

Abstract
The relevance of simian/human immunodeficiency virus (SHIV) infection of macaques to HIV-1 infection in humans depends on how closely SHIVs mimic HIV-1 transmission, pathogenesis, and diversity. Circulating HIV-1 strains are predominantly subtypes C and A and overwhelmingly require CCR5 for entry, yet most SHIVs incorporate CXCR4-using subtype B envelopes (Envs). While pathogenic subtype C-based SHIVs have been constructed, the subtype A-based SHIVs (SHIV-As) constructed to date have been unable to replicate in macaque cells. To understand the barriers to SHIV-A replication in macaque cells, HIVAQ23/SIVvif was constructed by engineering a CCR5-tropic subtype A provirus to express SIV vif, which counters the macaque APOBEC3G restriction. HIVAQ23/SIVvif replicated poorly in pig-tailed macaque (Ptm) lymphocytes, but viruses were adapted to Ptm lymphocytes. Two independent mutations in gp120, G312V (V3 loop) and A204E (C2 region), were identified that increased peak virus levels by >100-fold. Introduction of G312V and A204E to multiple subtype A Envs and substitution of G312 and A204 with other residues increased entry into Ptm cells by 10- to 100-fold. G312V and A204E Env variants continued to require CCR5 for entry but were up to 50- and 200-fold more sensitive to neutralization by IgG1b12 and soluble CD4 and had a 5- to 50-fold increase in their ability to utilize Ptm CD4 compared to their wild-type counterparts. These findings identify the inefficient use of Ptm CD4 as an unappreciated restriction to subtype A HIV-1 replication in Ptm cells and reveal amino acid changes to gp120 that can overcome this barrier.
INTRODUCTION
Nonhuman primate (NHP) models of HIV/AIDS are important tools for studying aspects of HIV-1 transmission, pathogenesis, and immunity. The most commonly used NHPs in these studies are Asian macaques, particularly rhesus macaques (Rhm; Macaca mulatta) and pig-tailed macaques (Ptm; Macaca nemestrina) (2). HIV-1 is thought to be able to utilize the macaque CD4 and CXCR4/CCR5 receptors for viral entry (13, 28); however, HIV-1 replication is restricted by other factors, most notably, TRIM5α in Rhm and the APOBEC3 family of cytidine deaminases in both species (15, 36, 58). Because of these species-specific restrictions, the direct study of HIV-1 infection and pathogenesis in macaques is not possible, and instead research has focused on the development of SIV/HIV chimeric viruses (SHIVs). SHIVs have traditionally been constructed by inserting the region of the HIV-1 genome encompassing the tat, rev, vpu, and env genes into the context of the pathogenic SIVmac239 backbone (55). More recently, directed approaches that rely on engineering HIV-1 to encode SIV gag and vif sequences to specifically counter macaque TRIM5α and APOBEC3 have led to the creation of minimal SHIVs that primarily encode HIV-1 proteins (16, 24). The env gene is a particularly important component of SHIVs, because the envelope (Env) determines viral tropism and is the target of neutralizing antibodies, which are considered an important part of the protective HIV-1 immune response (reviewed in reference 37).
If SHIVs are to be effective as predictors of human disease and vaccine efficacy, they should closely mimic the transmitted strains in human infection. The Envs from most circulating strains of HIV-1 require CCR5 as a coreceptor for entry, whereas many of the current SHIVs make use of envelopes from CXCR4-tropic or dual-tropic clones, such as NL4-3, HXB2, HIVSF33, and HIV89.6 (31, 35, 51, 55). Moreover, the Envs encoded by these SHIVs are highly sensitive to neutralization compared to circulating HIV-1 variants (4, 5, 38, 60). Among subtype B CCR5-tropic SHIVs (14, 41, 45), SHIVSF162 passaged isolates are the most commonly used; however, the SF162 Env encoded by these SHIVs is also extremely sensitive to neutralization (54, 63). Thus, current SHIVs do not provide a realistic benchmark for neutralizing antibody protection from circulating strains of HIV-1, and many also do not model the dominant CCR5-mediated mode of transmission.
The worldwide epidemic is comprised of very diverse HIV-1 genotypes, termed clades or subtypes. In sub-Saharan Africa, which carries the highest burden of new HIV-1 infections and HIV-1-related deaths, subtypes C and A predominate (17). SHIVs that are infectious to macaques have been generated using subtype C env sequences (9, 56, 57), as well as env sequences from the circulating recombinant CRF_AE, which is the most common HIV-1 subtype in Southeast Asia (20, 26). Despite the relative prominence of subtype A strains in the most afflicted regions of the world, attempts to make subtype A-based SHIVs (SHIV-As) have thus far been unsuccessful, as the SHIV-As tested to date failed to replicate in macaque cells (19).
In order to gain further insight into barriers to SHIV-A replication in macaque cells, we created HIVAQ23/SIVvif, a minimal SHIV encoding the vif gene from SIVmac239 in the context of the Q23-17 provirus, which is a CCR5-tropic subtype A HIV-1 molecular clone obtained soon after seroconversion (48). This minimal SHIV approach takes advantage of the fact that the APOBEC3-mediated restriction to HIV-1 replication in Ptm cells can be countered by SIV Vif (15) and the fact that, in contrast to rhmTRIM5α, the ptmTRIM5 isoforms and TRIMCyp do not antagonize HIV-1 infection (6, 7, 33, 62). In this study, the replicative properties and adaptation of HIVAQ23/SIVvif to Ptm cells were explored. Two adaptive mutations were identified that, when introduced into different subtype A Envs, permit much more efficient usage of ptmCD4, resulting in a dramatic increase in the infectivity of Ptm cells. These findings identify the inefficient use of ptmCD4 as a previously uncharacterized barrier to subtype A HIV-1 replication in Ptm cells and provide approaches to increase SHIV-A infection in macaque cells.
MATERIALS AND METHODS
Construction of HIVAQ23/SIVvif.
HIVAQ23/SIVvif, a full-length replication-competent clone expressing vif from SIVmac239, was created from Q23Δvif (46). Q23Δvif was derived from the Q23-17 full-length molecular clone (48) and was engineered with unique SalI and MluI restriction sites at the 5′ and 3′ ends of vif, causing a frameshift in the endogenous vif gene and allowing for the insertion and expression of different vif variants (53). To make HIVAQ23/SIVvif, the entire SIVmac239 vif open reading frame was amplified from SIVmac239Δenv (a gift from David Evans) by using forward primer 5′-GAAGGTCGACATGGAGGAGGAAAAGA-3′ and reverse primer 5′-AGTGACGCGTTCATGCCAGTATTCCCAA-3′ (restriction sites are underlined). The PCR product was then digested with the SalI and MluI restriction enzymes, ligated into Q23Δvif, and verified by sequencing.
Env clones and mutagenesis.
In addition to Q23ENV.17 (50) (referred to here as Q23-17), the following plasmids expressing subtype A Envs were used in the study: QF495.23 M.ENV.A3 (4), referred to here as QF495.A3; BG505.W6M.ENV.B1 and MG505.W0M.ENV.H3 (64), referred to here as BG505.B1 and MG505.H3, respectively; and Q259.D2.26 and Q259.D2.17 (34). Additionally, the SF162P3 clone, constructed by inserting the predominant V1-V5 sequence from the SHIVSF162P3 isolate into the HIV-1SF162 Env clone (21) (a gift from Cecilia Cheng-Mayer) and the SIV Mne CL8 Env clone (47) were used.
Mutations were introduced to the subtype A Env clones by site-directed mutagenesis using primers designed using the QuikChange site-directed mutagenesis kit (Stratagene; primer sequences are available upon request) to amplify 25 ng of plasmid with Pfu Turbo (Invitrogen) under the following reaction conditions: 95°C for 5 min, followed by 18 cycles of 95°C for 30 s, 55°C for 1 min, and 68°C for 16 min. The Env mutants were sequenced through the entirety of the env open reading frame to verify that no undesired nucleotide changes had occurred.
Construction of other full-length molecular clones.
Chimeric full-length molecular clones were constructed by digesting the subtype B pNL-DT5R (24) (a gift from Malcolm Martin) and HIVAQ23/SIVvif with EcoRI (restriction site located in vpr) and XhoI (restriction site located in nef) and ligating the heterologous fragments. The resulting chimeras were named Q/Nvpr-nef and N/Qvpr-nef to indicate the origin of the vpr-to-nef portion in each clone (Fig. 1a).
Fig. 1.

Infection of human PBMCs and Ptm lymphocytes with minimal SHIVs. (a) Schematic representation of the contribution of Q23-17, NL4-3, and SIVmac239 sequences to the minimal SHIVs used for replication experiments. E and X represent the EcoRI and XhoI sites used to make N/Qvpr-nef and Q/Nvpr-nef. (b) p24gag levels shown as a function of time postinfection in human PBMCs and Ptm lymphocytes. The data points represent the average measurements from duplicate infected cultures. The figure key is shown in the top plot, and viruses with an asterisk were used at a 10-fold-higher MOI for infection of Ptm cells. The results are representative of at least three independent experiments.
The HIVAQ23/SIVvif provirus was engineered to express different env genes of interest by using a previously described method (49). In short, HIVAQ23/SIVvif was digested with SmaI (restriction site located in vpr) and XhoI to excise an ∼3-kb sequence encompassing the Q23-17 env gene. Heterologous env genes were then introduced into HIVAQ23/SIVvif by digesting the env clones of interest with SmaI and XhoI and ligating the fragment into HIVAQ23/SIVvif.
Virus production and titration.
HEK 293T cells (referred to throughout as 293Ts) were used to produce all virus preparations and were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS) and 2 mM l-glutamine.
Full-length replication-competent viruses were produced by transfecting 293T cells using polyethyleneimine (PEI; Polysciences). Briefly, 2 × 106 293Ts were plated 24 h prior to transfection in a T75 tissue culture flask. The next day 6 μg of DNA was incubated with 60 μg of PEI in 600 μl of serum-free DMEM for 10 min before adding the mixture to cells. Viral supernatants were collected 72 h after transfection, filtered through a 0.22-μm filter (Millipore Corporation), and stored at −80°C until use.
The following env-deficient HIV-1 proviruses were used to generate pseudoviruses: Q23Δenv and pLai3ΔenvLuc2 (a gift from Michael Emerman), both of which have been described previously (34, 65), and Q23Δenv-GFP, which was constructed by subcloning enhanced green fluorescent protein (eGFP) from pEGFPN1 (Clontech) into Q23Δenv. For this purpose, BamHI and NotI sites were introduced at nucleotides 31 and 63 in the nef open reading frame of Q23Δenv, and the eGFP sequence was introduced using these same restriction sites.
Pseudoviruses were produced by cotransfecting 293T cells with one of the plasmids encoding env-deficient proviruses described above and plasmids encoding env clones at a 2:1 mass ratio. To do this, 2.5 × 105 293T cells were plated in each well of a six-well dish 24 h prior to transfection. For each well, 1 μg of total DNA was mixed with 10 μg of PEI in 100 μl of serum-free DMEM. In some cases, 3 μl of Fugene 6 (Roche) was used in place of PEI per the manufacturer's instructions. Pseudoviruses lacking Env [Env(−)] were used to determine the background levels of some assays and were generated by cotransfecting the empty pCI-neo (Promega) mammalian expression vector in place of an env-expressing plasmid. Viral supernatants were harvested 48 to 72 h posttransfection and cleared of cellular debris by centrifuging at 1,300 rpm for 5 min. In some cases, cleared supernatants were concentrated ∼20- to 50-fold by using Amicon Ultra 10K filters (Millipore Corporation). All viral preparations were frozen at −80°C until use.
Viral titers were determined by infecting TZM-bl reporter cells (NIH AIDS Research and Reference Reagent Program) with thawed cell-free virus in the presence of 10 μg/ml of DEAE-dextran. Forty-eight hours later, the cells were fixed and stained for beta-galactosidase activation, and blue foci were counted to obtain infectious titers. Infectious titers were reported as the number of infectious particles (IP)/ml (5).
Virus replication assays.
Full-length proviral clones were assessed for the production of infectious virus in human peripheral blood mononuclear cells (PBMCs), primary Ptm PBMCs, and immortalized Ptm lymphocytes. PBMCs from HIV-negative donors were isolated by the Ficoll gradient method, activated for 72 h with 10 U of phytohemagglutinin M/ml (Roche), and maintained in RPMI 1640 medium (Invitrogen) with 10% heat-inactivated FCS, 2 mM l-glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 10 U/ml of interleukin-2 (Roche) for 48 h prior to infection and thereafter. Ptm PBMCs were isolated using a 95% Ficoll gradient, activated for 40 h in with 4 U of phytohemagglutinin M/ml, and maintained in RPMI 1640 medium with 25 mM HEPES, 20% heat-inactivated FCS, 2 mM l-glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 100 U/ml of recombinant human interleukin-2 (Roche). The immortalized Ptm lymphocytes used in this study have been described previously (40) (a gift from Nina Munoz and Hans-Peter Kiem). Ptm lymphocytes were maintained in Iscove's modified Dulbecco's medium (Invitrogen) with 10% heat-inactivated FCS, 2 mM l-glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 100 U of interleukin-2/ml (Chiron Corporation).
Viral stocks were mixed with either 3 × 106 stimulated donor human PBMCs, 2 × 106 Ptm PBMCS, or 1 × 106 immortalized Ptm lymphocytes at a multiplicity of infection (MOI) of 0.02 or 0.2 in a final volume of 250 μl. After spinoculation (43) at room temperature for 2 to 3 h at 1,200 × g, cells were washed three times in 1.5 ml of the appropriate medium and resuspended into duplicate 600-μl cultures in a 48-well dish. Cultures were maintained for 15 days, with approximately two-thirds of the medium being replaced every 3 days. The p24gag levels were determined by measuring cleared culture supernatants with a p24gag antigen enzyme-linked immunosorbent assay kit (ZeptoMetrix, Buffalo, NY).
Luciferase assays.
Infections of 8 × 104 immortalized Ptm lymphocytes were performed in triplicate by spinoculation with luciferase reporter viruses for 2 h at an MOI of 0.2 in a final volume of 100 μl. Infections were allowed to proceed for 72 h before lysis with Brite-Glo reagent (Invitrogen) according to the manufacturer's guidelines. Luciferase activity was immediately read on a Fluoroskan Ascent FL luminometer (Thermo LabSystems) with a 1,000-ms integration period. Infections with Env(−) pseudoviruses were used to determine background levels for the assay.
Long-term culturing of HIVAQ23/SIVvif and sequencing of outgrowth variants.
Cultures of 1 × 106 immortalized Ptm lymphocytes were infected with HIVAQ23/SIVvif at an MOI of 0.2 in an initial volume of 1 ml in 12-well dishes. Cells were maintained at a concentration of 0.8 × 106 to 3 × 106 cells/ml without discarding cells and with at least two-thirds of the medium being replenished every 4 to 5 days. Supernatant was assessed for the presence of virus every 4 to 5 days by infecting TZM-bl cells.
Using the Qiagen blood DNA kit, total DNA was extracted from cells in cultures in which viral outgrowth was identified. The number of integrated proviral copies was determined using a previously described real-time PCR assay (3). To sequence viral variants, 1,000 copies of the integrated provirus were amplified with four nested PCRs, resulting in overlapping amplicons spanning the entirety of the HIV-1 genome. The primers and conditions used for this are available upon request. The nested PCRs were performed in triplicate, and the PCR products were detected as single prominent bands by gel electrophoresis. All reaction mixtures were treated with ExoSap (Amersham Biosciences), and the amplicons were sequenced directly without gel purification.
Infection of Ptm lymphocytes in the presence of TAK779.
TAK779-treated immortalized Ptm lymphocytes were preincubated with 1 μM TAK779 (NIH AIDS Reference and Reagent Program) for 2 h at 37°C and maintained thereafter in 1 μM TAK779. Triplicate infections of 8 × 104 TAK779-treated or untreated cells were then performed by spinoculation for 2 h with pseudotyped luciferase reporter viruses at an MOI of 0.2 in a final volume of 100 μl. Infected cultures were maintained for 72 h before lysis and subsequent measurement of luciferase activity. The percent inhibition by TAK779 was determined by comparing relative luciferase levels in TAK779-treated cells and untreated cells.
Neutralization assays.
Neutralization assays were performed using the TZM-bl neutralization assay as described previously (5). Approximately 500 IP of Q23Δenv-derived pseudoviruses was incubated with six serial 3-fold dilutions of IgG1b12 (8) (referred to here as b12) or soluble CD4 (sCD4) in a 96-well plate. One hour later, 1 × 104 TZM-bl cells were added to each well with DEAE-dextran to a final concentration of 10 μg/ml. The relative levels of infection were determined by assessing β-galactosidase activity in triplicate wells after 48 h. Median inhibitory concentrations (IC50s) were defined as the concentration of b12 or sCD4 that resulted in 50% inhibition of β-galactosidase, and these values were calculated using the linear fit model.
Construction and transient transfection of CD4 and CCR5 expression plasmids.
Human CCR5 and Ptm CCR5 (referred to here as huCCR5 and ptmCCR5, respectively) in the pBABE-puro vector were described previously (11, 25). To clone human and Ptm CD4 (referred to here as huCD4 and ptmCD4, respectively), total RNA was isolated from human PBMCs and Ptm lymphocytes by using the Qiagen RNeasy minikit. Total cDNA was obtained by reverse transcribing 2 μg of RNA with SuperScript II reverse transcriptase (Invitrogen) using oligo(dT) primers. The CD4 open reading frame was PCR amplified using forward primer 5′-GATGGATCCATGAACCGGGGAGTCCC-3′ with reverse primer 5′-GGTGTCGACTCAAATGGGGCTACATG-3′ for huCD4 and forward primer 5′-GATGGATCCATGAACCGGGGAATCCC-3′ with the same reverse primer for ptmCD4. The PCR products were digested with BamHI and SalI (underlined in the primers), cloned into the pBABE-puro plasmid (39), and verified by sequencing.
CD4 and CCR5 expression plasmids were cotransfected in equal amounts into 293T cells using Fugene 6 at a ratio of 3 μl of transfection reagent to 1 μg of DNA as per the manufacturer's protocol. In some cases, transfections were performed with 6 μg of DNA in T75 tissue culture flasks with 18 μl of Fugene in a total volume of 200 μl of serum-free DMEM.
Analysis of receptor expression levels by flow cytometry.
CD4 and CCR5 expression levels were determined by flow cytometry using allophycocyanin-conjugated mouse anti-human CD4 antibody (catalog number 551980; BD Biosciences) and phycoerythrin-conjugated mouse anti-human CCR5 (catalog number 550632; BD Biosciences). Briefly, 1 × 105 to 2 × 105 cells were washed in phosphate-buffered saline (PBS)–2% FBS and incubated in ∼100 μl of PBS–2% FBS with 2 μl of the anti-CD4 antibody and/or 5 μl of the anti-CCR5 antibody at room temperature for 30 min. The cells were then washed once in 1 ml of PBS–2% FBS and resuspended in 200 μl PBS–2% FBS for analysis by flow cytometry.
RESULTS
Replication of HIVAQ23/SIVvif in immortalized Ptm lymphocytes.
HIVAQ23/SIVvif, a minimal SHIV derived from the subtype A CCR5-tropic Q23-17 provirus, was tested for its ability to replicate in human PBMCs and immortalized Ptm lymphocytes. NL-DT5R, a CXCR4-tropic minimal SHIV that is derived from NL4-3 and encodes the vif gene and the cyclophilin A (CypA)-binding loop from SIVmac239, was used as a positive control for replication in Ptm cells (24) (Fig. 1a). In human PBMCs, HIVAQ23/SIVvif achieved peak levels of ∼300 ng p24gag/ml, slightly higher than NL-DT5R, which reached peak levels of ∼100 ng p24gag/ml. In Ptm cells, however, while NL-DT5R reached levels of ∼300 ng p24gag/ml, HIVAQ23/SIVvif reached peak levels that were ∼300-fold lower, at ∼1 ng p24gag/ml, despite the fact that infections with HIVAQ23/SIVvif were performed at a 10-fold higher MOI (Fig. 1b).
To determine which regions of the HIVAQ23/SIVvif genome were responsible for its impaired infection of Ptm lymphocytes, reciprocal chimeras between HIVAQ23/SIVvif and NL-DT5R were constructed and evaluated for their abilities to replicate in human and Ptm cells (Fig. 1a). In human PBMCs, both chimeric viruses replicated to levels of p24gag that were within the same range as HIVAQ23/SIVvif and NL-DT5R (Fig. 1b). In Ptm lymphocytes, Q/Nvpr-nef replicated to similar levels as NL-DT5R, indicating that the 5′-long terminal repeat and gag-pol region of HIVAQ23/SIVvif functioned for virus replication in Ptm cells. Conversely, N/Qvpr-nef reached a peak level of p24gag that was >2 logs lower than NL-DT5R, even with infections performed at a 10-fold-higher MOI (Fig. 1b). This suggested that the 3′ region of HIVAQ23/SIVvif, encompassing the entirety of tat, rev, vpu, and env, as well as parts of vpr and nef, was responsible for the low levels of replication in Ptm cells.
Entry of viruses bearing subtype A Envs into immortalized Ptm lymphocytes.
Given that the viral determinant for impaired replication of HIVAQ23/SIVvif in Ptm cells included the env gene,the Q23-17 Env was examined for its ability to mediate entry into Ptm cells. The Q23-17 Env was compared to the SF162P3 Env, which is derived from SHIVSF162P3, a CCR5-tropic isolate known to establish persistent spreading infection in Ptms (21). Pseudotyped luciferase reporter viruses, which were normalized based on MOI, infected Ptm lymphocytes ∼30-fold less efficiently when carrying the Q23-17 Env than those pseudotyped with the SF162P3 Env (Fig. 2).
Fig. 2.
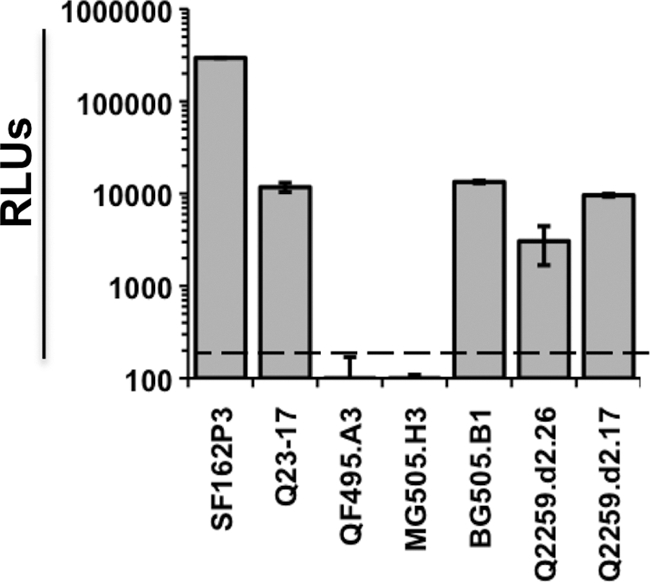
Single-cycle infection of Ptm lymphocytes with luciferase pseudoviruses bearing subtype A Envs. The y axis shows relative light units (RLUs) in cells infected with the viruses indicated on the x axis. The dashed line represents the background RLU level for the experiment in cells infected with Env(−) pseudovirus. Error bars represent the standard deviations obtained from triplicate wells. The results are representative of at least three independent experiments.
To determine whether the Q23-17 Env was typical of subtype A Envs, five additional CCR5-tropic subtype A Envs, obtained from recently infected individuals, were tested for their ability to infect immortalized Ptm lymphocytes. Viruses carrying the BG505.B1, Q259.d2.26, and Q259.d2.17 Envs mediated entry into Ptm cells to levels that were comparable to Q23-17, while the infectivities of viruses carrying the QF495.A3 and MG505.H3 Envs were below the background levels of the assay (Fig. 2). Overall, these results suggested that inefficient entry into Ptm cells is a common characteristic of subtype A Envs.
Adaptation of HIVAQ23/SIVvif to immortalized Ptm lymphocytes and identification of adaptive amino acid changes.
To determine if HIVAQ23/SIVvif could be adapted to replicate in immortalized Ptm lymphocytes, the virus was maintained in multiple long-term cultures in two independent experiments. In the first experiment, viral outgrowth was seen in one of nine cultures after approximately 35 days in culture, with viral supernatant reaching titers of >106 IP/ml in TZM-bl cells (data not shown). Sequencing of the integrated proviral genome in triplicate from cells in this culture revealed only a single G-to-T mutation, which was clearly present in all sequences and encoded a glycine-to-valine change at position 312 of the Env surface unit protein gp120 (G312V; HXB2 numbering). Introduction of the G-to-T mutation to HIVAQ23/SIVvif resulted in virus replication that achieved a >2-log increase in p24gag levels compared to wild type (Fig. 3a). However, the HIVAQ23/SIVvif G312V molecular clone showed intermediate levels of replication compared to the viral quasispecies, with the peak p24gag level of the adapted viral quasispecies being ∼2 logs higher than the G312V molecular clone. This indicated that the G312V amino acid change may only confer a portion of the full replication potential of the adapted viral quasispecies in Ptm cells. While no other dominant mutation was identified in the genome of the adapted virus that could readily explain these differences, the possibility that other compensatory mutations arose in the viral quasispecies over the 15-day period of the replication assays cannot be ruled out.
Fig. 3.

Infection of human PBMCs and immortalized Ptm lymphocytes with HIVAQ23/SIVvif carrying the G312V change (a) or the A204E change (b). The p24gag levels are shown as a function of time postinfection in human PBMCs (top graphs) and Ptm lymphocytes (bottom graphs). The data points represent the average measurements from duplicate cultures. The figure key is shown in the top plots, and Adapt. Sup. refers to the uncloned adapted viral quasispecies obtained by long-term culturing from which the mutations were isolated. The results are representative of at least three independent experiments. (c) Infection of primary Ptm PBMCs from two different donors with parental HIVAQ23/SIVvif (light gray diamonds), HIVAQ23/SIVvif G312V (dark gray triangles), or HIVAQ23/SIVvif A204E (black diamonds). The p24gag levels are shown as a function of time postinfection, and data points represent the average measurements from duplicate cultures.
In a second experiment, replicating virus was found in 2 out of 30 cultures after approximately 32 days of culturing, with titers of >105 IP/ml (data not shown). Sequencing of the integrated proviral genome from cells in each of the two independent cultures harboring outgrowth virus revealed an identical adaptive mutation: a single C-to-A nucleotide change, resulting in a predicted alanine-to-glutamic acid change at position 204 of gp120 (A204E; HXB2 numbering). Introduction of this same C-to-A nucleotide change into HIVAQ23/SIVvif resulted in a >4-log increase in p24gag levels compared to the wild-type virus (Fig. 3b), which was similar to the levels of replication observed with the adapted viral quasispecies, thus indicating that the A204E amino acid change is responsible for the increased ability of the second adapted virus to infect Ptm cells.
Viruses bearing the envelope protein with the G312V and A204E amino acid changes were also tested for replication in primary Ptm PBMCs from two different donor animals. Peak replication was achieved at day 3 or 6 for the wild-type HIVAQ23/SIVvif and at day 6 for the G312V and A204E variants. The G312V change increased peak p24gag levels by approximately 4-fold compared to wild type, and the A204E p24gag levels increased by approximately 10-fold compared to wild type (Fig. 3c). The levels of replication of these variants were considerably lower (∼10- to 100-fold) than the replication levels of the CXCR4-tropic minimal SHIV, NL-DT5R (data not shown).
Effect of amino acid changes at G312 and A204 on the ability of the Q23-17 Env to mediate entry into immortalized Ptm lymphocytes.
To examine how the G312V and A204E changes were affecting entry into Ptm cells, reporter pseudoviruses carrying the Q23-17 Env with the G312V or A204E change were used to infect Ptm lymphocytes. Levels of entry were ∼100-fold higher than wild type for the Q23-17 G312V variant and ∼50-fold higher for the Q23-17 A204E variant. These levels were generally slightly higher than those of the SF162P3 Env, in the case of the G312V substitution, and similar to the SF162P3 Env in the case of the A204E substitution (Fig. 4). Additionally, a double mutant was generated to determine whether there were any synergistic effects between the G312V and A204E substitutions. However, pseudoviruses bearing the Q23-17 double mutant Env were not infectious in TZM-bl cells and so were not examined for entry into Ptm cells (data not shown).
Fig. 4.
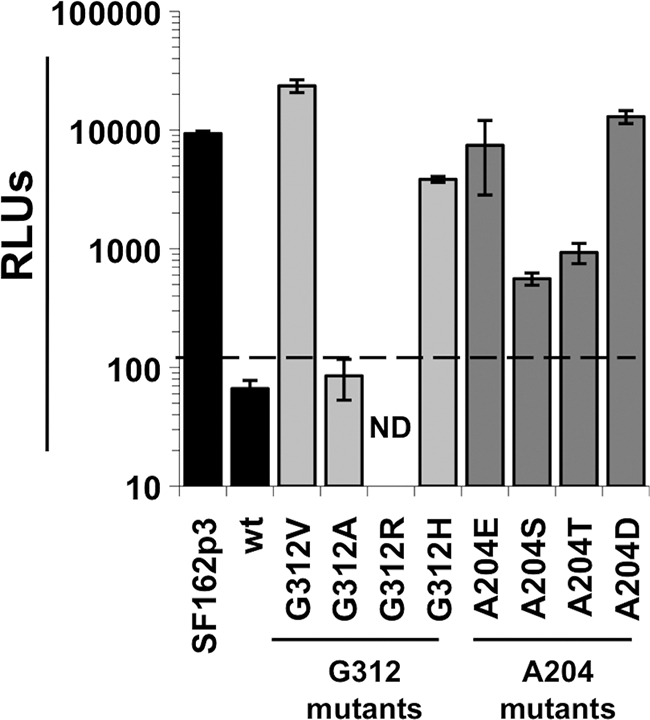
Single-cycle infection of Ptm lymphocytes with luciferase pseudoviruses bearing Q23-17 Env mutants. The y axis shows relative light units (RLUs) in cells infected with the virus variants indicated on the x axis. The dashed line represents background RLU levels observed in cells infected with Env(−) pseudovirus. Error bars represent the standard deviations obtained from triplicate wells. wt, wild-type Q23-17 envelope; ND, not determined due to insufficient titer. The results are representative of at least two independent experiments.
The adaptive A204E and G312V changes that were observed in culture are rare among HIV-1 sequences, with only three examples of the G312V substitution and seven examples of the A204E substitution observed in the >10,000 HIV-1/SIVcpz Env sequences surveyed (http://www.hiv.lanl.gov). Furthermore, changes to G312 and A204 have not been reported in any infectious SHIVs constructed to date (data not shown). The G312 residue, which is the first amino acid of the GPG(R/Q) motif located at the tip of the V3 loop (30), is conserved in >92% of Env sequences from all HIV-1 subtypes and >96% in subtype A Envs. The most common amino acid substitutions at G312 are alanine, arginine and, specific to subtype A, histidine. The A204 residue, which is located adjacent to the β3-strand of the bridging sheet in the C2 region of gp120 (52), is conserved in >98% of all HIV-1 subtypes and is invariant in subtype A. The most common substitutions for A204 are serine and threonine.
A panel of mutants encoding the most common amino acid substitutions at G312 and A204 was made to examine how these substitutions compared to the adaptive G312V and A204E substitutions for their ability to mediate entry into Ptm cells. The titers of luciferase reporter pseudoviruses carrying the mutant Q23-17 Envs were comparable to those carrying the wild-type Q23-17 in HeLa-derived TZM-bl cells, with the exception of the G312R variant, whose limited infectivity precluded it from further study (data not shown). In Ptm cells, the level of entry of the G312A mutant was comparable to wild type, whereas the G312H mutant had a >15-fold-increased level of entry. Substitutions of serine and threonine at A204 had more modest outcomes; the A204S and A204T mutants mediated entry into Ptm lymphocytes at levels that were only 3- to 5-fold greater than wild-type Q23-17 (Fig. 4).
To see if the A204E change was eliciting its effect due to the introduction of a negative charge, A204 was substituted with aspartic acid (A204D), which also bears a negative charge. Notably, the viruses carrying the A204D Env variants typically showed an approximately 100-fold decrease in infectivity on TZM-bl cells compared to the wild-type Q23-17 Env (data not shown). However, when equal MOIs of the A204D virus were used to infect Ptm cells, luciferase levels were comparable to the A204E variant that was adapted in culture (Fig. 4). All together these results indicate that it is not the presence of a specific amino acid at position 204 (E) or G312 (V) that confers increased entry into Ptm lymphocytes; other amino acid changes at these positions can also impact infectivity in Ptm cells.
Effects of G312V and A204E amino acid changes in other subtype A Envs on entry and replication in Ptm cells.
To examine whether the effects of the G312V and A204E changes were context specific, the changes were introduced individually to each of the subtype A Envs tested in Fig. 2 and the mutated variants were assayed for their ability to mediate entry into Ptm lymphocytes. Most of the reporter viruses carrying the G312V and A204E Envs had infectious titers that were comparable to the parental wild type in TZM-bl cells. This was not true, however, for the Q259.d2.26 G312V and A204E variants, which were 10- to 20-fold less infectious than the wild type, and for the Q259.d2.17 G312V and A204E variants, whose low titers precluded them from use in the assay (data not shown). In all cases where the mutants retained infectivity in human cells, introduction of the G312V and A204E changes increased entry into Ptm cells compared to the wild-type Envs. These increases in entry ranged from a 10-fold increase, in the case of the Q259.d2.26 A204E variant, to a >100-fold increase in the case of the MG505.H3 A204E variant (Fig. 5a). Despite the increase in entry, the G312V and A204E variants did not all mediate entry to the same degree as the SF162P3 Env, with levels of entry that were as much as 10-fold less, as in the case of Q259.d2.26 (Fig. 5a).
Fig. 5.

(a) Single-cycle infection of Ptm lymphocytes with luciferase pseudoviruses bearing G312V and A204E Env variants. The y axis shows relative light units (RLUs) in cells infected with the viruses indicated on the x axis. The dashed line represents the background RLU level observed in cells infected with Env(−) pseudovirus. Error bars represent the standard deviations obtained from triplicate wells. (b) Infection of human PBMCs and Ptm lymphocytes with HIVAQ23/SIVvif expressing BG505.B1 (left panel) and Q259.D2.26 (right panel) Env variants. The p24gag levels are shown as a function of time postinfection in human PBMCs (top graphs) and Ptm lymphocytes (bottom graphs). The data points represent the average measurements from duplicate cultures. The figure key is shown in the top plots. wt, the wild-type Env to which the G312V or A204E change was introduced. Data in both panels a and b are representative of three independent experiments.
To determine whether increases in entry predicted increased replication in Ptm lymphocytes, full-length molecular clones of HIVAQ23/SIVvif expressing the BG505.B1 or Q259.d2.26 Envs and their associated A204E and G312V variants were tested. Replication-competent viruses harboring the BG505.B1 Env and its variants were able to establish levels of infection reaching ∼300 ng p24gag/ml in human PBMCs, similar to those seen previously with HIVAQ23/SIVvif. The results in Ptm cells were also reminiscent of HIVAQ23/SIVvif, with viruses harboring the BG505.B1 G312V and A204E variants, reaching peak p24gag levels that were more than 3 logs greater than the wild type (Fig. 5b, left panel). However, the results for Q259.d2.26 and its G312V and A204E variants did not follow this trend. The G312V and A204E amino acid changes resulted in a 1.5- to 2-log reduction in p24gag levels in human PBMCs compared to the virus expressing the wild-type Q259.d2.d26 Env (Fig. 5b, right panel). These results were mirrored in Ptm lymphocytes, where the G312V and A204E changes did not appreciably increase spreading infection compared to the wild-type virus, reaching maximum p24gag levels of only 1 ng/ml.
Influence of the G312V and A204E amino acid changes on coreceptor usage.
To establish if the G312V and A204E amino acid changes were exerting their effects by causing a change in coreceptor usage, Ptm lymphocytes were treated with saturating amounts of the CCR5 antagonist TAK779 and then infected with luciferase pseudoviruses carrying the G312V and A204E Env variants. Infection mediated by the CCR5-tropic positive-control SF162P3 was 100% inhibited by TAK779 and, as expected for a CXCR4-tropic Env, treatment with TAK779 had relatively little effect on infection mediated by the NL-DT5R Env (Table 1). Much like SF162P3, entry by the G312V and A204E variants was greatly inhibited (98 to 100%) by TAK779 (Table 1), thus indicating that Envs with the G312V and A204E changes continued to require CCR5 as a coreceptor for entry into Ptm lymphocytes.
Table 1.
Inhibition of G312V and A204E variants by TAK779
| Clone | Residue change | % inhibitiona |
|---|---|---|
| Q23-17 | G312V | 100 ± 0 |
| A204E | 98 ± 1 | |
| QF495.A3 | G312V | 99 ± 1 |
| A204E | 100 ± 0 | |
| MG505.H3 | G312V | 100 ± 0 |
| A204E | 100 ± 0 | |
| BG505.B1 | G312V | 100 ± 0 |
| A204E | 99 ± 0 | |
| Q259.D2.26 | G312V | 100 ± 0 |
| A204E | 100 ± 0 | |
| SF162P3 | 100 ± 0 | |
| NL-DT5R | 15 ± 21 |
Percent inhibition of luciferase activity in cells treated with 1 μM TAK779, compared to untreated cells (averages ± standard deviations from two independent experiments).
Neutralization of the G312V and A204E variants by b12 and sCD4.
The b12 monoclonal antibody and sCD4 were used to probe differences that the G312V and A204E variants may be causing in Env conformation and interaction with CD4. None of the viruses bearing the wild-type Envs were sensitive to neutralization by b12 (IC50, >50 μg/ml), as had been observed previously (5, 64). The G312V and A204E amino acid changes had variable effects on neutralization by b12, depending on the viral context (Table 2). For example, the QF495.A3, Q259.d2.26, and MG505.H3 G312V and A204E Env variants were susceptible to neutralization, with IC50s ranging from ∼0.6 μg/ml for the QF494.A3 A204E variant to ∼8.6 μg/ml for the MG505.H3 G312V variant. Conversely, much like their wild-type counterparts, the G312V and A204E variants of Q23-17 and BG505.B1 were resistant to b12 neutralization.
Table 2.
Neutralization of G312V and A204E variants by b12 and sCD4
| Envelope clone | Residue change | IC50 (μg/ml)a |
|
|---|---|---|---|
| b12 | sCD4 | ||
| Q23-17 | >50b | >50b | |
| G312V | >50b | 0.8 ± 0.7 | |
| A204E | >50b | 0.3 ± 0.2c | |
| QF495.A3 | >50b | 48 ± 2.8d | |
| G312V | 1.4 ± 0.1 | <0.2e | |
| A204E | 0.6 ± 0.0 | <0.2e | |
| MG505.H3 | >50b | >50b | |
| G312V | 8.6 ± 0.4 | 6.9 ± 2.0 | |
| A204E | 1.5 ± 0.4 | 0.7 ± 0.0 | |
| BG505.B1 | >50b | >50b | |
| G312V | >50b | <0.2e | |
| A204E | >50b | 0.3 ± 0.2c | |
| Q259.D2.26 | >50b | 47 ± 4.5d | |
| G312V | 2.0 ± 0.6 | 0.2 ± 0.1c | |
| A204E | 1.8 ± 0.5 | <0.2e | |
Data are averages ± standard deviations for two experiments unless otherwise noted.
IC50s from duplicate experiments were both >50.
An IC50 value of <0.2 from one experiment was set to 0.1, and the average ± standard deviation is reported.
An IC50 value of >50 from one experiment was set to 50, and the average ± standard deviation is reported.
The IC50s from duplicate experiments were both <0.2.
The effects of the amino acid changes on sensitivity to sCD4 were more dramatic and consistent. The wild-type Envs were all relatively insensitive to sCD4, with IC50s of >40 μg/ml. Introduction of the G312V and A204E changes rendered each Env highly susceptible to neutralization by sCD4, with IC50s ranging from ∼8 μg/ml to <0.2 μg/ml, the latter representing a greater-than-200-fold increase in susceptibility compared to the wild type (Table 2). There did not appear to be a correlation between the sensitivities of the G312V and A204E variants to b12 and to sCD4. This was exemplified by the Q23-17 and BG505.B1 variants, which were insensitive to b12, but exquisitely sensitive to sCD4.
Use of Ptm CD4 and CCR5 by G312V and A204E variants.
The G312V and A204E Env variants were next examined to see if they showed any differences in their ability to mediate entry using Ptm CD4 or CCR5. To do this, 293T cells were transiently transfected with CD4 and CCR5 expression plasmids in all possible combinations: huCD4/huCCR5, huCD4/ptmCCR5, ptmCD4/ptmCCR5, and ptmCD4/huCCR5 (Fig. 6a). In a given experiment, 20 to 50% of the cells were found to be double positive for CD4 and CCR5 expression, and the levels of expression for the receptors were similar across the four receptor combinations (Fig. 6a). The cells were infected with GFP reporter pseudoviruses carrying different Envs, and GFP-positive cells were counted in triplicate wells, with the resulting titers being normalized to the huCD4/huCCR5 combination. Viruses bearing the SF162P3 Env, as well as those bearing the Env from SIV Mne CL8, a molecular clone that establishes persistent infection in Ptms (44), were used as positive controls. These positive-control viruses were able to infect cells expressing any of the four combinations of CD4 and CCR5 to comparable levels (Fig. 6b).
Fig. 6.

(a) Transient expression of human and Ptm CD4 and CCR5 in 293T cells. The y axis represents CD4 expression, and the x axis represents CCR5 expression as determined by flow cytometry. The CD4/CCR5 combination is denoted at the top of each plot, and pBabe-puro denotes cells transfected with the empty pBabe-puro vector. These plots are representative of three replicate experiments. (b) Single-cycle infection of 293T cells expressing human versus Ptm CD4 and CCR5 with GFP reporter pseudoviruses bearing wild-type subtype A Envs and G312V and A204E variants. The y axis represents viral infection relative to the huCD4/huCCR5 combination, which was set to 1 for reference. The name of the parental virus is indicated at the top of each plot, and the amino acid change is indicated on the x axis (wt refers to the wild-type Env variant). In general 20 to 300 GFP-positive cells were enumerated per well, depending on the dilution, the virus, and the CD4/CCR5 pair. No GFP-positive cells were observed when cells were infected with Env(−) pseudoviruses (data not shown). Three replicates were performed and each replicate included all of the receptor pairings, with the exception of one in which the ptmCD4/huCCR5 pair was not included. The final replicate experiment for the ptmCD4/huCCR5 pair was later performed in a separate experiment. The error bars represent the standard deviations of the means from all of the data amassed in the replicate experiments.
Viruses carrying each of the five wild-type Envs infected cells expressing ptmCCR5 to similar levels as those expressing huCCR5 but showed a 10- to 30-fold decrease in infection in cells expressing ptmCD4 versus cells expressing huCD4 (Fig. 6b). The G312V and A204E Env variants were also unchanged in their ability to mediate infection when we compared ptmCCR5 to huCCR5, but they mediated increased levels of infection via ptmCD4 by as much as 50-fold, typically reaching levels of infection that were comparable to those in cells expressing huCD4 (Fig. 6b). An exception to this was the MG505.H3 G312V variant, which increased ptmCD4-mediated entry by as little as 5-fold, still ∼10-fold lower than entry mediated by huCD4. Overall, however, these findings suggested that the subtype A Envs used in this study were limited in their capacity to infect cells expressing ptmCD4 and that the G312V and A204E variants increased the efficiency with which the Envs are able to utilize ptmCD4.
Expression of CD4 on immortalized Ptm lymphocytes.
To determine whether differences in the level of CD4 expression may contribute to differences in infectivity of the adapted HIVAQ23/SIVvif variants in Ptm versus human lymphocytes, the expression of CD4 in the immortalized Ptm lymphocytes was compared to the expression of CD4 in primary Ptm PBMCs and in human PBMCs. Expression of CD4 was equivalent between all of the cells types (Fig. 7a), suggesting that differences in infectivity cannot be explained by differences in cell surface CD4 levels. There are differences in amino acid sequence between the human and Ptm CD4 molecules, including differences in regions that play a role in envelope binding, as shown in Fig. 7b.
Fig. 7.

(a) CD4 expression on human PBMCs, Ptm PBMCs, and immortalized Ptm lymphocytes. Live cells were gated on CD4 expression, and CD4-positive cells are shown in the histogram. Shaded curves represent unstained cells and, in the PBMC expression profiles, the black and gray curves represent profiles of cells from two different donors. (b) Amino acid alignments of the D1 and D2 domains of human and Ptm CD4. Triangles above the alignment denote the beginning residue of the D1, D2, or D3 (not labeled) immunoglobin-like domains. Dots in the Ptm sequence indicate conserved residues, and in positions where the Ptm and human sequences differ, the corresponding residue encoded by Ptms is shown. The residues that are highlighted in gray were previously found to be important for gp120 binding (1).
DISCUSSION
In this study, the limited ability of subtype A Envs to use ptmCD4 for entry was identified as a major barrier to the replication of SHIV-As in macaque cells. A minimal SHIV-A, HIVAQ23/SIVvif, replicated poorly in Ptm lymphocytes, but variants displaying increased replication in Ptm lymphocytes were selected after long-term culturing. Two independent adaptive amino acid changes in Env, G312V and A204E, conferred more efficient entry into Ptm cells when introduced into the Q23-17 parental Env. These same changes conferred high levels of replication in Ptm lymphocytes when introduced into the parental HIVAQ23/SIVvif proviral clone. Importantly, increased entry was also observed when either of these changes was independently introduced into multiple subtype A Envs, suggesting that these amino acid positions have a general impact on the interactions between subtype A Env and ptmCD4. Env variants encoding G312V and A204E maintained CCR5 tropism but were much more sensitive to neutralization by sCD4 and mediated more efficient entry by ptmCD4. These findings implicate Env/CD4 interactions in the restriction of SHIV-A replication in macaque cells and provide insight into specific amino acid positions in gp120 that can enhance these interactions.
Prior studies with subtype B minimal SHIVs suggested that the inclusion of the vif gene from SIVmac239 in HIVAQ23/SIVvif would be sufficient for the virus to evade APOBEC3-mediated restriction, thus allowing for replication in Ptm cells (15). However, HIVAQ23/SIVvif replicated poorly in immortalized Ptm lymphocytes, in contrast to NL-DT5R, a CXCR4-tropic minimal SHIV that encodes both the vif gene and the CypA-binding loop of SIVmac239 (24), which achieved high levels of replication. Similar results were observed when these SHIVs were used to infect primary Ptm lymphocytes (data not shown). The SIV CypA binding loop is required to evade Rhm TRIM5α but should not be necessary for replication in Ptm lymphocytes, because the capsid-directed TRIM5 isoforms and TRIM Cyp expressed by Ptms do not restrict HIV-1 (6, 7, 15, 33, 62). Indeed, the lack of restriction to the HIV-1 capsid in Ptm cells was further verified here by using chimeras between NL-DT5R and HIVAQ23/SIVvif, which showed that the HIVAQ23/SIVvif capsid supported replication in Ptm lymphocytes. Instead, the chimeras demonstrated that it was the 3′ portion of HIVAQ23/SIVvif, including tat, rev, vpu, and env, that limited the replication of the SHIV-A in Ptm lymphocytes. These findings were consistent with previous studies using traditional SHIVs encoding the 3′ elements of HIV-1, which implicated env as the main determinant for infection of macaque cells (19). The identification of the Q23-17 Env as the reason for reduced replication in Ptm cells was definitively shown when mutations encoding the G312V or A204E amino acid changes to gp120, which were identified in viruses selected for increased replication in immortalized Ptm lymphocytes, were introduced into the parental clone, and the resulting viruses replicated in immortalized Ptm lymphocytes to levels that were comparable to NL-DT5R. Viruses bearing the G312V or A204E mutations also showed increased replication compared to the parental virus in primary Ptm PBMCs, but the differences were much more modest than those observed in immortalized CD4 lymphocytes, perhaps reflecting the fact that CD4/CCR5-positive lymphocytes comprise only a fraction of the PBMCs.
The increases in entry into Ptm lymphocytes by the G312V and A204E variants in luciferase reporter assays were not necessarily directly correlated with increases in virus spread, as exemplified by the G312V change in the context of Q23-17 and BG505.B1. The G312V changes conferred similar increases in entry in the single-cycle reporter assay as the A204E mutants, yet in the viral replication assays, peak replication levels were much lower for a full-length clone encoding the Q23-17 G312V Env compared to one encoding the BG505.B1 G312V Env. The same immortalized Ptm lymphocytes were used for both the single-cycle and replication assays, ruling out the possibility of differences in cell targets. This suggests that differences in infectivity may be due to differences in how the viruses were generated. Depending on the Env being expressed, pseudoviruses have been shown to express artificially large amounts of Env, both in the processed and unprocessed forms (18), and this can result in higher levels of infectivity compared to replication-competent virus (49).
The G312 residue, located at the tip of the V3 loop, and the A204 residue, located adjacent to the β3-strand of the bridging sheet, are in regions of gp120 that are known to mediate interaction with the coreceptor (22, 52). However, the increased ability of the G312V and A204E variants to infect Ptm cells was not due to a change in coreceptor use or an increased ability to mediate infection by ptmCCR5. Surprisingly, subtype A Envs were deficient in their ability to mediate infection of cells expressing ptmCD4, and the G312V and A204E amino acid changes rescued this deficiency. Flow cytometric analyses demonstrated that the differences in infectivity could not be explained by differences in cell surface CD4 expression. These results argue that the barrier to HIVAQ23/SIVvif replication in Ptm lymphocytes was due to inefficient use of ptmCD4 and that the G312V and A204E changes arose to circumvent this barrier.
A notable property of the G312V and A204E variants was their increased sensitivity to sCD4, with both mutations conferring increased sensitivity to sCD4 in all Envs tested, in most cases by more than 100-fold. This finding suggests that sensitivity to sCD4 may predict how well a particular envelope uses ptmCD4 for entry. If this is the case, then most circulating HIV-1 Env variants, which tend to be relatively insensitive to sCD4 (5, 32), may not promote efficient entry via ptmCD4. However, these findings also raise the interesting possibility that sensitivity to sCD4 may provide a means to identify representative transmitted Env variants that would be the best candidates for a successful SHIV. In support of this hypothesis, it is significant that many of the existing CCR5-tropic SHIVs incorporate HIV-1 Envs that are relatively sensitive to soluble CD4, including the SF162 lineage (10, 27), ADA (60), BaL (12), and the subtype C isolate HIV2873i (56).
One model that may explain how changes to G312 and A204 elicit their effect is by increasing exposure of the CD4-binding site, which could then allow for better usage of ptmCD4. G312 is found in the V3 loop, which has been shown to be a determinant for increased sensitivity to sCD4 (23, 42, 59), implying that changes to the V3 loop may participate in quaternary interactions in Env that could lead to a more exposed CD4-binding site. A204 is adjacent to the highly conserved C205 residue, whose replacement has been shown to increase the susceptibility of the HIV-1 Env to sCD4, presumably by abrogating a highly conserved disulfide bond, resulting in a more open Env conformation (61). It is possible that changes to A204, particularly the introduction of a negative charge, as in the A204E and A204D variants, may also serve to modulate this disulfide bond or may open up the Env structure via other steric interactions. The increased exposure of the CD4-binding site in the G312V and A204E variants is further supported by the sensitivity that some of the variants display to the b12 monoclonal antibody, whose epitope overlaps the CD4-binding site (8, 66). The high degree of conservation of the G312 and A204 residues may indicate that they play some role in maintaining the structural integrity of the envelope trimer. This may explain why the G312V/A204E double mutant and, in some contexts, the single G312V or A204E changes (for example, in the case of the Q259.D2.17 Env) result in an envelope that does not support efficient entry or viral replication.
Little is known regarding how differences in macaque and human CD4 impact the ability of HIV-1 Envs to mediate infection of macaque cells, although one study concluded that ptmCD4 is not a barrier to HIV-1 infection (13). The lab-adapted LAI IIIb strain of HIV-1 was used in that previous study, and the results are consistent with observations that CXCR4-tropic subtype B SHIVs are infectious in macaque cells. The differences between that study and the data presented here most likely reflect differences in the biology of lab-adapted strains compared to CCR5-tropic variants cloned directly from infected individuals, and this serves as a reminder of the difficulty of extrapolating findings with lab-adapted HIV variants to more relevant, circulating strains of HIV-1.
The primary determinants of the HIV Env-CD4 interaction have been mapped to the D1 and D2 domains of human CD4, and human and ptmCD4 differ at 17 amino acid positions in these domains. Structural studies indicate that F43 and R58 from CD4 form contacts with gp120 (29). There is an amino acid change at R58 (K in Ptm) that could alter Ptm CD4-Env interactions, but it is a conservative amino acid difference. There are other, nonconservative amino acid differences at S23 (N in Ptm) and N52 (S in Ptm). Although these residues have not been recognized as contact residues in structural analyses, they have been identified in mutagenesis studies as being critical for gp120 binding (1). Thus, disruption of CD4-gp120 binding, either by modulation of direct amino acid interactions or indirectly, through effects on binding affinity, may explain the differences in HIV-mediated entry between human and Ptm CD4 observed here.
Prior studies showed that SHIV-As are not infectious in Rhm cells (19), and our findings may provide an explanation for these results. The published sequence of Rhm CD4 is identical to the Ptm CD4 at each of the four residues noted above (data not shown). Further studies are needed to determine whether Rhm CD4 presents a similar barrier to infection as ptmCD4 and whether amino acid changes at positions A204 and G312, which are highly conserved across all subtypes, can overcome this barrier. Such studies may also help define the precise changes that alter HIV Env-macaque CD4 interactions.
The usefulness of SHIV models, particularly as tools to examine the biology of HIV-1 transmission and strategies to prevent infection, depends on how well they mimic HIV-1 transmission and early infection in humans. There are numerous barriers to HIV-1 infection in macaques, and the SHIV proviruses tested to date have required further adaptation in the animal to increase replication. The improved understanding of host restriction factors has permitted more targeted approaches to developing infectious SHIVs, although the initial chimeric viruses did not replicate to high levels in infected animals (15, 16, 24). Here, we have identified differences in CD4 as a barrier to HIV-1 infection of pig-tailed macaque cells and showed that a single amino acid change in Env is sufficient to surmount this limitation, at least for subtype A Envs. These data indicate it may be possible to develop SHIVs that are derived from more relevant HIV-1 variants with only minor modifications. However, in using these findings to develop more relevant SHIV models, it will be important to consider how changes that permit efficient CD4 interaction impact other key biological properties of the envelope, such as sensitivity to neutralization. Identifying inefficient use of CD4 as a barrier to HIV-1 infection of macaque cells provides a critical first step in the process of designing SHIVs based on biologically relevant HIV-1 variants.
ACKNOWLEDGMENTS
We thank Catherine Blish, Dara Lehman, Erica Lovelace, and Shiu-Lok Hu for many helpful discussions; Jason Kimata for comments on the manuscript; and Stephanie Rainwater for constructing Q23Δenv-GFP.
This study was supported by NIH grants AI38518 and R01 HD058304 to J.O.
Footnotes
Published ahead of print on 16 February 2011.
REFERENCES
- 1. Arthos J., et al. 1989. Identification of the residues in human CD4 critical for the binding of HIV. Cell 57:469–481 [DOI] [PubMed] [Google Scholar]
- 2. Baroncelli S., Negri D. R., Michelini Z., Cara A. 2008. Macaca mulatta, fascicularis and nemestrina in AIDS vaccine development. Expert Rev. Vaccines 7:1419–1434 [DOI] [PubMed] [Google Scholar]
- 3. Benki S., et al. 2006. Quantification of genital human immunodeficiency virus type 1 (HIV-1) DNA in specimens from women with low plasma HIV-1 RNA levels typical of HIV-1 nontransmitters. J. Clin. Microbiol. 44:4357–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blish C. A., et al. 2009. Cross-subtype neutralization sensitivity despite monoclonal antibody resistance among early subtype A, C, and D envelope variants of human immunodeficiency virus type 1. J. Virol. 83:7783–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blish C. A., Nedellec R., Mandaliya K., Mosier D. E., Overbaugh J. 2007. HIV-1 subtype A envelope variants from early in infection have variable sensitivity to neutralization and to inhibitors of viral entry. AIDS 21:693–702 [DOI] [PubMed] [Google Scholar]
- 6. Brennan G., Kozyrev Y., Hu S. L. 2008. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. U. S. A. 105:3569–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brennan G., Kozyrev Y., Kodama T., Hu S. L. 2007. Novel TRIM5 isoforms expressed by Macaca nemestrina. J. Virol. 81:12210–12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burton D. R., et al. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027 [DOI] [PubMed] [Google Scholar]
- 9. Chen Z., et al. 2000. Enhanced infectivity of an R5-tropic simian/human immunodeficiency virus carrying human immunodeficiency virus type 1 subtype C envelope after serial passages in pig-tailed macaques (Macaca nemestrina). J. Virol. 74:6501–6510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chung H. K., Suschak J., Galmin L., Rose N., Pal R. 2010. Characterization of a SHIV162P3 variant evolved in an infected rhesus macaque with persistent plasma viremia. Virus Res. 151:229–234 [DOI] [PubMed] [Google Scholar]
- 11. Deng H., et al. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666 [DOI] [PubMed] [Google Scholar]
- 12. Dey B., Del Castillo C. S., Berger E. A. 2003. Neutralization of human immunodeficiency virus type 1 by sCD4-17b, a single-chain chimeric protein, based on sequential interaction of gp120 with CD4 and coreceptor. J. Virol. 77:2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fomsgaard A., et al. 1995. Receptor function of CD4 structures from African green monkey and pig-tail macaque for simian immunodeficiency virus, SIVsm, SIVagm, and human immunodeficiency virus type-1. Viral Immunol. 8:121–133 [DOI] [PubMed] [Google Scholar]
- 14. Harouse J. M., et al. 2001. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIVSF162P3. J. Virol. 75:1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hatziioannou T., et al. 2009. A macaque model of HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 106:4425–4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hatziioannou T., et al. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. [DOI] [PubMed] [Google Scholar]
- 17. Hemelaar J., Gouws E., Ghys P. D., Osmanov S. 2006. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 20:W13–W23 [DOI] [PubMed] [Google Scholar]
- 18. Herrera C., et al. 2005. The impact of envelope glycoprotein cleavage on the antigenicity, infectivity, and neutralization sensitivity of Env-pseudotyped human immunodeficiency virus type 1 particles. Virology 338:154–172 [DOI] [PubMed] [Google Scholar]
- 19. Himathongkham S., et al. 2002. Species tropism of chimeric SHIV clones containing HIV-1 subtype-A and subtype-E envelope genes. Virology 298:189–199 [DOI] [PubMed] [Google Scholar]
- 20. Himathongkham S., et al. 2000. Simian-human immunodeficiency virus containing a human immunodeficiency virus type 1 subtype-E envelope gene: persistent infection, CD4(+) T-cell depletion, and mucosal membrane transmission in macaques. J. Virol. 74:7851–7860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu M., et al. 2003. Increased mucosal transmission but not enhanced pathogenicity of the CCR5-tropic, simian AIDS-inducing simian/human immunodeficiency virus SHIVSF162P3 maps to envelope gp120. J. Virol. 77:989–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang C. C., et al. 2007. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317:1930–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hwang S. S., Boyle T. J., Lyerly H. K., Cullen B. R. 1992. Identification of envelope V3 loop as the major determinant of CD4 neutralization sensitivity of HIV-1. Science 257:535–537 [DOI] [PubMed] [Google Scholar]
- 24. Kamada K., et al. 2006. Generation of HIV-1 derivatives that productively infect macaque monkey lymphoid cells. Proc. Natl. Acad. Sci. U. S. A. 103:16959–16964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimata J. T., et al. 1999. Coreceptor specificity of temporal variants of simian immunodeficiency virus Mne. J. Virol. 73:1655–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klinger J. M., Himathongkham S., Legg H., Luciw P. A., Barnett S. W. 1998. Infection of baboons with a simian immunodeficiency virus/HIV-1 chimeric virus constructed with an HIV-1 Thai subtype E envelope. AIDS 12:849–857 [DOI] [PubMed] [Google Scholar]
- 27. Kozak S. L., et al. 1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol. 71:873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kunstman K. J., et al. 2003. Structure and function of CC-chemokine receptor 5 homologues derived from representative primate species and subspecies of the taxonomic suborders Prosimii and Anthropoidea. J. Virol. 77:12310–12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwong P. D., et al. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LaRosa G. J., et al. 1990. Conserved sequence and structural elements in the HIV-1 principal neutralizing determinant. Science 249:932–935 [DOI] [PubMed] [Google Scholar]
- 31. Li J. T., et al. 1995. Persistent infection of macaques with simian-human immunodeficiency viruses. J. Virol. 69:7061–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li M., et al. 2006. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in southern Africa. J. Virol. 80:11776–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liao C. H., Kuang Y. Q., Liu H. L., Zheng Y. T., Su B. 2007. A novel fusion gene, TRIM5-cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS 21(Suppl. 8):S19–S26 [DOI] [PubMed] [Google Scholar]
- 34. Long E. M., Rainwater S. M., Lavreys L., Mandaliya K., Overbaugh J. 2002. HIV type 1 variants transmitted to women in Kenya require the CCR5 coreceptor for entry, regardless of the genetic complexity of the infecting virus. AIDS Res. Hum. Retrovir. 18:567–576 [DOI] [PubMed] [Google Scholar]
- 35. Luciw P. A., Pratt-Lowe E., Shaw K. E., Levy J. A., Cheng-Mayer C. 1995. Persistent infection of rhesus macaques with T-cell-line-tropic and macrophage-tropic clones of simian/human immunodeficiency viruses (SHIV). Proc. Natl. Acad. Sci. U. S. A. 92:7490–7494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mariani R., et al. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31 [DOI] [PubMed] [Google Scholar]
- 37. Mascola J. R., Montefiori D. C. 2010. The role of antibodies in HIV vaccines. Annu. Rev. Immunol. 28:413–444 [DOI] [PubMed] [Google Scholar]
- 38. Moore J. P., Ho D. D. 1995. HIV-1 neutralization: the consequences of viral adaptation to growth on transformed T cells. AIDS 9(Suppl. A):S117–S136 [PubMed] [Google Scholar]
- 39. Morgenstern J. P., Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Munoz N. M., Trobridge G. D., Kiem H. P. 2009. Ex vivo expansion and lentiviral transduction of Macaca nemestrina CD4+ T cells. J. Med. Primatol. 38:438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishimura Y., et al. 2010. Generation of the pathogenic R5-tropic simian/human immunodeficiency virus SHIVAD8 by serial passaging in rhesus macaques. J. Virol. 84:4769–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Brien W. A., Chen I. S., Ho D. D., Daar E. S. 1992. Mapping genetic determinants for human immunodeficiency virus type 1 resistance to soluble CD4. J. Virol. 66:3125–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Doherty U., Swiggard W. J., Malim M. H. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74:10074–10080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Overbaugh J., Rudensey L. M., Papenhausen M. D., Benveniste R. E., Morton W. R. 1991. Variation in simian immunodeficiency virus env is confined to V1 and V4 during progression to simian AIDS. J. Virol. 65:7025–7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pal R., et al. 2003. Characterization of a simian human immunodeficiency virus encoding the envelope gene from the CCR5-tropic HIV-1 Ba-L. J. Acquir. Immune Defic. Syndr. 33:300–307 [DOI] [PubMed] [Google Scholar]
- 46. Piantadosi A., Humes D., Chohan B., McClelland R. S., Overbaugh J. 2009. Analysis of the percentage of human immunodeficiency virus type 1 sequences that are hypermutated and markers of disease progression in a longitudinal cohort, including one individual with a partially defective Vif. J. Virol. 83:7805–7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pineda M. J., Orton B. R., Overbaugh J. 2007. A TRIM5α-independent post-entry restriction to HIV-1 infection of macaque cells that is dependent on the path of entry. Virology 363:310–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poss M., Overbaugh J. 1999. Variants from the diverse virus population identified at seroconversion of a clade A human immunodeficiency virus type 1-infected woman have distinct biological properties. J. Virol. 73:5255–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Provine N. M., Puryear W. B., Wu X., Overbaugh J., Haigwood N. L. 2009. The infectious molecular clone and pseudotyped virus models of human immunodeficiency virus type 1 exhibit significant differences in virion composition with only moderate differences in infectivity and inhibition sensitivity. J. Virol. 83:9002–9007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rainwater S. M., et al. 2007. Cloning and characterization of functional subtype A HIV-1 envelope variants transmitted through breastfeeding. Curr. HIV Res. 5:189–197 [DOI] [PubMed] [Google Scholar]
- 51. Reimann K. A., et al. 1996. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J. Virol. 70:6922–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rizzuto C. D., et al. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949–1953 [DOI] [PubMed] [Google Scholar]
- 53. Sakurai A., et al. 2004. Functional analysis of HIV-1 vif genes derived from Japanese long-term nonprogressors and progressors for AIDS. Microbes Infect. 6:799–805 [DOI] [PubMed] [Google Scholar]
- 54. Seaman M. S., et al. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84:1439–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shibata R., et al. 1991. Generation of a chimeric human and simian immunodeficiency virus infectious to monkey peripheral blood mononuclear cells. J. Virol. 65:3514–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Siddappa N. B., et al. 2009. Neutralization-sensitive R5-tropic simian-human immunodeficiency virus SHIV-2873Nipm which carries env from an infant with recent HIV clade C infection. J. Virol. 83:1422–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Song R. J., et al. 2006. Molecularly cloned SHIV-1157ipd3N4: a highly replication-competent, mucosally transmissible R5 simian-human immunodeficiency virus encoding HIV clade C Env. J. Virol. 80:8729–8738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stremlau M., et al. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 59. Sullivan N., et al. 1998. Determinants of human immunodeficiency virus type 1 envelope glycoprotein activation by soluble CD4 and monoclonal antibodies. J. Virol. 72:6332–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sullivan N., Sun Y., Li J., Hofmann W., Sodroski J. 1995. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line-passaged human immunodeficiency virus type 1 isolates. J. Virol. 69:4413–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Anken E., et al. 2008. Only five of 10 strictly conserved disulfide bonds are essential for folding and eight for function of the HIV-1 envelope glycoprotein. Mol. Biol. Cell 19:4298–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Virgen C. A., Kratovac Z., Bieniasz P. D., Hatziioannou T. 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. U. S. A. 105:3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Walker L. M., et al. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu X., et al. 2006. Neutralization escape variants of human immunodeficiency virus type 1 are transmitted from mother to infant. J. Virol. 80:835–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamashita M., Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J. Virol. 78:5670–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou T., et al. 2007. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]