

Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), caused by influenza A virus H5N1 and severe acute respiratory syndrome coronavirus (SARS-CoV), supposedly depend on activation of the oxidative-stress machinery that is coupled with innate immunity, resulting in a strong proinflammatory host response. Inflammatory cytokines, such as interleukin 1β (IL-1β), IL-8, and IL-6, play a major role in mediating and amplifying ALI/ARDS by stimulating chemotaxis and activation of neutrophils. To obtain further insight into the pathogenesis of SARS-CoV-associated ALI, we compared SARS-CoV infections in two different nonhuman primate species, cynomolgus macaques and African green monkeys. Viral titers in the upper and lower respiratory tract were not significantly different in SARS-CoV-infected macaques and African green monkeys. Inflammatory cytokines that play a major role in mediating and amplifying ALI/ARDS or have neutrophil chemoattractant activity, such as IL-6, IL-8, CXCL1, and CXCL2, were, however, induced only in macaques. In contrast, other proinflammatory cytokines and chemokines, including osteopontin and CCL3, were upregulated in the lungs of African green monkeys to a significantly greater extent than in macaques. Because African green monkeys developed more severe ALI than macaques, with hyaline membrane formation, some of these differentially expressed proinflammatory genes may be critically involved in development of the observed pathological changes. Induction of distinct proinflammatory genes after SARS-CoV infection in different nonhuman primate species needs to be taken into account when analyzing outcomes of intervention strategies in these species.
INTRODUCTION
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), major causes of respiratory failure with high morbidity and mortality, are observed under multiple pathogenic conditions, among which are sepsis, gastric acid aspiration, and infections with influenza A virus H5N1 and severe acute respiratory syndrome coronavirus (SARS-CoV) (21, 55). Increased permeability of the alveolar-capillary barrier, resulting in pulmonary edema, hypoxia, and influx of neutrophils observed under these conditions, may be followed by a fibroproliferative phase with alveolar hyaline membranes and various degrees of interstitial fibrosis that may ultimately progress or resolve (52). The onset of severe lung injury supposedly depends on activation of the oxidative-stress machinery that is coupled with innate immunity and activates transcription factors, such as NF-κB, resulting in a proinflammatory host response (21). As similar ARDS symptoms, triggered by different stimuli, can be observed in multiple species, a common ARDS “injury pathway” has been proposed (21). Inflammatory cytokines, such as interleukin 1β (IL-1β), IL-8, and IL-6, play a major role in this pathway, mediating and amplifying ALI/ARDS by stimulating chemotaxis and activation of neutrophils (52, 55).
SARS-CoV infection in humans causes lower respiratory tract disease, and 20 to 30% of patients develop severe inflammation of the lung, characterized by ARDS (36, 37). Children younger than 12 years of age are relatively unaffected by the disease (18, 28, 57), whereas elderly patients have a poor prognosis, with mortality rates of up to ∼50% (36, 37). The pulmonary damage is thought to be caused by a disproportionate immune response, with elevated levels of cytokines, such as CXCL10, CCL2, IL-6, IL-8, IL-1β, and gamma interferon (IFN-γ) (3, 19, 24, 41, 51, 56, 58).
Although a wide range of animals can be infected with SARS-CoV, among them rodents (mice and hamsters), carnivores (ferrets and cats), and nonhuman primates (macaques and African green monkeys), none of the current animal models has fully reproduced all features of SARS (16, 49). SARS-CoV-infected adult mice show no clinical signs of disease, although viral titers peak early after infection and reach relatively high levels in the respiratory tract, and the infection results in mild inflammatory changes in the respiratory tract. Aged mice and mice infected with certain adapted SARS-CoV variants do show signs of clinical disease and significant pathological changes in the lungs, which correlate with higher virus titers and/or a stronger host response, with differential expression of IL-6, CCL3, CXCL10, IL-1β, and tumor necrosis factor alpha (TNF-α) (42–44, 49). SARS-CoV infection of nonhuman primates results in multiple foci of diffuse alveolar damage, with flooding of the alveoli with edema fluid and influx of macrophages, neutrophils, and lymphocytes and extensive loss of epithelium from alveolar and bronchial walls (15, 16, 27). Analysis of gene signatures in SARS-CoV-infected macaques revealed induction of a strong innate immune response characterized by the induction of antiviral signaling pathways and the stimulation of various proinflammatory cytokine genes, including those for IL-1β, CCL2, IL-6, IL-8, and CXCL10 (7, 46). Aged macaques develop more severe pathology upon SARS-CoV infection than young adult macaques, which could be explained only by stronger innate host responses with an increase in differential expression of genes associated with inflammation, including IL-8 (46). These data are in line with previous observations in ALI/ARDS caused by multiple nonviral pathogenic conditions (52, 55).
Despite intensive studies of the pathogenesis of SARS-CoV, the molecular mechanisms of virus-induced acute lung injury are still largely uncharacterized. Age is a predisposing factor for severe SARS-CoV-associated disease, and young adults show only mild symptoms and pathology upon infection. Previous studies in SARS-CoV-infected young adult African green monkeys revealed a more severe course of the infection than in young adult macaques (32). To determine which pathogenic pathways operate in these two related nonhuman primate host species upon SARS-CoV infection, comparative host genomic analyses of SARS-CoV-infected young adult African green monkeys and macaques were performed. The observed commonalities and differences provide new insights into SARS-CoV pathogenesis and virus-induced acute lung injury.
MATERIALS AND METHODS
Animal studies.
Approval for animal experiments was obtained from the Institutional Animal Welfare Committee. Four cynomolgus macaques (Macaca fascicularis) and four African green monkeys (Chlorocebus aethiops), 3 to 5 years old with active temperature transponders in the peritoneal cavity, were inoculated with 5 × 106 50% tissue culture infective doses (TCID50) of SARS-CoV strain HKU39849 suspended in 5 ml phosphate-buffered saline (PBS). Four milliliters was applied intratracheally, 0.5 ml intranasally, and 0.25 ml on each conjunctiva, as described previously (7, 12, 15, 27, 46). The four SARS-CoV-infected young adult cynomolgus macaques have been described previously (46). For reverse transcription (RT)-PCR and immunohistochemistry, data from previously SARS-CoV-infected aged cynomolgus macaques were used (46). Two young adult macaques and African green monkeys (age range, 3 to 9 years old) were mock (PBS) infected. The animals were checked daily for clinical signs. Just before infection and at days 2 and 4 postinfection, animals were anesthetized with ketamine, and oral, nasal, and rectal swabs were taken and placed in 1 ml Dulbecco's modified Eagle's medium supplemented with 100 IU penicillin/ml and 100 μg of streptomycin/ml (virus transport medium). The swabs were frozen at −70°C until RT-PCR analysis was performed. The animals were euthanized 4 days after infection by exsanguination under ketamine anesthesia.
Necropsies were performed according to a standard protocol. For semiquantitative assessment of gross pathology, the percentage of affected lung tissue from each lung lobe was determined at necropsy and recorded on a schematic diagram of the lung, and the area of affected lung tissue was subsequently calculated (gross pathology score). One lung from each monkey was inflated with 10% neutral-buffered formalin by intrabronchial intubation and suspended in 10% neutral-buffered formalin overnight. Samples were collected in a standard manner (from the cranial, medial, and caudal parts of the lung), embedded in paraffin, cut at 3 μm, and used for titration (see below) or immunohistochemistry (see below) or stained with hematoxylin and eosin (H&E). The lung, liver, spleen, kidney, intestine, trachea, and tracheobronchial lymph node H&E sections were examined by light microscopy.
For semiquantitative assessment of SARS CoV infection-associated inflammation in the lung, two H&E-stained slides per animal (cranial and caudal lobes) were examined for inflammatory foci by light microscopy using a 10× objective. Each focus was scored for size (1, smaller than or equal to the area of the 10× objective; 2, larger than the area of the 10× objective and smaller than or equal to the area of the 2.5× objective; 3, larger than the area of the 2.5× objective) and degree of inflammation (1, low density of inflammatory cells; 2, intermediate density of inflammatory cells; 3, high density of inflammatory cells). The cumulative scores for the inflammatory foci provided the total score per animal. Sections were examined without knowledge of the identities of the animals.
Three lung tissue samples (one from the cranial part of the lung, one from the medial part, and one from the caudal part) were homogenized in 2 ml virus transport medium, using Polytron PT2100 tissue grinders (Kinematica). After low-speed centrifugation, the homogenates were frozen at −70°C until they were inoculated on Vero E6 cell cultures in 10-fold serial dilutions. The identity of the isolated virus was confirmed as SARS-CoV by RT-PCR of the supernatant (15).
Immunohistochemistry.
Serial 3-μm lung sections were stained using mouse anti-SARS nucleocapsid IgG2a (clone Ncap4; Imgenex; 1:1,600), mouse anti-osteopontin (clone AKm2A1; Santa Cruz), rabbit anti-ACE2 (angiotensin 1-converting enzyme 2) (ab15348; Abcam), or isotype control antibodies (clones 11711 and 20102; R&D) according to standard protocols (15, 27). Quantitative assessment of SARS-CoV antigen expression in the lungs was performed as described previously (15). For phenotyping (to confirm cell types), we used a destaining-restaining technique (8). Briefly, the red precipitate (aminoethylcarbazole [AEC]) used for visualization of osteopontin antigen staining was dissolved in graded 100% to 70% alcohols. To detach the primary antibody and red immunohistochemistry signals, slides were soaked in eluting buffer (5 ml 0.1 M HCl, 95 ml 0.1 M NaCl containing 1 M glycine) for 2 h. The sections were treated for two 5-min intervals by heating them in citric acid buffer, pH 6.0, to denature any undetached primary antibody. The slides were then incubated with mouse anti-human CD68 (clone KP1; Dako) 1/200 in PBS-0.1% bovine serum albumin (BSA) for 1 h at room temperature. After being washed, the sections were incubated with horseradish peroxidase-labeled goat-anti-mouse IgG1 (Southern Biotech) 1/100 in PBS-0.1% BSA for 1 h at room temperature. Peroxidase activity was revealed by incubating the slides in 3,3′-diaminobenzidine-tetrachlorhydrate (DAB) (Sigma) for 3 to 5 min, resulting in a brown precipitate, followed by counterstaining with hematoxylin.
RNA extraction and quantitative RT-PCR.
RNA from 200 μl of swabs was isolated with the Magnapure LC total nucleic acid isolation kit (Roche) external lysis protocol and eluted in 100 μl. SARS-CoV RNA was quantified on the ABI prism 7700, with use of the TaqMan Reverse Transcription Reagents and TaqMan PCR Core Reagent kit (Applied Biosystems), using 20 μl isolated RNA, 1× TaqMan buffer, 5.5 mM MgCl2, 1.2 mM deoxynucleoside triphosphates (dNTPs), 0.25 U AmpliTaq gold DNA polymerase, 0.25 U Multiscribe reverse transcriptase, 0.4 U RNase inhibitor with 200 nM primers and 100 nM probe specific for the SARS-CoV nucleocapsid protein gene (7, 46). The amplification parameters were 30 min at 48°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. RNA dilutions isolated from a SARS-CoV stock were used as a standard. Average results (± standard errors of the mean [SEM]) for macaque (n = 4) and African green monkey (n = 4) groups were expressed as SARS-CoV equivalents per ml swab medium.
Lung tissue samples (0.3 to 0.5 g) were taken for RT-PCR and microarray analysis in RNA-later (Ambion, Inc.). RNA was isolated from homogenized postmortem tissue samples using Trizol reagent (Invitrogen) and the RNeasy minikit (Qiagen). cDNA synthesis was performed with ∼1 μg total RNA and Superscript III reverse transcriptase (Invitrogen) with oligo(dT), according to the manufacturer's instructions. Semiquantitative RT-PCR was performed as described previously to detect SARS-CoV mRNA and to validate cellular gene expression changes as detected with microarrays of SPP1, CCL3, and CCL20 (Applied Biosystems; Hs00167093_m1, Rh02788104_gH, and Rh02788116_m1) and IL-8, IL-6, and IFN-β (7). Differences in gene expression are represented as the fold change in gene expression relative to a calibrator and normalized to a reference, using the 2−ΔΔCt method (29). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an endogenous control to normalize quantification of the target gene. The samples from the mock-infected macaques or African green monkeys were used as a calibrator. Average results (±SEM) for groups were expressed as the fold change compared to PBS-infected animals (29).
Statistical analysis.
Data (RT-PCR, gross pathology, and histology scores for SARS-CoV-infected animals) were compared using Student's t test. Differences were considered significant at a P value of <0.05.
RNA labeling, microarray hybridization, scanning, and data preprocessing.
Pooled total RNA (2.4 μg) from one to three separate lung pieces per animal, with substantial SARS-CoV replication (>105-fold change), was labeled using the One-Cycle Target Labeling Assay (Affymetrix) and hybridized onto Affymetrix GeneChip Rhesus Macaque Genome Arrays (Affymetrix) according to the manufacturer's recommendations. Image analysis was performed using Gene Chip Operating Software (Affymetrix). Microarray Suite version 5.0 software (Affymetrix) was used to generate .dat and .cel files for each experiment. The data were normalized using a variance stabilization algorithm (VSN) (20). Transformed probe values were summarized into one value per probe set by the median polish method (53). Primary data are available at http://www.virgo.nl in accordance with proposed minimum information about a microarray experiment (MIAME) standards.
Microarray data analysis.
Principal-component analysis (PCA) was performed on a single normalized data set, including all African green monkey and cynomolgus macaque samples. For PCA, 1,147 probe sets were included that deviated ≥1.5-fold from the geometrical mean (absolute values) for at least one of the samples. Differentially expressed mRNA transcripts were identified using LIMMA (version 2.12.0) (47). Correction for multiple testing was achieved by requiring a false-discovery rate (FDR) of 0.05, calculated with the Benjamini-Hochberg procedure (1). To understand the gene functions and the biological processes represented in the data and to obtain differentially expressed molecular and cellular functions, Ingenuity Pathways Knowledge Base (Ingenuity Systems) was used. Heat maps of proinflammatory pathways were generated using complete linkage and Euclidian distance in Spotfire DecisionSite for Functional Genomics version 9.1 (Spotfire; Tibco) and Ingenuity Pathways Knowledge Base (Ingenuity Systems), using log (base 2)-transformed expression values, with minimum and maximum values of the color range of −4 and 4, respectively.
RESULTS
Clinical signs and gross pathology in SARS-CoV-infected African green monkeys.
Cynomolgus macaques develop pathology upon SARS-CoV infection, and the severity is associated with aging (46). In this study, we compared SARS-CoV infection in two related nonhuman primate host species, young adult cynomolgus macaques and African green monkeys, focusing on the latter. Four young adult African green monkeys were infected with SARS-CoV HKU39849, and as a control, two were PBS infected. During the 4-day experiment, all animals remained free of clinical symptoms. Their body temperatures, measured by transponders in the peritoneal cavity, demonstrated a rhythmic pattern, with temperatures fluctuating between 35°C at night and 39°C during the day, prior to infection (Fig. 1A). Between days 1 and 2 after SARS-CoV infection, an increase in body temperature was recorded during the night in all animals. In addition, some African green monkeys showed an increase in body temperature up to 40°C during the first 2 days after SARS-CoV infection (Fig. 1A). Even at days 3 and 4 postinfection, elevated temperatures were still observed in some African green monkeys. At gross necropsy, 4 days postinfection, the lungs of African green monkeys showed (multi)focal pulmonary consolidation (Fig. 1B and C). The consolidated lung tissue was gray-red, firm, level, and less buoyant than normal. Strikingly, one animal had severe lesions, with up to 30% of total lung tissue affected (Fig. 1B). Four young adult cynomolgus macaques infected with SARS-CoV HKU39849 and two PBS-infected cynomolgus macaques were used for comparative analyses (46). In contrast to infected African green monkeys, no increase in body temperature of up to 40°C during the first 2 days after SARS-CoV infection was observed in macaques (46). Between days 1 and 2 after SARS-CoV infection, an increase in body temperature was recorded during the night in three out of four animals (46). Temperatures returned to normal from day 3 postinfection onward (46). The lungs of young adult macaques showed small, patchy, macroscopic lesions (Fig. 1C) (46).
Fig. 1.
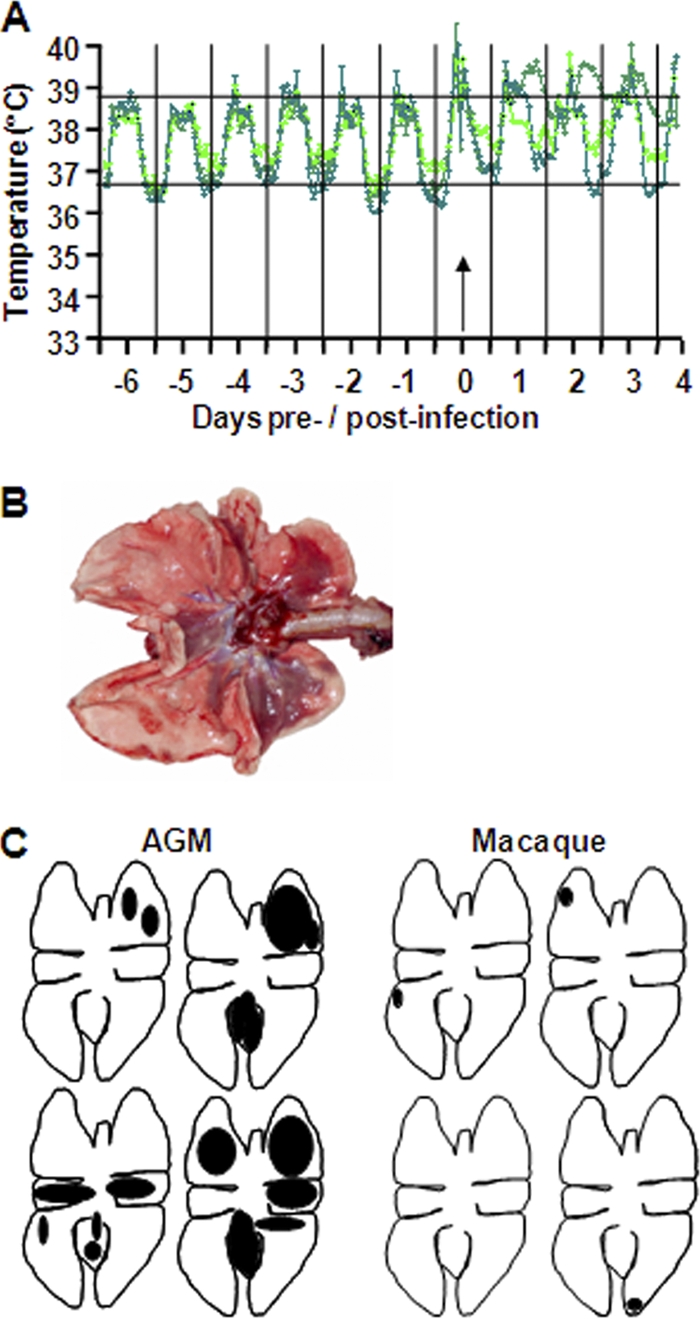
African green monkeys are more prone to develop SARS-CoV-associated disease than young adult macaques. (A) Fluctuations in body temperatures measured by transponders in the peritoneal cavity in three out of four SARS-CoV-infected African green monkeys. Temperatures are shown from day 6 prior to infection until 4 days postinfection. The arrow indicates day zero, when the animals were infected. The black horizontal lines mark the average range of temperature fluctuations prior to infection. (B) Macroscopic appearance of lung tissue of SARS-CoV-infected African green monkeys at day 4 postinfection, with dark-red consolidation. (C) Schematic diagrams of the lungs showing gross pathology lesions of SARS-CoV-infected young adult African green monkeys (AGM) and macaques.
Histopathology in SARS-CoV-infected African green monkeys.
The lesions in the lungs of African green monkeys consisted of acute exudative diffuse alveolar damage characterized by thickened alveolar septa with infiltration of moderate numbers of neutrophils and macrophages, lymphocytes, and plasma cells, and necrosis with karyolysis, karyorrhexis, and loss of cellular detail (Fig. 2A). Multifocally, moderate hypertrophy and hyperplasia of type II pneumocytes was observed (Fig. 2A and B). A variable amount of eosinophilic proteinaceous fluid (edema and exudate) with fibrin and eosinophilic hyaline membrane formation (Fig. 2A and B), few syncytia, and moderate numbers of foamy alveolar macrophages, neutrophils, and erythrocytes (hemorrhage) was seen in alveolar lumina. Mild necrosis, hypertrophy, and hyperplasia of bronchiolar epithelium were encountered (Fig. 2A). Peribronchiolar, peribronchial, and perivascular infiltrations of moderate numbers of lymphocytes, plasma cells, macrophages, and neutrophils with mild to moderate edema were seen (Fig. 2A). There was a mild to moderate proliferation of the bronchus-associated lymphoid tissue (BALT), with focal nodular lymphocytic proliferation and formation of follicles with additional exocytosis of neutrophils in the tracheal epithelium (Fig. 2A). A mild multifocal lymphoplasmacytic tracheobronchoadenitis, characterized by a multifocal mild infiltration of lymphocytes, macrophages, plasma cells, and neutrophils in and surrounding the bronchial seromucous glands with mild necrosis of the epithelium, was observed in all African green monkeys (Fig. 2A). Two animals showed evidence of multifocal mild lymphoplasmacytic pleuritis consisting of a mild multifocal distention of the pleura with infiltration of a few lymphocytes, plasma cells, macrophages, and neutrophils and mild edema. The lymphoid organs were activated, with proliferation of the tracheobronchial lymph node and spleen, characterized by increased numbers of neutrophils and histiocytes and distinct germinal centers in the lymphoid follicles. No significant lesions were seen in other tissues examined or in the tissues of negative-control animals. The histopathological lesions in the respiratory tracts of SARS-CoV-infected cynomolgus macaques were largely similar in character to those described for the African green monkeys (46). However, hyaline membrane formation was not observed, and the lesions in macaques were significantly less severe and extensive than those in African green monkeys (Fig. 3A), which corroborated the gross pathology scores (Fig. 3B).
Fig. 2.

Histological analyses of lungs from SARS-CoV-infected African green monkeys. (A) Alveoli of PBS-infected (right) and SARS-CoV-infected (left) African green monkeys showing diffuse alveolar damage, characterized by distension of alveolar walls with influx of inflammatory cells, necrosis, type II pneumocyte hyperplasia, intra-alveolar edema, and hyaline membrane formation. The bronchioles of PBS-infected (right) and SARS-CoV-infected (left) African green monkeys show infiltration of inflammatory cells, intraluminal edema fluid, and hypertrophy and hyperplasia of bronchiolar epithelium. The tracheas of PBS-infected (right) and SARS-CoV-infected (left) African green monkeys show multifocal lymphoplasmacytic tracheobronchoadenitis with infiltration of inflammatory cells surrounding the bronchial seromucous glands. (B) Lesions of SARS-CoV-infected African green monkeys are shown in more detail, with hyaline membrane formation (left) and type II pneumocyte hyperplasia (right). The sections were stained with H&E. Original magnifications, ×20 and ×40.
Fig. 3.
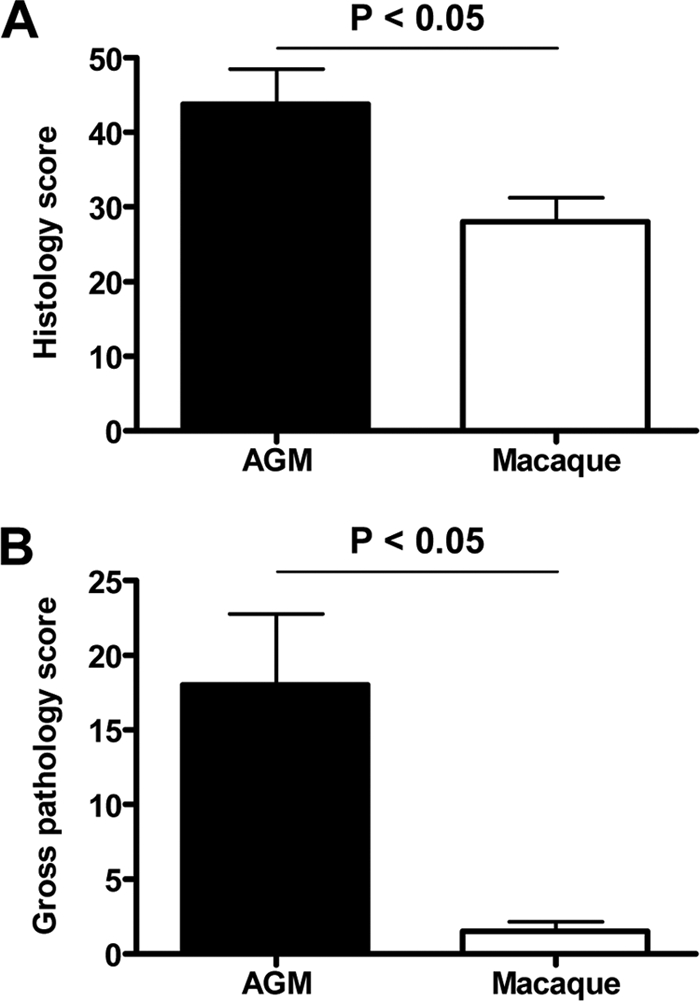
African green monkeys display more severe pathology than macaques. (A) Histology scores of the lungs of African green monkey (AGM) and macaque groups were determined and averaged (plus SEM). (B) Gross pathology scores of AGM and macaque groups were determined by visual inspection of the lungs during necropsy and averaged (plus SEM).
Virus replication and tropism in SARS-CoV-infected African green monkeys.
SARS-CoV mRNA levels were detected in throat (Fig. 4A) and nasal (Fig. 4B) swabs from African green monkeys on days 2 and 4 postinfection and in rectal swabs on day 4 postinfection (data not shown). In addition, virus replication in the lungs was demonstrated by RT-PCR and titration, and immunohistochemistry revealed SARS-CoV antigen expression in the lungs of African green monkeys within or directly adjacent to areas with lesions (Fig. 4C and D). In alveoli of African green monkeys, cells that resembled type I and type II pneumocytes and rarely alveolar macrophages expressed SARS-CoV antigen (Fig. 5A). SARS-CoV antigen was not detected in bronchioles or bronchi but was observed in ciliated epithelial cells in the tracheas of two African green monkeys (Fig. 5A). The differences in the percentages of SARS-CoV-infected cells in the lungs or virus titers in the throat, noses, and lungs of African green monkeys and macaques were not statistically significant (Fig. 4). SARS-CoV, however, was not detected in ciliated epithelial cells in the trachea on day 4 postinfection in young adult macaques (Fig. 5) or in rectal swabs. In both PBS-infected African green monkeys and macaques, we could show expression of ACE2, which is the receptor for SARS-CoV, in cells morphologically resembling type II epithelial cells (Fig. 5B). Significant differences between ACE2 expression levels in African green monkeys and macaques could not be observed. Overall, African green monkeys displayed more severe pathology than macaques upon infection with SARS-CoV, which correlated with a broader range of cell types infected at day 4 postinfection, but not with the extent of virus replication.
Fig. 4.

Viral replication levels in the respiratory tracts of SARS-CoV-infected African green monkeys and macaques. (A and B) SARS-CoV replication in the throats (A) and noses (B) of SARS-CoV-infected African green monkeys (black bars) and macaques (white bars) at days 2 and 4 postinfection as determined by real-time RT-PCR. Viral-RNA levels are displayed as TCID50 equivalents (eq.)/ml swab medium (plus SEM). (C) Average fold change in SARS-CoV mRNA levels (plus SEM) in the lungs of African green monkeys and macaques compared to PBS-infected animals as determined by real-time RT-PCR and depicted on a log scale. In addition, SARS-CoV titration of lung homogenates is shown in TCID50 per gram lung tissue. (D) Quantitative assessment of SARS-CoV-infected cells in the lungs of African green monkeys and macaques depicted as a percentage of SARS-CoV-positive fields (plus SEM).
Fig. 5.

Lungs of African green monkeys and cynomolgus macaques showing SARS-CoV (A) and ACE2 (B) antigen expression in the alveoli and/or trachea from SARS-CoV and PBS-infected animals, respectively. Sections were stained with immunoperoxidase and counterstained with hematoxylin. Original magnifications, ×20.
Host response to SARS-CoV in African green monkeys and macaques.
To understand why SARS-CoV infection in African green monkeys results in more severe pathology than in young adult macaques, we generated genome-wide mRNA expression profiles from lung tissue with substantial SARS-CoV replication (Fig. 6A). Principal-component analysis was performed on a single normalized data set of African green monkey samples, together with macaque samples. This unsupervised method clearly separated SARS-CoV-infected lung mRNA expression profiles from PBS control expression profiles for both species. In addition, the infected and uninfected expression profiles of the two species also separated well. Species- and infection-related expression profile differences coincided with principal components 1 and 2, respectively (Fig. 6B). These results show that species-associated mRNA expression profile differences are maintained upon SARS-CoV infection, suggesting that African green monkeys and macaques respond differently to SARS-CoV infection.
Fig. 6.
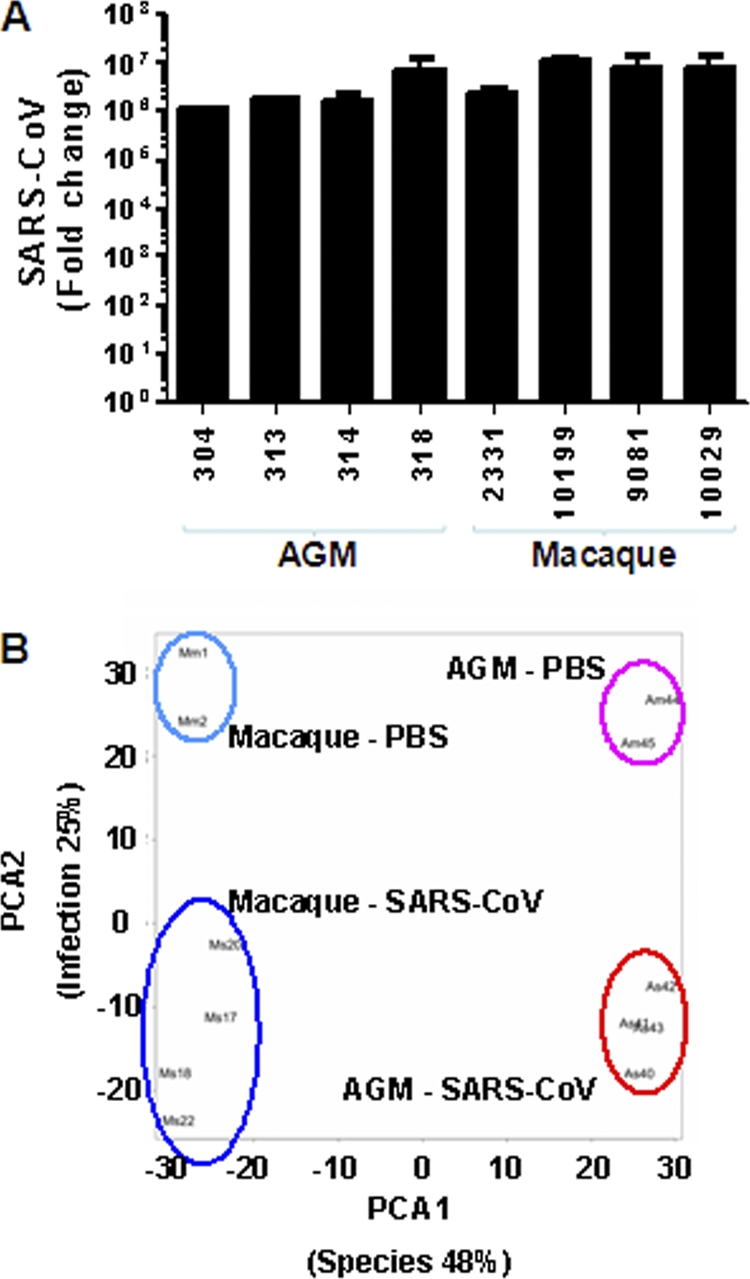
Global gene expression profiles of individual African green monkeys and macaques. (A) Average fold change in SARS-CoV mRNA levels (plus SEM) in the lungs of African green monkeys and macaques compared to PBS-infected animals as determined by real-time RT-PCR and depicted on a log scale. (B) PCA of transcriptional-profiling data. Each combination of letters and numbers (Am44, Am45, Mm1, Mm2, Am40, Am41, Am42, Am43, Mm17, Mm18, Mm20, and Mm22) represents an individual PBS- or SARS-CoV-infected animal plotted in two dimensions using their projections onto the first two principal components, species and infection. The colored ovals represent the groups of PBS-infected and SARS-CoV-infected African green monkeys and macaques. The first two principal components account for 48% and 25% of variation in the data, respectively.
To analyze the host response to infection in African green monkeys and macaques in more detail, mRNA transcripts differentially expressed between infected and uninfected lung samples were identified using the analysis of variance (ANOVA)-like method LIMMA (47) for both species separately. To obtain a maximum view of infection-induced host response differences between African green monkeys and macaques, the microarray data for the two species were normalized separately. Infected African green monkeys expressed 556 gene transcripts differently from PBS-infected animals. These differently expressed genes were analyzed within the context of molecular pathways, using a functional-analysis approach with biologically related genes (Ingenuity). The most significantly regulated molecular/cellular functions in African green monkeys compared to PBS-infected animals were associated with a proinflammatory response and included cellular growth and proliferation, cell death, cell movement, and cell-to-cell signaling (Fig. 7A). Upon analysis of the gene transcripts within the context of biological pathways using Ingenuity Knowledge Base, the top gene interaction network in African green monkeys showed a strong antiviral response with differentially expressed type I interferon-stimulated genes and genes associated with the induction of type I interferons (Fig. 7B), including ISG15, ISG20, DDX58, DHX58, IFI/IFITs, MX1, MX2, IRF7, IRF9, OAS1, and OAS2. In addition, the network contained NF-κB (Fig. 7B), a transcription factor implicated in proinflammatory host responses and development of ALI/ARDS (10, 17, 21). Differential expression of genes associated with acute lung injury, inflammation, and/or hypoxia signaling in African green monkeys included C3AR1, CEBPD, CCL2, CCL3, IL1RN, TIMP1, CDKN1A, SPARC, VEGFA, SPP1, CSF1, CSF3R, CD86, KIT, CCL8, CXCL10, PML, SP100, and SOD2 genes. Some of these genes are also upregulated in SARS patient sera or in patients with ARDS (51, 55). Young adult SARS-CoV-infected macaques expressed 475 gene transcripts differently from PBS-infected animals. These genes were associated with the same molecular/cellular functions observed in African green monkeys, with similar numbers of differentially expressed genes per function (Fig. 7A). Analysis of the gene transcripts in the context of biological pathways revealed differential expression of genes in both antiviral and proinflammatory responses (Fig. 7C). These data suggest that African green monkeys and macaques display similar types of responses upon infection, with strong induction of antiviral and proinflammatory pathways, which has been described for SARS-CoV-infected cynomolgus macaques previously but based on regulation of a different array of individual genes within the antiviral and proinflammatory pathways (7, 46).
Fig. 7.

Innate host response to SARS-CoV infection in African green monkeys. (A) Numbers of differentially expressed genes in African green monkeys and macaques with functions in, for example, cellular growth and proliferation, cell movement, cell death, or cell-to-cell signaling and interaction obtained from the Ingenuity Pathways Knowledge Base compared to those in their respective PBS-infected animals. (B) African green monkeys present a “top” network, with genes involved in the antiviral response and proinflammatory response. A network is a group of biologically related genes that is derived from known relationships present in the Ingenuity Pathways Knowledge Base. The diagram represents the interactions, both direct (solid lines) and indirect (dashed lines), between differentially expressed genes and gene products identified on day 4 postinfection in African green monkeys. Red indicates upregulated genes, whereas downregulated genes are depicted in green. (C) Macaques also present a “top” network, with genes involved in the antiviral response and proinflammatory response. The diagram represents the interactions, both direct (solid lines) and indirect (dashed lines), between differentially expressed genes and gene products identified on day 4 postinfection in macaques. Red indicates upregulated genes, whereas downregulated genes are depicted in green.
As the principal-component analysis suggested that the host response to SARS-CoV infection was different in African green monkeys and macaques, the gene expression profiles of SARS-CoV-infected African green monkeys and macaques (n = 4) were directly compared using LIMMA (8 samples normalized in a single set). A total of 1,607 gene transcripts were differentially expressed. Upon analysis of these 1,607 gene transcripts within the context of biological processes and pathways using Ingenuity Pathways Knowledge Base, this subset of genes showed indications of an innate host response to viral infection. Among the top significantly differentially regulated (P < 0.005) functional categories were inflammatory disease, cell movement, and cell-to-cell signaling and interaction, which included genes like those for CCL20, CXCL1, CXCL2, DEFB1, IL1RL1, IL1RN, MMP7, MMP9, IL-8, SERPINE1, SPP1, TFPI2, and VCAM1, which were either up- or downregulated, depending on the gene (Fig. 8A to C), indicating that they are upregulated in either African green monkeys or macaques. These data suggest that both host species and SARS-CoV infection are factors involved in determining the transcription of cellular genes and that significant differences exist in the proinflammatory host response to SARS-CoV infection, corresponding to the host species.
Fig. 8.

Innate host responses to SARS-CoV infection in African green monkeys and macaques. (A to C) Gene expression profiles showing differentially expressed genes involved in development of ARDS (A), NF-κB target genes (B), and cytokine/chemokine genes (C) of African green monkeys and macaques compared to their PBS controls and from the direct contrast of SARS-CoV-infected African green monkeys and macaques (AGM-Mac). Gene sets were obtained from the Ingenuity Pathways Knowledge Base and were changed ≥2-fold (absolute) in at least one of the African green monkey or macaque groups compared to their respective PBS-infected controls. The data presented are error-weighted fold change averages for four animals per group. The genes shown in red were upregulated, those in green were downregulated, and those in gray were not significantly differentially expressed in infected animals relative to PBS-infected animals (log [base 2]-transformed expression values; the minimum and maximum values of the color range were −4 and 4). The gene accession numbers are available in Table S1 in the supplemental material.
In order to understand the host responses in the context of host species, we directly compared lung samples from PBS-infected African green monkeys (n = 2) and macaques (n = 2). LIMMA analysis revealed that 2,198 gene transcripts were differentially expressed (change, ≥2-fold; P < 0.05), with categories such as inflammatory disease, cell death, cell movement, and cellular growth and proliferation among the top significantly differentially regulated functions (P < 0.005). Of the 2,198 differentially expressed genes, 917 were also differentially expressed in the direct comparison of SARS-CoV-infected African green monkeys and macaques, but they did not include genes for cytokines/chemokines, such as CCL20, CCL3, and SPP1. Our data indicate that significant differences exist in the basal gene expression levels of African green monkeys and macaques, which may partly explain why differences in pathology were observed after SARS-CoV infection.
To obtain more insight into the differences in the host responses to SARS-CoV infection of African green monkeys and macaques, mRNA expression profiles for specific molecular pathways of infected animals of both species were compared to those of their PBS controls, but also directly against each other. Heat maps were generated for differentially regulated genes involved in ARDS (Fig. 8A), NF-κB signaling (Fig. 8B), and cytokine/chemokine signaling (Fig. 8C), which indicated that the host responses to infection with respect to these pathways in African green monkeys and macaques are different. Surprisingly, inflammatory cytokines that play a major role in mediating and amplifying ALI/ARDS or have neutrophil chemoattractant activity, such as IL-6, IL-8, CXCL1, and CXCL2 (10, 25, 55), were differentially expressed in macaques, but not in African green monkeys (Fig. 8C). TaqMan analysis confirmed that IL-6 and IL-8, key players in ALI/ARDS, were differentially expressed at significantly lower levels in African green monkeys than in young adult macaques (Fig. 9A and B), in contrast to the IFN-β levels, which were not significantly different (Fig. 9C). Moreover, in the direct comparison of the microarray data on SARS-CoV-infected African green monkeys and macaques, IL-8, CXCL1, and CXCL2 were also differentially expressed (Fig. 8A to C). These observations suggest that classical pathways known to be involved in development of ALI/ARDS are differently regulated in these two nonhuman primate species. From previous studies, we know that in aged cynomolgus macaques, IL-8 gene expression is further upregulated (Fig. 9A) (46) and correlated with significantly more severe pathology. Because African green monkeys showed even more pathology than young adult macaques, other pathogenic pathways besides the induction of genes such as those for IL-6 and IL-8 are most likely involved in the observed pathological changes (21, 46).
Fig. 9.

Quantitative RT-PCR confirmation of cytokine/chemokine mRNA levels. Quantitative RT-PCR for IL-8 (A), IL-6 (B), IFN-β (C), SPP1 (D), CCL3 (E), and CCL20 (F) was performed on one to three separate lung samples per animal with substantial virus replication. The data presented are error-weighted (plus SEM) averages of the fold change in SARS-CoV-infected African green monkeys and macaques compared to their respective PBS-infected controls.
Interestingly, CCL3, SPP1, and CCL20 were induced strongly in African green monkeys, but not in young adult macaques (Fig. 8A to C and 9D to F). This observation was corroborated by the direct comparison of SARS-CoV-infected African green monkeys with macaques, which showed that CCL20 and SPP1 were differentially expressed (Fig. 8B and C). It is notable that both these genes were also statistically significantly upregulated in aged macaques compared to young adult macaques (Fig. 9D to F) (46). CCL3 is involved in the acute inflammatory state in the recruitment and activation of polymorphonuclear leukocytes, whereas CCL20 is strongly chemotactic for lymphocytes but weakly attracts neutrophils. Osteopontin, encoded by SPP1, has been implicated in a broad range of disease processes, including chronic obstructive pulmonary disease, asthma, multiple sclerosis, rheumatoid arthritis, and cardiovascular disease (34, 54). Osteopontin has both matrix protein-like and cytokine-like properties and is expressed by different cell types of the immune system (34, 54). To determine which cell types are responsible for the osteopontin expression, immunohistochemical detection of the protein was performed on lung sections of SARS-CoV-infected African green monkeys (Fig. 10). SARS-CoV-infected African green monkeys showed marked staining of osteopontin in the alveoli in areas with lesions. Strong expression of osteopontin was observed in infiltrating cells, and staining with an antibody directed against CD68 indicated that the infiltrating cells that expressed osteopontin were mainly macrophages (Fig. 9). Similar staining was performed on the lungs of young adult and aged macaques, also showing that mainly macrophages were stained for osteopontin expression (data not shown). The immunohistochemistry data, however, do not allow quantification of the amounts of osteopontin expression in the different species.
Fig. 10.

Osteopontin expression in macrophages in SARS-CoV-infected African green monkeys. The same lung sections of SARS-CoV-infected African green monkeys were stained for expression of osteopontin (A) or CD68 (B), a marker for macrophages. The sections were stained with immunoperoxidase and counterstained with hematoxylin. Original magnifications, ×20.
DISCUSSION
The acute onset of severe lung injury supposedly depends on activation of the oxidative-stress machinery that is coupled with innate immunity and activates transcription factors, resulting in a proinflammatory host response (21). Proinflammatory cytokines, such as IL-1β, IL-8, CXCL2, and IL-6, play a major role in mediating and amplifying ALI/ARDS by stimulating chemotaxis and activation of neutrophils (10, 52, 55). Our previous studies with regard to SARS-CoV-associated ALI/ARDS in young adult and aged macaques are in line with these observations. The acute phase of SARS is characterized by infiltration of inflammatory cells, edema, the formation of hyaline membranes, and proliferation characterized by type II pneumocyte hyperplasia (16, 46). In addition, severe pathology in macaques was associated with increased expression of proinflammatory cytokines, including CXCL10, CCL2, IL-6, and IL-8, which was also observed in human SARS cases (3, 19, 24, 41, 46, 51, 56, 58). Although great advances have been made in understanding ALI/ARDS disease processes, the different molecular mechanisms that control the outcome are still poorly understood. By comparing and contrasting SARS-CoV infections in different but related host species (African green monkeys and cynomolgus macaques), we can now further delineate host factors involved in disease outcome.
The nature of acute lung injury observed after SARS-CoV infection in young adult African green monkeys is in general similar to the pathology observed after SARS-CoV infection in young adult macaques (46). Moreover, virus replication levels were not significantly different between the two species. SARS-CoV-infected African green monkeys, however, developed more severe pathology, with hyaline membranes, than young adult macaques, for which hyaline membranes were not observed (46). African green monkeys seem to be the only animal species that shows relatively severe pathology when infected with SARS-CoV as young adults. These data are in line with previous observations in SARS-CoV-infected African green monkeys (32). Comparative gene expression analyses in African green monkeys and macaques upon SARS-CoV infection revealed that both antiviral and proinflammatory pathways are strongly induced upon infection. However, many individual genes within these pathways are differently induced by African green monkeys and macaques. To our surprise, inflammatory cytokines that play a major role in mediating and amplifying ALI/ARDS or have neutrophil chemoattractant activity, such as IL-8, IL-6, CXCL1, and CXCL2 (10, 25, 55), were upregulated in young adult macaques, but not in African green monkeys. Aged macaques display an even stronger proinflammatory host response to infection than young adult macaques, with significantly increased expression of inflammatory genes, including IL-8, suggesting that exacerbated expression of proinflammatory cytokines may result in development of severe pathology (46). Thus, the host response in African green monkeys upon SARS-CoV infection is not similar to the host response in young adult or aged macaques.
Although induction of classical proinflammatory mediators, such as IL-8 and IL-6, known to be involved in development of ALI/ARDS, was not observed, other proinflammatory cytokines and chemokines, including SPP1, CCL20, and CCL3, were upregulated to a significantly greater extent in African green monkeys than in young adult macaques. Previous studies revealed that CCL3 plays a critical role in promoting lung inflammation and fibrosis, that significantly elevated levels of CCL3 were observed in mice infected with highly pathogenic influenza A virus compared to those in mice infected with low-pathogenic virus, and that CCL3 has been associated with fatal outcomes in human H5N1 influenza A virus infections (6, 38, 40, 45). Osteopontin is involved in the development of various inflammatory diseases, including multiple sclerosis, rheumatoid arthritis, and atherosclerosis (4, 13, 33, 34, 39). Moreover, osteopontin has been recognized as a key cytokine involved in immune cell recruitment and type 1 (Th1) cytokine expression, and it plays a role in the development of lung fibrosis (2, 9, 34). In addition, osteopontin regulates infiltration and activation of fibroblasts, considered the main mediators of tissue fibrosis. (35). In both murine lung fibrosis and idiopathic pulmonary fibrosis, osteopontin is among the most prominently expressed cytokines (34), and osteopontin is strongly expressed by alveolar macrophages in the lungs of acute respiratory distress syndrome patients (50). This observation was underscored by our finding that osteopontin was mainly expressed in infiltrating macrophages in SARS-CoV-infected African green monkeys. The SPP1 promoter is responsive to many agents, including cytokines, growth factors, hormones, and angiotensin II (30, 34). The latter is of interest, as angiotensin II is a component of the renin-angiotensin system and promotes disease pathogenesis, induces lung edema, and impairs lung function (22). ACE2, the SARS-CoV receptor, cleaves angiotensin II and is a negative regulator of the system. In mice, it was shown that recombinant SARS-CoV spike protein downregulates ACE2 expression and thereby promotes lung injury (26). These data provide a potential link between SARS-CoV infection, SPP1 gene expression, and lung injury. Given the fact that both SPP1 and CCL20 genes were upregulated in aged SARS-CoV-infected macaques (46), activation of these genes does not seem to be restricted per se to African green monkeys and may be involved in the exacerbated SARS-CoV disease in aged macaques.
Several recent studies have provided evidence for a critical role of macrophages in SARS. Depletion of alveolar macrophages before inoculation with a mouse-adapted version of SARS-CoV results in enhanced T-cell responses and protection of BALB/c mice from lethal infection (59, 60). Moreover, SARS-CoV-infected STAT1−/− mice exhibited dysregulation of T-cell and macrophage differentiation, leading to development of alternatively activated macrophages that mediate a profibrotic environment, suggesting that macrophage dysregulation causes formation of prefibrotic lesions in the lungs of SARS-CoV-infected mice (61). Based on our comparative analysis of SARS-CoV-associated acute lung injury in African green monkeys and macaques, we hypothesize that what is viewed as ARDS, consisting of infiltration of inflammatory cells, edema, the formation of hyaline membranes, and type II pneumocyte hyperplasia, may be caused by regulation of the transcription of a different array of proinflammatory genes. Besides a common ARDS “injury pathway” with neutrophil influx and expression of IL-1β, IL-8, and IL-6, other pathways that can amplify ARDS may exist (21). Possibly, a prominent influx of macrophages expressing proinflammatory cytokines, such as osteopontin, may play a role in the development of ALI/ARDS, as well. In this respect, it is notable that an influx of neutrophils and macrophages was observed in SARS-CoV-infected aged macaques on day 4 postinfection (45), whereas it seemed that mainly macrophages were infiltrating the lungs in African green monkeys.
The fact that closely related nonhuman primates show intrinsic differences in the host response to virus infection is not unprecedented. African green monkeys infected with simian immunodeficiency virus (SIV) do not develop chronic immune activation and AIDS, in contrast to humans infected with human immunodeficiency virus or macaques infected with SIV (11, 23, 31, 48). The nonpathogenic SIV infections differ from pathogenic infections by their benign clinical course, maintenance of peripheral CD4+ T-cell counts at normal levels, absence of chronic immune activation by controlling levels of type I interferon, and preservation of normal levels of mucosal T helper type 17 cells (11, 23, 31, 48). One may speculate that the evolutionary adaptations in the innate immune system that allow coexistence of primate lentiviruses with the host without causing AIDS also result in a different host response to other acute viral infections, such as that observed for SARS-CoV infection in African green monkeys. Alternatively, in both macaques and African green monkeys, SARS-CoV antigen was detected in tracheal and bronchiolar epithelial cells early on day 1 or 2 postinfection (reference 32 and our unpublished observations). African green monkeys, however, may display more severe pathology than macaques upon infection with SARS-CoV as a consequence of the broader range of cell types infected at day 4 postinfection. Studies in mice revealed that differences in tropism are associated with the outcome of the SARS-CoV infection (44).
Our studies provide evidence that the use of (closely) related host species reveals valuable insights into virus-host interactions (14). If our assumption that different injury pathways can lead to development of ALI/ARDS is correct, it is important to realize that interventions that ameliorate pathology by modulating the host response in one host species may be ineffective in another species. Osteopontin induction by mediators of acute inflammation, such as TNF-α, IL-1β, angiotensin II, and NF-κB (30), may be sensitive to the inhibitory action of type I IFN (5) and partly explain why type I interferon treatment of aged SARS-CoV-infected macaques resulted in mitigation of pathology (46). Ultimately, these studies need to be extended to the human host in order to identify markers of host susceptibility and severity of disease that enable the rational design of multipotent biological response modifiers to combat ALI/ARDS induced by different agents.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Dias d'Ullois, M. A. Bijl, and F. Zaaraoui-Boutahar for technical assistance.
This work was funded by NIH grant HL080621-01A1 and the Virgo consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative, and partially funded by the Dutch government (BSIK 03012), Netherlands.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 16 February 2011.
REFERENCES
- 1. Benjamini Y., Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Soc. B 57:289–300 [Google Scholar]
- 2. Berman J. S., et al. 2004. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L1311–L1318 [DOI] [PubMed] [Google Scholar]
- 3. Cameron M. J., et al. 2007. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 81:8692–8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chabas D., et al. 2001. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294:1731–1735 [DOI] [PubMed] [Google Scholar]
- 5. Chen M., et al. 2009. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T cells in MS. Eur. J. Immunol. 39:2525–2536 [DOI] [PubMed] [Google Scholar]
- 6. de Jong M. D., et al. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12:1203–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Lang A., et al. 2007. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV-infected macaques. PLoS Pathog. 3:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng R., et al. 2008. Distinctly different expression of cytokines and chemokines in the lungs of two H5N1 avian influenza patients. J. Pathol. 216:328–336 [DOI] [PubMed] [Google Scholar]
- 9. Eferl R., et al. 2008. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc. Natl. Acad. Sci. U. S. A. 105:10525–10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan J., Ye R. D., Malik A. B. 2001. Transcriptional mechanisms of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L1037–L1050 [DOI] [PubMed] [Google Scholar]
- 11. Favre D., et al. 2009. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 5:e1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fouchier R. A., et al. 2003. Aetiology: Koch's postulates fulfilled for SARS virus. Nature 423:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gravallese E. M. 2003. Osteopontin: a bridge between bone and the immune system. J. Clin. Invest. 112:147–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haagmans B. L., Andeweg A. C., Osterhaus A. D. 2009. The application of genomics to emerging zoonotic viral diseases. PLoS Pathog. 5:e1000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haagmans B. L., et al. 2004. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat. Med. 10:290–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haagmans B. L., Osterhaus A. D. 2006. Nonhuman primate models for SARS. PLoS Med. 3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haddad J. J. 2002. Science review: redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for nuclear factor-kappaB. Crit. Care 6:481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hon K. L., et al. 2003. Clinical presentations and outcome of severe acute respiratory syndrome in children. Lancet 361:1701–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang K. J., et al. 2005. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 75:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huber W., von Heydebreck A., Sultmann H., Poustka A., Vingron M. 2002. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18(Suppl. 1):S96–S104 [DOI] [PubMed] [Google Scholar]
- 21. Imai Y., et al. 2008. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imai Y., et al. 2005. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacquelin B., et al. 2009. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119:3544–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Y., et al. 2005. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 171:850–857 [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi Y. 2006. Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 26:307–316 [DOI] [PubMed] [Google Scholar]
- 26. Kuba K., et al. 2005. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11:875–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuiken T., et al. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leung C. W., Chiu W. K. 2004. Clinical picture, diagnosis, treatment and outcome of severe acute respiratory syndrome (SARS) in children. Paediatr. Respir. Rev. 5:275–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 30. Lorenzen J. M., et al. 2010. Angiotensin II receptor blocker and statins lower elevated levels of osteopontin in essential hypertension—results from the EUTOPIA trial. Atherosclerosis 209:184–188 [DOI] [PubMed] [Google Scholar]
- 31. Mandl J. N., et al. 2008. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 14:1077–1087 [DOI] [PubMed] [Google Scholar]
- 32. McAuliffe J., et al. 2004. Replication of SARS coronavirus administered into the respiratory tract of African Green, rhesus and cynomolgus monkeys. Virology 330:8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morimoto J., Kon S., Matsui Y., Uede T. 2010. Osteopontin as a target molecule for the treatment of inflammatory diseases. Curr. Drug Targets 11:494–505 [DOI] [PubMed] [Google Scholar]
- 34. O'Regan A. 2003. The role of osteopontin in lung disease. Cytokine Growth Factor Rev. 14:479–488 [DOI] [PubMed] [Google Scholar]
- 35. Pardo A., et al. 2005. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2:e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peiris J. S., et al. 2003. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361:1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peiris J. S., Guan Y., Yuen K. Y. 2004. Severe acute respiratory syndrome. Nat. Med. 10:S88–S97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perrone L. A., Plowden J. K., Garcia-Sastre A., Katz J. M., Tumpey T. M. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4:e1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petrow P. K., et al. 2000. Expression of osteopontin messenger RNA and protein in rheumatoid arthritis: effects of osteopontin on the release of collagenase 1 from articular chondrocytes and synovial fibroblasts. Arthritis Rheum. 43:1597–1605 [DOI] [PubMed] [Google Scholar]
- 40. Quintero P. A., Knolle M. D., Cala L. F., Zhuang Y., Owen C. A. 2010. Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J. Immunol. 184:1575–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reghunathan R., et al. 2005. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roberts A., et al. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roberts A., et al. 2005. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J. Virol. 79:5833–5838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rockx B., et al. 2009. Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J. Virol. 83:7062–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith R. E., et al. 1995. A role for C-C chemokines in fibrotic lung disease. J. Leukoc. Biol. 57:782–787 [DOI] [PubMed] [Google Scholar]
- 46. Smits S. L., et al. 2010. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 6:e1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smyth G. K. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 48. Sodora D. L., et al. 2009. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat. Med. 15:861–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Subbarao K., Roberts A. 2006. Is there an ideal animal model for SARS? Trends Microbiol. 14:299–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takahashi F., et al. 2004. Osteopontin is strongly expressed by alveolar macrophages in the lungs of acute respiratory distress syndrome. Lung 182:173–185 [DOI] [PubMed] [Google Scholar]
- 51. Tang N. L., et al. 2005. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin. Chem. 51:2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsushima K., et al. 2009. Acute lung injury review. Intern. Med. 48:621–630 [DOI] [PubMed] [Google Scholar]
- 53. Tukey J. W. 1977. Some thoughts on clinical trials, especially problems of multiplicity. Science 198:679–684 [DOI] [PubMed] [Google Scholar]
- 54. Wang K. X., Denhardt D. T. 2008. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19:333–345 [DOI] [PubMed] [Google Scholar]
- 55. Ware L. B., Matthay M. A. 2000. The acute respiratory distress syndrome. N. Engl. J. Med. 342:1334–1349 [DOI] [PubMed] [Google Scholar]
- 56. Wong C. K., et al. 2004. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 136:95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wong G. W., Li A. M., Ng P. C., Fok T. F. 2003. Severe acute respiratory syndrome in children. Pediatr. Pulmonol. 36:261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Y., et al. 2004. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect. Immun. 72:4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhao J., Zhao J., Perlman S. 2010. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 84:9318–9325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao J., Zhao J., Van Rooijen N., Perlman S. 2009. Evasion by stealth: inefficient immune activation underlies poor T cell response and severe disease in SARS-CoV-infected mice. PLoS Pathog. 5:e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zornetzer G. A., et al. 2010. Transcriptomic analysis reveals a mechanism for a pre-fibrotic phenotype in STAT1 knockout mice during SARS-coronavirus infection. J. Virol. 84:11297–11309 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.