-dead p15G2A myristoylation-minus construct was employed as a wild-type control. The p15G2A mutant allows extended incubations in the absence of syncytium formation to improve detection of p15 localized to the plasma membrane, and it traffics to the cell surface in a manner similar to that for authentic p15 (15). To partially deplete intracellular pools of p15 (whose fluorescence intensity masks the fluorescence of plasma membrane localized p15), cells were treated for 3 h with cycloheximide to stop translation and allow trafficking of the already-translated p15 proteins to the cell surface, as previously reported (15). As shown by confocal fluorescence microscopy, using identical laser intensities and image capture parameters, a clearly detected ring of fluorescence surrounding transfected cells was evident in cells expressing GFP-tagged p15G2A and all of the truncated p15 TMD constructs (Fig. 4B). In contrast, fluorescence in cells expressing EGFP alone was broadly distributed throughout the cytoplasm and nucleus. Truncation of the p15 TMD therefore does not grossly alter p15 trafficking to the plasma membrane. Since 3- to 5-fold decreases in surface expression of the p10 or p14 FAST proteins only delay the kinetics of syncytium formation (2), it is unlikely that minor changes in surface expression of the truncated p15 constructs undetectable by fluorescence microscopy would account for the complete absence of cell-cell fusion activity of these constructs. The p15 TMD therefore specifically influences membrane fusion activity independent of the role of the TMD as a signal-anchor or of any role in protein trafficking to the plasma membrane.
Transmembrane glycine and serine residues are essential for p15-mediated fusion.
The TMDs of several viral fusion proteins contain a higher proportion of glycine residues than do nonfusion proteins, and in some cases these glycine residues are important for fusion activity (11). In the case of the FAST proteins, substitution analysis indicates that TMD glycine residues are not required for p14-induced cell-cell fusion (10), while a triglycine motif in the N-terminal half of the ARV p10 TMD is essential for fusion activity (46). To determine the functional importance of the two glycine residues present in the p15 TMD, these glycines were individually replaced with alanine residues to create p15G27A and p15G33A (Fig. 5 A). Each construct was transfected into QM5 cells, and the relative fusion capabilities were determined by syncytial indexing. Both glycine substitutions had a dramatic effect on membrane fusion. At 10 h posttransfection, when syncytium formation induced by authentic p15 was approaching maximal levels, p15G27A produced no visible syncytia, while the syncytiogenic activity of p15G33A was reduced by ∼70% (Fig. 5B). By 24 h posttransfection, p15G27A did induce the formation of a limited number of very small syncytia, while the syncytia induced by p15G33A expanded in size but had still not completely fused the monolayer (data not shown). The TMD glycine residues are therefore not essential, but their absence substantially decreases the fusion activity of the p15 TMD.
Fig. 5.

Transmembrane glycine and serine residues are essential for p15-mediated fusion. (A) Linear representation of the p15 TMD and the locations of alanine substitutions of the glycine and serine residues in the indicated substitution constructs. (B) The percent syncytium formation relative to that for authentic p15 at 10 h posttransfection was determined by comparing the average number of syncytial nuclei per field in Giemsa-stained QM5 monolayers transfected with the indicated glycine and serine point substitutions. Results are the means ± standard deviations (SD) from a representative experiment done in triplicate. (C) Dual-color pore formation assay. QM5 cells were cotransfected with pEGFP and pcDNA3, authentic p15, p15S23A, p15S23/24A, p15S23-25A, p15G27A, or p15G33A and overlaid with calcein-labeled Vero cells at 8 h posttransfection. The extent of pore formation was determined at 22 h posttransfection by FACS analysis with gating for GFP-positive cells as described above. The results are corrected for background dye transfer in cells transfected with empty vector, and the means ± SD are from a representative experiment in triplicate.
Constructs were also created in which alanine residues were cumulatively substituted for the serine residues in the N-proximal triserine motif that is unique to the p15 TMD (Fig. 5A). Replacement of just one serine residue (p15S23A) inhibited p15-induced syncytiogenesis by >80% at 10 h posttransfection, and each accumulated substitution resulted in a corresponding further decrease (p15S23/24A) and eventual elimination (p15S23-25A) of syncytium formation (Fig. 5B). Pore formation induced by p15 cannot be assessed beyond 10 h posttransfection (the size of the syncytia is incompatible with FACS analysis). However, when the fusion-defective p15 constructs bearing alanine substitutions of the glycine or serine residues were subjected to the dual-color pore formation assay at 22 h posttransfection, the rank order of syncytiogenic activities of these mutants (i.e., p15G33A > p15S23A > p15G27A > p15S23/24A > p15S23-25A) was paralleled by their relative pore formation activities (Fig. 5 B and C). The transmembrane glycine residues and the triserine motif therefore directly contribute to the role of the p15 TMD in pore formation and syncytiogenesis.
The p15 TMD requires hydrophobic, β-branched residues at the N terminus for fusion.
The truncation and alanine replacement results indicated the importance of specific N-terminal residues in the p15 TMD. In addition to being hydrophobic, the N-terminal residues of the p15 TMD are also β-branched amino acids. Truncation, replacement, or substitution analysis of several viral fusion proteins, such as baculovirus gp64, HIV gp41, VSV G, and parainfluenza virus 5 F protein, did not indicate a role for N-terminal TMD β-branched amino acids (5, 26, 33, 54). However, studies conducted using synthetic TMD peptides have implicated terminal β-branched residues in the role of TMDs in membrane fusion, possibly due to their helix-destabilizing activity (18, 23, 25, 37). The p15 protein provided an opportunity to test whether N-terminal hydrophobic and/or β-branched TMD residues are required for the cell-cell membrane fusion activity of this FAST protein. The N-terminal residues of the p15 TMD were replaced either with threonine, a β-branched and hydrophilic residue, or with leucine, a hydrophobic and non-β-branched residue (Fig. 6 A). Western blotting indicated that all of these mutant constructs were expressed as well as authentic p15 (Fig. 6B). To examine the effect of these substitutions on membrane fusion, the kinetics of syncytium formation were obtained from transfected QM5 monolayers that were fixed and Giemsa stained from 6 to 16 h posttransfection (Fig. 6C). Authentic p15 rapidly induced cell-cell fusion, with the onset of syncytium formation occurring within 4 to 6 h posttransfection, and these syncytia rapidly expanded in size by 8 h posttransfection. By 10 h posttransfection, syncytial nuclei present in p15-transfected monolayers were too numerous to count. Substitutions of the N-terminal isoleucine residue with either threonine or leucine led to dramatic decreases in both the onset and extent of syncytiogenesis (Fig. 6C). At 8 h posttransfection, when p15-transfected monolayers were becoming extensively fused, there was little if any evidence of syncytia in cells transfected with p15I21T or p15I21L. However, both of these constructs containing single substitutions of the N-terminal isoleucine residue induced small syncytia by 10 h posttransfection, and these syncytia continued to develop to significant levels by 16 h posttransfection (Fig. 6C). In contrast, replacement of both the N-terminal isoleucine and valine residues with either threonine or leucine residues resulted in a complete loss of syncytium formation, even at later time points (data not shown).
Fig. 6.

The fusion activity of the p15 TMD requires hydrophobic and β-branched residues at the N terminus. (A) Linear representation of the p15 TMD and locations and identities of the β-branched substitution constructs (121T, IV21/22T, I21L, IV21/22L, I21V, and V22I). (B) Lysates from QM5 cells transfected with p15 or the indicated p15 mutant constructs were analyzed by SDS-PAGE and immunoblotting using polyclonal anti-p15 C-terminal antibody or antiactin antibody as a loading control. (C) The average number of syncytial nuclei present in QM5 cells transfected with p15, p15I21L, or p15I21T at 6 to 16 h posttransfection was quantified by microscopic examination of five random fields of Giemsa-stained monolayers. The number of syncytial nuclei induced by authentic p15 exceeded countable levels after 8 h posttransfection. Results are the means ± SD from a representative experiment done in triplicate. (D) QM5 cells transfected with the indicated p15 β-branched substitution constructs were assessed for cell-cell pore formation at 18 h posttransfection using a dual-color FACS-based fluorescent pore formation assay, as described for Fig. 1. Cells transfected with authentic p15 cannot be assessed for pore formation at this late stage due to extensive syncytium formation. Results are the means ± SD from a representative experiment done in triplicate, indicating the percentage of donor cells coexpressing EGFP and the indicated p15 construct that acquired the calcein red fluorescent cytoplasmic marker from target cells. (E) The average number of syncytial nuclei present in QM5 cells transfected with p15, p15I21V, or p15V22I at 6 to 10 h posttransfection was quantified by microscopic examination of Giemsa-stained monolayers. The number of syncytial nuclei induced by authentic p15 exceeded countable levels after 8 h posttransfection. Results are the means ± SD from a single experiment done in triplicate.
To determine whether the reduced syncytiogenic activities of the threonine- and leucine-substituted constructs were paralleled by their pore formation activities, these constructs were analyzed using the dual-color pore formation assay. At 8 to 9 h posttransfection, when pore formation induced by authentic p15 was approaching the maximal levels that can be quantified by FACS analysis (Fig. 1C), the p15 constructs containing N-terminal TMD substitutions induced no detectable pore formation above background levels (data not shown), consistent with the absence of significant levels of syncytium formation at this time (Fig. 6C). Extended incubation (18 h posttransfection) of the p15 constructs containing single N-terminal substitutions (p15I21L or p1521T) resulted in detectable levels of pore formation that exceeded background levels (Fig. 6D), the extent of which mirrored the relative levels of syncytium formation (i.e., p15I21T induced slightly more pore formation and syncytiogenesis than p15I21L). The constructs containing double N-terminal substitutions (p15IV21/22T and p15IV21/22L), both of which were completely inactive for syncytium formation (data not shown), failed to induce pore formation that significantly exceeded background levels (Fig. 6D).
The terminal substitution results suggested that the functionality of the p15 TMD is dependent on N-terminal, hydrophobic, β-branched residues. To determine if there was any preference for a particular hydrophobic β-branched residue at either of these two locations, the Ile-Val sequence was converted to Ile-Ile or Val-Val (p15I21V and p15V22I, respectively). A kinetic analysis of syncytium formation indicated the syncytiogenic activity of p15I21V was indistinguishable from that of authentic p15, while the p15V22I construct displayed only a slight lag in the rate of syncytium formation (Fig. 6E). The fusion activity of p15 is therefore intimately associated with the presence of hydrophobic, β-branched residues at the N terminus of the TMD.
Nonfusogenic point substitutions in the TMD do not affect p15 clustering.
The TMDs of other viral fusion proteins such as influenza virus HA may self-associate to form a proteinaceous ring that restricts lipid flow at the fusion site. TMDs are also known to promote the clustering of fusion proteins at the fusion site via association with membrane lipid microdomains (8, 41, 48), and the p14 FAST protein has been shown to associate with detergent-resistant membranes (13). Based on the above considerations, it was conceivable that the loss of fusion activity resulting from p15 TMD substitutions could reflect disruption of p15 clustering at the fusion site, mediated either by disruption of potential TMD-TMD interactions or by alteration of TMD-dependent recruitment to detergent-resistant membrane microdomains. To examine whether p15 self-associates, due to either multimer formation or association with detergent-resistant membranes, authentic p15 was coexpressed with a p15 construct carrying two C-terminal HA tags separated by a flexible linker (p15HAC). Transfected cells were lysed at 4°C in either RIPA buffer or 0.5% Triton X-100, conditions known to either disrupt or maintain, respectively, p14 association with detergent-resistant membrane microdomains (13). The cell lysates were immunoprecipitated using an anti-HA monoclonal antibody, and the immune complexes were analyzed by SDS-PAGE and Western blotting using anti-p15 antiserum. The HA antibody precipitated p15 from cells expressing p15HAC and empty vector but not from cells cotransfected with untagged p15 and empty vector, confirming the specificity of the immunoprecipitation (Fig. 7 A, lanes 5 and 6 versus lanes 2 and 3). In cells coexpressing p15 and p15HAC, the HA antibody precipitated both the tagged and untagged versions of p15, which were easily distinguished by the gel mobility shift associated with the presence of the double HA epitope tag (Fig. 7A, lane 9). This coprecipitation occurred efficiently from cell lysates prepared with Triton X-100 but not from cell lysates disrupted with the more stringent RIPA detergent treatment (Fig. 7A, lane 9 versus lane 8). These results suggested that p15 polypeptides coassociate in cells. The disruption of this coassociation by RIPA buffer but not Triton X-100 suggests either that p15 clusters in detergent-resistant membranes or that p15 forms multimeric interactions that are subject to disruption by the more stringent detergent conditions.
Fig. 7.

TMD alterations that abrogate p15 fusion activity do not disrupt p15 clustering. (A) QM5 cells were cotransfected with untagged p15 and empty vector (v:p15), HA epitope-tagged p15 and empty vector (v:HA), or untagged and HA epitope-tagged p15 (p15:HA) and then lysed with either RIPA or Triton X-100 lysis buffer at 10 h posttransfection. The cell lysates were immunoprecipitated using mouse monoclonal anti-HA antibody. Lysates (lys) and RIPA (RIP) or Triton X-100 (TIP) immunoprecipitates were analyzed by SDS-PAGE and Western blotting with polyclonal anti-p15 antibody. The locations of the HA-tagged (p15HAC) and untagged (p15) are indicated on the right. A background band that migrated slightly slower than wild-type p15 is indicated (*). (B) The same experimental approach as outlined for panel A was used for immunoprecipitation with anti-HA monoclonal antibody of Triton X-100 cell lysates prepared from cells cotransfected with various combinations of the following plasmids: empty vector (vec); HA-tagged authentic p15 (HA); and untagged versions of the IV/L, S23/24A, G33A, and p15TM14 (TM14) p15 mutant constructs. Cells were doubly transfected in the indicated combinations (e.g., IV/L:vec cells were cotransfected with the p15IV/L construct and empty vector, while IV/L:HA cells were cotransfected with p15IV/L and HA-tagged p15). Lysates (L) and immunoprecipitates (IP) were analyzed by Western blotting using anti-p15 C-terminal antibody. The locations of the HA-tagged wild-type p15 (p15HAC) and the untagged mutant versions of the p15 constructs (p15mut) are indicated on the right.
To determine whether the loss of fusion activity due to p15 TMD substitutions was associated with disrupted clustering of p15, the coprecipitation assay was repeated with representatives of the nonfunctional p15 constructs. TMD substitutions that replaced N-terminal β-branched residues (p15IV21/22L), disrupted the triserine motif (p15S23/24A), or replaced a glycine residue (p15G33A) and the nonfunctional p15TM14 chimera were all tested. Cell lysates were prepared using the Triton X-100 conditions that maintained p15 coassociation. Appropriate controls, as described above, using cells cotransfected with each p15 construct and empty vector confirmed the specificity of the coimmunoprecipitations (Fig. 7B, lanes 1, 2, 3, 4, 7, 8, 11, 12, 15, and 16). Most importantly, in each case where the untagged p15 substitution construct was coexpressed with p15HAC, immunoprecipitation using the anti-HA antibody also coprecipitated the untagged p15 polypeptide containing the TMD substitution (Fig. 7B, lanes 6, 10, 14, and 18), including the p15TM14 construct containing the heterologous p14 TMD (Fig. 7B, lane 18). The loss of membrane fusion activity due to substitution or alteration of the p15 TMD is therefore unlikely to reflect altered p15 clustering at the fusion site.
DISCUSSION
The TMDs of many fusion proteins, both viral and cellular, have recently been identified as motifs that actively contribute to protein-mediated membrane fusion (25). While current hypotheses suggest a role for TMDs in the enhancement of pore formation and enlargement, little is known about the possible mechanisms that might be employed to facilitate such a task. Similarly to the p14 protein (10), and independent of its role as a signal-anchor or in protein trafficking, the p15 TMD plays a direct role in the fusion reaction, functioning at or prior to the formation of stable fusion pores. However, unlike the case for p14, the fusion function of the p15 TMD is highly sensitive to truncation or to amino acid substitutions of internal glycine residues, the triserine motif unique to the p15 TMD, or the N-terminal, hydrophobic, β-branched residues. These stringent sequence requirements of the p15 TMD are reflected by the inability of heterologous FAST protein TMDs to functionally replace the TMD of p15. Thus, while a funnel-shaped architecture may be the primary feature needed for a TMD to support p14-induced membrane fusion, such is not the case for p15. The p15 FAST protein is still, however, a modular fusogen as shown by the ability of the p15 TMD and endodomain to function in conjunction with the p14 ectodomain. The context-dependent modularity of the FAST proteins suggests that the different FAST proteins have assembled specific combinations of distinctive ecto-, endo-, and transmembrane domain features that function together in a coordinated manner to drive membrane merger.
Despite numerous differences in the repertoire and arrangement of structural motifs within members of the FAST protein family, a commonality is emerging in that the FAST protein TMDs play an active role in the fusion reaction. Mutations of the p15 TMD that result in a loss of fusion activity do not alter p15 membrane insertion, trafficking to the plasma membrane, or clustering into multimers and/or membrane microdomains, implying that the p15 TMD is directly involved in mediating membrane fusion. The direct correlation between the syncytiogenic and pore-forming activities of the various p15 constructs further implies that the TMD is required for stable pore formation. Whether this reflects a direct role for the p15 TMD in pore formation or in membrane merger events that precede pore formation is unclear, since assays and conditions that allow detection of hemifused structures induced by influenza virus HA are not applicable to the FAST proteins and/or are unable to detect a hemifusion intermediate (9). While no conclusive role for the TMDs of fusion proteins has emerged, several hypotheses converge upon a model in which elastic stresses generated by conformational rearrangements in the ectodomain are transferred to the membrane by the TMD, with the resulting membrane tensions serving to rupture the hemifusion diaphragm and drive pore formation (21, 28, 31). This model does not provide a role for the TMD in the hemifusion process, and it is not compatible with the FAST proteins, since their small ectodomains are unlikely to function using mechanical energy provided by dramatic ectodomain conformational changes to promote membrane tension and fusion.
While truncation studies of several enveloped virus fusion proteins converge on a minimum functional TMD length of just 16 to 18 residues, the p15 TMD was highly sensitive to N- and C-terminal truncations. For influenza virus HA, baculovirus gp64, and HIV gp41, truncation of these 23- to 27-residue TMDs to 16 to 18 residues did not eliminate their ability to mediate stable pore formation (1, 26, 43). The minimum TMD length requirement of the enveloped virus fusion proteins may reflect the need for a proteinaceous anchor that fully spans the membrane, providing sufficiently stable membrane anchoring to withstand and transmit the stresses arising from conformational rearrangements of the ectodomains (1, 25, 31). In contrast, deletion of just one N-terminal residue from the p15 TMD or four C-terminal residues abolished fusion activity. These results do not preclude the possibility that a p15 TMD shorter than 23 residues might still be functional, since the N-terminal truncations removed essential β-branched residues and the C-terminal truncation removed two aromatic residues that contribute to the funnel shape of the p15 TMD (10). Internal deletions of the p15 TMD might further address this length issue, as was done with the baculovirus gp64 protein (26). However, an alanine scan revealed no specific sequence requirements for the gp64 TMD (26), while the p15 TMD is highly sensitive to substitution at several locations, which complicates further truncation analysis to determine whether the p15 TMD has a minimal length requirement independent of specific sequence requirements.
The specific action of the TMD in the p15-mediated fusion reaction correlates with the presence of residues that typically destabilize α-helices. A comparison of TMD sequences from a variety of viral fusion proteins revealed a tendency for glycine or serine residues to cluster in the middle of a TMD, consistent with the hypothesis that helix flexibility may allow the TMD to withstand stresses during the membrane distortions that accompany pore formation (11). In contrast, the fusion-promoting function of the p15 TMD correlates with an essential role for N-proximal helix-destabilizing residues. The entire N-terminal half of the p15 TMD is composed of residues that are not generally considered to prefer an α-helical conformation. In addition to the N-proximal triserine motif, substitution of the TMD glycine residues also impaired p15 fusion activity, with substitution of the N-proximal glycine 27 residue exerting a more pronounced inhibitory effect on fusion activity than substitution of the centrally located glycine 33 residue (Fig. 5). Similarly, replacements of the N-terminal TMD isoleucine residue with alanine eliminated p15 fusion activity (Fig. 3), while replacements with a polar β-branched residue (threonine) or a hydrophobic non-β-branched residue (leucine) both dramatically impaired the rate of p15-induced syncytiogenesis and pore formation to approximately the same degree (Fig. 6). Combining these N-terminal substitutions with similar substitutions of the penultimate N-terminal valine residue eliminated p15 fusion activity. In contrast, p15 fusion activity was maintained following exchange of the N-terminal β-branched residues (i.e., Ile-Val was converted to Ile-Ile or Val-Val), indicating a specific requirement for hydrophobic, β-branched residues at the two N-terminal TMD positions. The p15 TMD is therefore highly sensitive to replacements of several N-proximal residues that could potentially alter the stability of a TMD helix. The predominance of helix-destabilizing residues in the p15 TMD may partially explain why other FAST protein TMDs are not functional in a p15 backbone, in that 65% of the p15 TMD is composed of β-branched, glycine, and serine residues while similar residues are present at only 32 to 37% in the p14 and p10 TMDs.
We are not aware of any other studies in the context of a fusion protein that directly implicate N-proximal β-branched residues in the fusion activity of these TMDs. For example, although the TMD of the parainfluenza virus 5 F protein contains an essential N-terminal isoleucine residue, substitution analysis correlated fusion activity with side chain hydrophobicity rather than β-branching (5). Peptide studies indicate that the fusion activity of a SNARE protein or the VSV G fusion protein TMDs correlates with their degree of TMD helix instability, with terminal β-branched residues being particularly pertinent for fusion (23, 24, 35, 37). Hydrogen/deuterium exchange studies with synthetic peptides suggest that TMD β-branched residues impart structural flexibility to α-helices, with a flexible backbone that fluctuates around an idealized conformation in the membrane correlating with the fusogenic activity of these TMD peptides (22, 34, 47). In addition to imparting a flexible structure to the N terminus of the TMD α-helix, which may partially disrupt lipid packing, the β-branched residues in the p15 TMD are located immediately adjacent to the triserine motif. The polar nature of the serine residues may serve to draw water into the outer leaflet to further destabilize the bilayer. A similar role was postulated for the serine residues in the HA TMD based on hydrogen/deuterium exchange studies of a TMD peptide, although the serines in the HA TMD are more centrally located and were solvent accessible due to formation of an oligomeric pore (51). Similar hydrogen/deuterium exchange studies conducted on the purified p15 FAST protein inserted into pure phospholipid bilayers could assess whether the combination of N-proximal helix instability and adjacent polar residues functions to partially hydrate the outer leaflet of the bilayer.
The p15 ectodomain contains only 19 residues and lacks the hydrophobic fusion peptide-like motifs present in the p10 and p14 ectodomains, although p15 contains a similar hydrophobic motif in its endodomain. A weakly fusogenic p15 ectodomain may require the TMD and endodomains to provide a greater contribution to the early lipid mixing stages of membrane fusion than is necessary for the p14 or p10 FAST proteins. In agreement with this prediction, the p15 TMD and endodomain function in concert with the p14 ectodomain, while a chimeric FAST protein containing the p15 TMD and ectodomain is nonfunctional (Fig. 2). We note that the former construct contains two hydrophobic patch motifs (present in the p14 ectodomain and p15 endodomain), while the latter, nonfunctional construct lacks such a motif in either the ecto- or endodomain. Similar chimeric studies of enveloped virus fusion proteins have yet to reveal how various domains function together to promote membrane merger. For example, the endodomain of influenza virus HA is dispensable for fusion activity but if present is functional only when the TMD and endodomain originate from the same protein (30), while the VSV G protein tolerates heterologous TMD and endodomains (33). There is also some suggestion that the TMD may directly interact with an ectodomain fusion peptide, as posited for influenza virus HA, where a glycine ridge on the fusion peptide may allow the peptide to slide down the side of the TMD, assisting in the formation of a fusion pore (49). This is unlikely to be the case for p15 given that the p15ect14 chimera is fully functional. The simplified, modular nature of the FAST proteins provides a suitable system to further define how different fusion modules interact to promote membrane fusion.
Based on the above considerations, we suggest a model in which the p15 TMD participates directly in both the initial and later steps of the fusion reaction. As with p14, reversible association of the p15 N-terminal myristoyl moiety with the donor membrane (Fig. 8 a) may alter lipid packing and/or the hydration layer between contacting bilayers, reducing hydration repulsion and making membrane merger a more favorable event (14). The structural flexibility conferred to the N-terminal region of the transmembrane helix by β-branched and glycine residues may be needed to further destabilize the donor membrane, as suggested by recent peptide studies showing that a dynamic TMD helix selectively promotes outer leaflet mixing (35). Interestingly, the addition of N-terminal acyl chains to TMD peptides stabilizes intrinsically dynamic helices (36). The essential N-terminal myristate moieties may confer a similar property on the p15 and p14 TMDs, stabilizing the helix until the close approach of a target membrane allows transfer of the myristate from the donor to the target membrane, triggering TMD helix instability and the onset of the fusion reaction. The region of the TMD that resides in the inner membrane leaflet is composed of bulky aromatic residues, while the region lying in the outer leaflet is composed primarily of residues with smaller side chains. Juxtaposition of the FAST protein TMDs due to ecto- or endodomain interactions through protein clustering would amplify the asymmetric distribution of the bulk of the TMD across the bilayer, promoting curvature of the donor membrane toward the target membrane (9, 10). Following close apposition of the target membrane, the concerted effects of the ectodomain and TMD on the lamellar structure of the outer leaflet would induce lipid mixing (Fig. 8b). The proximity of the p15 TMD triserine motif to the interface between the membrane and interstitial space may allow for hydrogen bonding of water molecules and/or lipid polar head groups, making a lipid emulsion a more energetically favorable intermediate during FAST protein-mediated membrane fusion. A disordered hemifusion intermediate was previously proposed for other viral and cellular fusogens (19, 50) and may be an attractive possibility for the FAST proteins, consistent with the inability of membrane curvature agents to detect this hemifusion state during p14-mediated cell-cell fusion (9). As with p14, the funnel shape of the p15 TMD could increase the positive curvature of the inner leaflet, promoting the direct transition from lipid mixing to fusion pore formation and completion of the membrane fusion reaction (Fig. 8c). This model integrates the disparate fusion activities of p14 and p15 TMD chimeras with the defined stringent sequence requirements of the p15 TMD and suggests that additional analysis of the FAST proteins may provide important insights into how these atypical, modular viral fusogens mediate membrane fusion.
Fig. 8.

Model for the role of the p15 TMD in membrane fusion. (a) The p15 ectodomain, which contains an N-terminal myristic acid (green squiggle) and a proline-rich region (blue oval), and the TMD (blue triangle) alter lipid packing and membrane hydration (water molecules shown as red and white spheres). The β-branched residues and triserine motif (blue coil) at the N terminus of the p15 TMD are predicted to induce helix instability, with the triserine motif acting as a hydrogen bond donor to draw water molecules further into the bilayer, increasing vertical fluctuation of lipid molecules and expanding the thickness of the interfacial region. Reversible donor membrane associations of the p15 ectodomain and the flexible helix structure of the N terminus of the TMD are depicted in blue and gray. The combination of these activities poises the donor membrane for interaction with a closely apposed target membrane. (b) Close approach of the target membrane triggers lipid mixing, which is depicted as a hemifused lipid emulsion rather than an ordered hourglass structure to reflect potential dynamic changes to the structure and hydration of the outer leaflet induced by the combined effects of the ectodomain and TMD described in panel a. The left side shows a side view of the p15 TMD modeled as an α-helix. The side chains are colored blue or green (aromatic residues), and the oxygens on polar side chains are colored red. The juxtaposition of three p15 TMDs due to homomeric p15 interactions (a trimer is shown merely for illustrative purposes) is represented with translucence on the right side. (c) The association of multiple p15 monomers at the fusion site is depicted (represented with translucence). The β-branched residues and triserine motif are omitted for simplicity. The funnel-shaped architecture of the p15 TMD is predicted to induce positive curvature of the inner leaflet, promoting the rapid transition from lipid mixing to pore formation.
ACKNOWLEDGMENTS
We thank Jingyun Shou for excellent technical assistance and Jolene Read for assistance with the mutant studies for Fig. 6E.
This work was supported by a grant to R.D. from the Canadian Institutes of Health Research (CIHR). E.K.C. was funded by scholarships from the Cancer Research Training Program (CRTP) with funding from the Dalhousie Cancer Research Program and the Nova Scotia Health Research Foundation (NSHRF).
Footnotes
Published ahead of print on 2 March 2011.
REFERENCES
- 1. Armstrong R. T., Kushnir A. S., White J. M. 2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 151:425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barry C., Duncan R. 2009. Multifaceted sequence-dependent and -independent roles for reovirus FAST protein cytoplasmic tails in fusion pore formation and syncytiogenesis. J. Virol. 83:12185–12195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barry C., Key T., Haddad R., Duncan R. 2010. Features of a spatially constrained cystine loop in the p10 FAST protein ectodomain define a new class of viral fusion peptides. J. Biol. Chem. 285:16424–16433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beltzer J. P., et al. 1991. Charged residues are major determinants of the transmembrane orientation of a signal-anchor sequence. J. Biol. Chem. 266:973–978 [PubMed] [Google Scholar]
- 5. Bissonnette M. L., Donald J. E., DeGrado W. F., Jardetzky T. S., Lamb R. A. 2009. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J. Mol. Biol. 386:14–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brosig B., Langosch D. 1998. The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci. 7:1052–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown C. W., et al. 2009. The p14 FAST protein of reptilian reovirus increases vesicular stomatitis virus neuropathogenesis. J. Virol. 83:552–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chazal N., Gerlier D. 2003. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 67:226–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clancy E. K., Barry C., Ciechonska M., Duncan R. 2010. Different activities of the reovirus FAST proteins and influenza hemagglutinin in cell-cell fusion assays and in response to membrane curvature agents. Virology 397:119–129 [DOI] [PubMed] [Google Scholar]
- 10. Clancy E. K., Duncan R. 2009. Reovirus FAST protein transmembrane domains function in a modular, primary sequence-independent manner to mediate cell-cell membrane fusion. J. Virol. 83:2941–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cleverley D. Z., Lenard J. 1998. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. U. S. A. 95:3425–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corcoran J. A., Duncan R. 2004. Reptilian reovirus utilizes a small type III protein with an external myristylated amino terminus to mediate cell-cell fusion. J. Virol. 78:4342–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Corcoran J. A., et al. 2006. The p14 fusion-associated small transmembrane (FAST) protein effects membrane fusion from a subset of membrane microdomains. J. Biol. Chem. 281:31778–31789 [DOI] [PubMed] [Google Scholar]
- 14. Corcoran J. A., et al. 2004. Myristoylation, a protruding loop, and structural plasticity are essential features of a nonenveloped virus fusion peptide motif. J. Biol. Chem. 279:51386–51394 [DOI] [PubMed] [Google Scholar]
- 15. Dawe S., Corcoran J. A., Clancy E. K., Salsman J., Duncan R. 2005. Unusual topological arrangement of structural motifs in the baboon reovirus fusion-associated small transmembrane protein. J. Virol. 79:6216–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dawe S., Duncan R. 2002. The S4 genome segment of baboon reovirus is bicistronic and encodes a novel fusion-associated small transmembrane protein. J. Virol. 76:2131–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duncan R., Corcoran J., Shou J., Stoltz D. 2004. Reptilian reovirus: a new fusogenic orthoreovirus species. Virology 319:131–140 [DOI] [PubMed] [Google Scholar]
- 18. Hofmann M. W., et al. 2004. De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 101:14776–14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jahn R., Grubmuller H. 2002. Membrane fusion. Curr. Opin. Cell Biol. 14:488–495 [DOI] [PubMed] [Google Scholar]
- 20. Kielian M., Rey F. A. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 4:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozerski C., Ponimaskin E., Schroth-Diez B., Schmidt M. F., Herrmann A. 2000. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74:7529–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langosch D., Arkin I. T. 2009. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 18:1343–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langosch D., Brosig B., Pipkorn R. 2001. Peptide mimics of the vesicular stomatitis virus G-protein transmembrane segment drive membrane fusion in vitro. J. Biol. Chem. 276:32016–32021 [DOI] [PubMed] [Google Scholar]
- 24. Langosch D., et al. 2001. Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J. Mol. Biol. 311:709–721 [DOI] [PubMed] [Google Scholar]
- 25. Langosch D., Hofmann M., Ungermann C. 2007. The role of transmembrane domains in membrane fusion. Cell Mol. Life Sci. 64:850–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Z., Blissard G. W. 2009. The Autographa californica multicapsid nucleopolyhedrovirus GP64 protein: analysis of transmembrane domain length and sequence requirements. J. Virol. 83:4447–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Z., Blissard G. W. 2008. Functional analysis of the transmembrane (TM) domain of the Autographa californica multicapsid nucleopolyhedrovirus GP64 protein: substitution of heterologous TM domains. J. Virol. 82:3329–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McNew J. A., et al. 2000. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150:105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melikyan G. B. 2008. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology 5:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melikyan G. B., Lin S., Roth M. G., Cohen F. S. 1999. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol. Biol. Cell 10:1821–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Melikyan G. B., White J. M., Cohen F. S. 1995. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 131:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyauchi K., et al. 2005. Role of the specific amino acid sequence of the membrane-spanning domain of human immunodeficiency virus type 1 in membrane fusion. J. Virol. 79:4720–4729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Odell D., Wanas E., Yan J., Ghosh H. P. 1997. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 71:7996–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ollesch J., et al. 2008. Secondary structure and distribution of fusogenic LV-peptides in lipid membranes. Eur. Biophys. J. 37:435–445 [DOI] [PubMed] [Google Scholar]
- 35. Poschner B. C., Fischer K., Herrmann J. R., Hofmann M. W., Langosch D. 2010. Structural features of fusogenic model transmembrane domains that differentially regulate inner and outer leaflet mixing in membrane fusion. Mol. Membr. Biol. 27:1–10 [DOI] [PubMed] [Google Scholar]
- 36. Poschner B. C., Langosch D. 2009. Stabilization of conformationally dynamic helices by covalently attached acyl chains. Protein Sci. 18:1801–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Poschner B. C., Quint S., Hofmann M. W., Langosch D. 2009. Sequence-specific conformational dynamics of model transmembrane domains determines their membrane fusogenic function. J. Mol. Biol. 386:733–741 [DOI] [PubMed] [Google Scholar]
- 38. Racine T., et al. 2009. Aquareovirus effects syncytiogenesis by using a novel member of the FAST protein family translated from a noncanonical translation start site. J. Virol. 83:5951–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salsman J., Top D., Barry C., Duncan R. 2008. A virus-encoded cell-cell fusion machine dependent on surrogate adhesins. PLoS Pathog. 4:e1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Salsman J., Top D., Boutilier J., Duncan R. 2005. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability. J. Virol. 79:8090–8100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheiffele P., Roth M. G., Simons K. 1997. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 16:5501–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schroth-Diez B., et al. 2000. The role of the transmembrane and of the intraviral domain of glycoproteins in membrane fusion of enveloped viruses. Biosci. Rep. 20:571–595 [DOI] [PubMed] [Google Scholar]
- 43. Shang L., Yue L., Hunter E. 2008. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 82:5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shmulevitz M., Duncan R. 2000. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 19:902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shmulevitz M., Epand R. F., Epand R. M., Duncan R. 2004. Structural and functional properties of an unusual internal fusion peptide in a nonenveloped virus membrane fusion protein. J. Virol. 78:2808–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shmulevitz M., Salsman J., Duncan R. 2003. Palmitoylation, membrane-proximal basic residues, and transmembrane glycine residues in the reovirus p10 protein are essential for syncytium formation. J. Virol. 77:9769–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stelzer W., Poschner B. C., Stalz H., Heck A. J., Langosch D. 2008. Sequence-specific conformational flexibility of SNARE transmembrane helices probed by hydrogen/deuterium exchange. Biophys. J. 95:1326–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takeda M., Leser G. P., Russell C. J., Lamb R. A. 2003. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. U. S. A. 100:14610–14617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tamm L. K. 2003. Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim. Biophys. Acta 1614:14–23 [DOI] [PubMed] [Google Scholar]
- 50. Tamm L. K., Crane J., Kiessling V. 2003. Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr. Opin. Struct. Biol. 13:453–466 [DOI] [PubMed] [Google Scholar]
- 51. Tatulian S. A., Tamm L. K. 2000. Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. Biochemistry 39:496–507 [DOI] [PubMed] [Google Scholar]
- 52. Top D., Barry C., Racine T., Ellis C. L., Duncan R. 2009. Enhanced fusion pore expansion mediated by the trans-acting endodomain of the reovirus FAST proteins. PLoS Pathog. 5:e1000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Top D., et al. 2005. Liposome reconstitution of a minimal protein-mediated membrane fusion machine. EMBO J. 24:2980–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vincent M. J., Raja N. U., Jabbar M. A. 1993. Human immunodeficiency virus type 1 Vpu protein induces degradation of chimeric envelope glycoproteins bearing the cytoplasmic and anchor domains of CD4: role of the cytoplasmic domain in Vpu-induced degradation in the endoplasmic reticulum. J. Virol. 67:5538–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. von Heijne G., Gavel Y. 1988. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 174:671–678 [DOI] [PubMed] [Google Scholar]
- 56. Wahlberg J. M., Spiess M. 1997. Multiple determinants direct the orientation of signal-anchor proteins: the topogenic role of the hydrophobic signal domain. J. Cell Biol. 137:555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. White J. M., Delos S. E., Brecher M., Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu Y., Zhang F., Su Z., McNew J. A., Shin Y. K. 2005. Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 12:417–422 [DOI] [PubMed] [Google Scholar]